
Resistance to CDK4/6 Inhibitors in Estrogen Receptor-Positive Breast Cancer



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
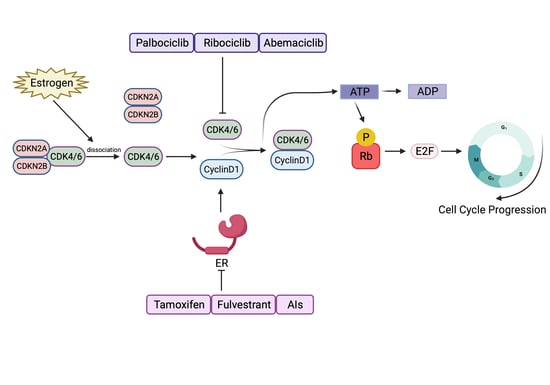
2. CDK4/6 Are Good Targets in ER+ Breast Cancer
3. The Three Approved CDK4/6 Inhibitors
3.1. Palbociclib
3.2. Ribociclib
3.3. Abemaciclib
4. Emergence of Intrinsic and Acquired Resistance to CDK4/6 Inhibitors
4.1. Intrinsic
4.2. Acquired
5. Mechanisms of Resistance to CDK4/6 Inhibitors
5.1. Cyclin D1–CDK4/6–Rb Activation
5.2. Cyclin E1/CDK2 Activation
5.3. PI3K–AKT Activation
5.4. KRAS Activation
5.5. Autophagy and Lysosomal Activity
5.6. FAT1 Loss
5.7. FGFR1 Activation
5.8. MDM2 Dysregulation
5.9. mTOR Activation
5.10. Increased G2M Activity: WEE1 and CDK7 Overexpression
6. Possible Therapies to Target Intrinsic or Acquired Resistance to CDK4/6 Inhibitors
6.1. Antiestrogens
6.2. PI3K Inhibitors
6.3. mTOR Inhibitors
6.4. FGFR Inhibitors
6.5. CDK2 Inhibitors
6.6. Chemotherapy
6.7. Autophagy Inhibitors
6.8. eIF4A Inhibitors
6.9. MDM2 Inhibitors
6.10. Immunotherapy
6.11. G2/M Checkpoint Inhibition
7. Preventing Resistance to CDK4/6 Inhibitors
8. Conclusions and Future Directions
9. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Salehiniya, H. Epidemiological Characteristics of and Risk Factors for Breast Cancer in the World. Breast Cancer 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Metzger-Filho, O.; Winer, E.P. The Natural History of Hormone Receptor-Positive Breast Cancer. Oncology 2012, 26, 688–694, 696. [Google Scholar]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 Inhibitor Resistance in ER-Positive Breast Cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Tan, E.P.; Duncan, F.E.; Slawson, C. The Sweet Side of the Cell Cycle. Biochem. Soc. Trans. 2017, 45, 313–322. [Google Scholar] [CrossRef]
- Langerak, P.; Russell, P. Regulatory Networks Integrating Cell Cycle Control with DNA Damage Checkpoints and Double-Strand Break Repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, C.; Herzog, F.; Kraft, C.; Pines, J. The Anaphase Promoting Complex/Cyclosome Is Recruited to Centromeres by the Spindle Assembly Checkpoint. Nat. Cell Biol. 2004, 6, 892–898. [Google Scholar] [CrossRef]
- Iolascon, A.; Giordani, L.; Moretti, A.; Tonini, G.P.; Lo Cunsolo, C.; Mastropietro, S.; Borriello, A.; Ragione, F.D. Structural and Functional Analysis of Cyclin-Dependent Kinase Inhibitor Genes (CDKN2A, CDKN2B, and CDKN2C) in Neuroblastoma. Pediatr. Res. 1998, 43, 139–144. [Google Scholar] [CrossRef]
- Green, J.L.; Okerberg, E.S.; Sejd, J.; Palafox, M.; Monserrat, L.; Alemayehu, S.; Wu, J.; Sykes, M.; Aban, A.; Serra, V.; et al. Direct Cdkn2 Modulation of Cdk4 Alters Target Engagement of Cdk4 Inhibitor Drugs. Mol. Cancer Ther. 2019, 18, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, H. Potential Biomarkers of Resistance to CDK4/6 Inhibitors: A Narrative Review of Preclinical and Clinical Studies. Transl. Breast Cancer Res. 2021, 2, 1–12. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of Cdk4 and Cdk6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Petrossian, K.; Kanaya, N.; Lo, C.; Hsu, P.-Y.; Nguyen, D.; Yang, L.; Yang, L.; Warden, C.; Wu, X.; Pillai, R.; et al. ERα-Mediated Cell Cycle Progression Is an Important Requisite for CDK4/6 Inhibitor Response in HR+ Breast Cancer. Oncotarget 2018, 9, 27736–27751. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (Lee011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef]
- Gul, A.; Leyland-Jones, B.; Dey, N.; De, P. A Combination of the PI3K Pathway Inhibitor plus Cell Cycle Pathway Inhibitor to Combat Endocrine Resistance in Hormone Receptor-Positive Breast Cancer: A Genomic Algorithm-Based Treatment Approach. Am. J. Cancer Res. 2018, 8, 2359–2376. [Google Scholar]
- Haricharan, S.; Punturi, N.; Singh, P.; Holloway, K.R.; Anurag, M.; Schmelz, J.; Schmidt, C.; Lei, J.T.; Suman, V.; Hunt, K.; et al. Loss of Mutl Disrupts Chk2-Dependent Cell-Cycle Control through Cdk4/6 to Promote Intrinsic Endocrine Therapy Resistance in Primary Breast Cancer. Cancer Discov. 2017, 7, 1168–1183. [Google Scholar] [CrossRef]
- Gao, A.; Sun, T.; Ma, G.; Cao, J.; Hu, Q.; Chen, L.; Wang, Y.; Wang, Q.; Sun, J.; Wu, R.; et al. LEM4 Confers Tamoxifen Resistance to Breast Cancer Cells by Activating Cyclin D-CDK4/6-Rb and ERα Pathway. Nat. Commun. 2018, 9, 4180. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor–Positive Advanced Breast Cancer? Oncology 2018, 32, 216–222. [Google Scholar]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, Her2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): A Randomised Phase 2 Study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Schwartz, G.K.; LoRusso, P.M.; Dickson, M.A.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; Courtney, R.; O’Dwyer, P.J. Phase I Study of PD 0332991, a Cyclin-Dependent Kinase Inhibitor, Administered in 3-Week Cycles (Schedule 2/1). Br. J. Cancer 2011, 104, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Lorusso, P.M.; Demichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, Dose-Escalation Trial of the Oral Cyclin-Dependent Kinase 4/6 Inhibitor PD 0332991, Administered Using a 21-Day Schedule in Patients with Advanced Cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Silvestri, M.; Cristaudo, A.; Morrone, A.; Messina, C.; Bennardo, L.; Nisticò, S.P.; Mariano, M.; Cameli, N. Emerging Skin Toxicities in Patients with Breast Cancer Treated with New Cyclin-Dependent Kinase 4/6 Inhibitors: A Systematic Review. Drug Saf. 2021, 44, 725–732. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting Cdk4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus Palbociclib versus Fulvestrant plus Placebo for Treatment of Hormone-Receptor-Positive, HER2-Negative Metastatic Breast Cancer That Progressed on Previous Endocrine Therapy (PALOMA-3): Final Analysis of the Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Lynce, F.; Blackburn, M.J.; Zhuo, R.; Gallagher, C.; Hahn, O.M.; Abu-Khalaf, M.; Mohebtash, M.; Wu, T.; Pohlmann, P.R.; Dilawari, A.; et al. Hematologic Safety of Palbociclib in Combination with Endocrine Therapy in Patients with Benign Ethnic Neutropenia and Advanced Breast Cancer. Cancer 2021, 127, 3622–3630. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for Hr-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Neven, P.; Chia, S.; Jerusalem, G.; Laurentiis, M.D.; Im, S.; Petrakova, K.; Bianchi, G.V.; Martín, M.; Nusch, A.; et al. Ribociclib plus Fulvestrant for Postmenopausal Women with Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer in the Phase III Randomized MONALEESA-3 Trial: Updated Overall Survival. Ann. Oncol. 2021, 32, 1015–1024. [Google Scholar] [CrossRef]
- Cheer, S.M.; Plosker, G.L.; Simpson, D.; Wagstaff, A.J. Goserelin: A Review of Its Use in the Treatment of Early Breast Cancer in Premenopausal and Perimenopausal Women. Drugs 2005, 65, 2639–2655. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Im, S.-A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus Endocrine Therapy for Premenopausal Women with Hormone-Receptor-Positive, Advanced Breast Cancer (MONALEESA-7): A Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. Monarch 2: Abemaciclib in Combination with Fulvestrant in Women with Hr+/Her2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. Monarch 3: Abemaciclib as Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. Monarch 1, a Phase Ii Study of Abemaciclib, a Cdk4 and Cdk6 Inhibitor, as a Single Agent, in Patients with Refractory Hr+/Her2− Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Wardley, A.M.; Zambelli, S.; Hilton, J.F.; Troso-Sandoval, T.A.; Ricci, F.; Im, S.-A.; Kim, S.-B.; Johnston, S.R.; Chan, A.; et al. Abemaciclib plus Trastuzumab with or without Fulvestrant versus Trastuzumab plus Standard-of-Care Chemotherapy in Women with Hormone Receptor-Positive, HER2-Positive Advanced Breast Cancer (MonarcHER): A Randomised, Open-Label, Phase 2 Trial. Lancet Oncol. 2020, 21, 763–775. [Google Scholar] [CrossRef]
- Corona, S.P.; Generali, D. Abemaciclib: A Cdk4/6 Inhibitor for the Treatment of Hr+/Her2− Advanced Breast Cancer. Drug Des. Devel. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Van Ommen-Nijhof, A.; Konings, I.R.; van Zeijl, C.J.J.; Uyl-de Groot, C.A.; van der Noort, V.; Jager, A.; Sonke, G.S. SONIA study steering committee Selecting the Optimal Position of CDK4/6 Inhibitors in Hormone Receptor-Positive Advanced Breast Cancer—The SONIA Study: Study Protocol for a Randomized Controlled Trial. BMC Cancer 2018, 18, 1146. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; André, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the Paloma-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Kong, T.; Xue, Y.; Cencic, R.; Zhu, X.; Monast, A.; Fu, Z.; Pilon, V.; Sangwan, V.; Guiot, M.-C.; Foulkes, W.D.; et al. Eif4a Inhibitors Suppress Cell-Cycle Feedback Response and Acquired Resistance to Cdk4/6 Inhibition in Cancer. Mol. Cancer Ther. 2019, 18, 2158–2170. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, X.; Wang, T.; Wang, J.; Yang, B.; He, Q.; Ding, L. Intrinsic and Acquired Resistance to CDK4/6 Inhibitors and Potential Overcoming Strategies. Acta Pharm. Sin. 2021, 42, 171–178. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of Cdk4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to Cdk4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 Amplification Promotes Breast Cancer Resistance to CDK4/6 Inhibitors and Loss of ER Signaling and Dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb Modulation as Common Events at Time of Resistance to Palbociclib in Hormone Receptor-Positive Breast Cancer. NPJ Breast Cancer 2018, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A Gene Expression Signature of Retinoblastoma Loss-of-Function Is a Predictive Biomarker of Resistance to Palbociclib in Breast Cancer Cell Lines and Is Prognostic in Patients with ER Positive Early Breast Cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef]
- Pandey, K.; An, H.-J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer: A Review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef]
- Guarducci, C.; Nardone, A.; Feiglin, A.; Migliaccio, I.; Malorni, L.; Bonechi, M.; Benelli, M.; Di Leo, A.; Hodgson, G.; Shapiro, G.; et al. Abstract PD7-12: Inhibition of CDK7 Overcomes Resistance to CDK4/6 Inhibitors in Hormone Receptor Positive Breast Cancer Cells. Cancer Res. 2019, 79, PD7-12. [Google Scholar]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. Neopalana: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.-C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C.; et al. MDM2 Antagonists Overcome Intrinsic Resistance to CDK4/6 Inhibition by Inducing P21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.-M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef]
- Song, G.; Ouyang, G.; Bao, S. The Activation of Akt/PKB Signaling Pathway and Cell Survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Lenihan, C.; Bouchekioua-Bouzaghou, K.; Shia, A.; Wilkes, E.; Casado-Izquierdo1, P.; Cutillas, P.; Schmid, P. Abstract P3-06-02: Characterization of Resistance to the Selective CDK4/6 Inhibitor Palbociclib in ER Positive Breast Cancer. Cancer Res. 2016, 76, P3-06-02. [Google Scholar]
- Liu, P.; Wang, Y.; Li, X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; Raimondi, F.M.; Pietranera, M.; Di Rocco, A.; Di Benedetto, L.; Miele, E.; Lazzeroni, R.; Cimino, G.; Spinelli, G.P. Assessment of Resistance Mechanisms and Clinical Implications in Patients with Kras Mutated-Metastatic Breast Cancer and Resistance to Cdk4/6 Inhibitors. Cancers 2021, 13, 1928. [Google Scholar] [CrossRef] [PubMed]
- Luangdilok, S.; Wanchaijiraboon, P.; Chantranuwatana, P.; Teerapakpinyo, C.; Shuangshoti, S.; Sriuranpong, V. Cyclin D1 Expression as a Potential Prognostic Factor in Advanced KRAS-Mutant Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2019, 8, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy Inhibition Augments the Anticancer Effects of Epirubicin Treatment in Anthracycline-Sensitive and -Resistant Triple-Negative Breast Cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Lanceta, L.; O’Neill, C.; Lypova, N.; Li, X.; Rouchka, E.; Waigel, S.; Gomez-Gutierrez, J.G.; Chesney, J.; Imbert-Fernandez, Y. Transcriptomic Profiling Identifies Differentially Expressed Genes in Palbociclib-Resistant Er+ Mcf7 Breast Cancer Cells. Genes 2020, 11, 467. [Google Scholar] [CrossRef]
- Fassl, A.; Brain, C.; Abu-Remaileh, M.; Stukan, I.; Butter, D.; Stepien, P.; Feit, A.S.; Bergholz, J.; Michowski, W.; Otto, T.; et al. Increased Lysosomal Biomass Is Responsible for the Resistance of Triple-Negative Breast Cancers to CDK4/6 Inhibition. Sci. Adv. 2020, 6, eabb2210. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Gong, Y.; Liang, X. Role of FAT1 in Health and Disease. Oncol. Lett. 2021, 21, 398. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the Fat1 Tumor Suppressor Promotes Resistance to Cdk4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed]
- Portman, N.; Milioli, H.H.; Alexandrou, S.; Coulson, R.; Yong, A.; Fernandez, K.J.; Chia, K.M.; Halilovic, E.; Segara, D.; Parker, A.; et al. MDM2 Inhibition in Combination with Endocrine Therapy and CDK4/6 Inhibition for the Treatment of ER-Positive Breast Cancer. Breast Cancer Res. 2020, 22, 87. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 Promotes the Rapid Degradation of P53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Laroche-Clary, A.; Chaire, V.; Algeo, M.-P.; Derieppe, M.-A.; Loarer, F.L.; Italiano, A. Combined Targeting of MDM2 and CDK4 Is Synergistic in Dedifferentiated Liposarcomas. J. Hematol. Oncol. 2017, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Dickson, M.A.; Antonescu, C.; Qin, L.-X.; Dooley, S.J.; Barlas, A.; Manova, K.; Schwartz, G.K.; Crago, A.M.; Singer, S.; et al. PDLIM7 and CDH18 Regulate the Turnover of MDM2 during CDK4/6 Inhibitor Therapy-Induced Senescence. Oncogene 2018, 37, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of MTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-Term Growth Inhibition in Estrogen Receptor-Positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef]
- Fallah, Y.; Demas, D.M.; Jin, L.; He, W.; Shajahan Haq, A.N. Targeting WEE1 Inhibits Growth of Breast Cancer Cells That Are Resistant to Endocrine Therapy and CDK4/6 Inhibitors. Front. Oncol. 2021, 11, 681530. [Google Scholar] [CrossRef]
- Oshi, M.; Takahashi, H.; Tokumaru, Y.; Yan, L.; Rashid, O.M.; Matsuyama, R.; Endo, I.; Takabe, K. G2M Cell Cycle Pathway Score as a Prognostic Biomarker of Metastasis in Estrogen Receptor (Er)-Positive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2921. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, S.; Ribas, R.; Simigdala, N.; Schuster, E.; Nikitorowicz-Buniak, J.; Ressa, A.; Gao, Q.; Leal, M.F.; Bhamra, A.; Thornhill, A.; et al. Tumour Kinome Re-Wiring Governs Resistance to Palbociclib in Oestrogen Receptor Positive Breast Cancers, Highlighting New Therapeutic Modalities. Oncogene 2020, 39, 4781–4797. [Google Scholar] [CrossRef]
- Martin, L.-A.; Pancholi, S.; Ribas, R.; Gao, Q.; Simigdala, N.; Nikitorowicz-Buniak, J.; Johnston, S.R.; Dowsett, M. Abstract P3-03-09: Resistance to Palbociclib Depends on Multiple Targetable Mechanisms Highlighting the Potential of Drug Holidays and Drug Switching to Improve Therapeutic Outcome. Cancer Res. 2017, 77, P3-03-09. [Google Scholar]
- Zhong, S.; Zhang, Y.; Yin, X.; Di, W. CDK7 Inhibitor Suppresses Tumor Progression through Blocking the Cell Cycle at the G2/M Phase and Inhibiting Transcriptional Activity in Cervical Cancer. Oncol. Targets Ther. 2019, 12, 2137–2147. [Google Scholar] [CrossRef]
- Malorni, L.; Curigliano, G.; Minisini, A.M.; Cinieri, S.; Tondini, C.A.; D’Hollander, K.; Arpino, G.; Bernardo, A.; Martignetti, A.; Criscitiello, C.; et al. Palbociclib as Single Agent or in Combination with the Endocrine Therapy Received before Disease Progression for Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer: TREnd Trial. Ann. Oncol. 2018, 29, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Cortes, J.; Ozyilkan, O.; Chen, S.-C.; Petrakova, K.; Manikhas, A.; Jerusalem, G.; Hegg, R.; Huober, J.; Chapman, S.C.; et al. Nextmonarch: Abemaciclib Monotherapy or Combined with Tamoxifen for Metastatic Breast Cancer. Clin. Breast Cancer 2021, 21, 181–190.e2. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting Activated PI3K/MTOR Signaling Overcomes Acquired Resistance to CDK4/6-Based Therapies in Preclinical Models of Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Enquist, J.; Iwig, J.; Binnerts, M.E.; Jamieson, G.; Fox, J.A.; Craig, A.R. Abstract C198: PDK1 Inhibitors SNS-229 and SNS-510 Cause Pathway Modulation, Apoptosis and Tumor Regression in Hematologic Cancer Models in Addition to Solid Tumors. Mol. Cancer Ther. 2015, 14, C198. [Google Scholar]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus Fulvestrant in PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer after a CDK4/6 Inhibitor (BYLieve): One Cohort of a Phase 2, Multicentre, Open-Label, Non-Comparative Study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Juric, D.; Turner, N.; Chia, S.; Drullinsky, P.; Prat, A.; Vázquez, R.V.; Akdere, M.; Arce, C.; et al. Abstract PD2-07: Alpelisib + Letrozole in Patients with PIK3CA-Mutated, Hormone-Receptor Positive (HR), Human Epidermal Growth Factor Receptor-2-Negative (HER2-) Advanced Breast Cancer (ABC) Previously Treated with a Cyclin-Dependent Kinase 4/6 Inhibitor (CDK4/6i) + fulvestrant: BYLieve study results. Cancer Res. 2021, 81, PD2-07. [Google Scholar]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef]
- Dhakal, A.; Antony Thomas, R.; Levine, E.G.; Brufsky, A.; Takabe, K.; Hanna, M.G.; Attwood, K.; Miller, A.; Khoury, T.; Early, A.P.; et al. Outcome of Everolimus-Based Therapy in Hormone-Receptor-Positive Metastatic Breast Cancer Patients after Progression on Palbociclib. Breast Cancer 2020, 14, 1178223420944864. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.M.; Al Rabadi, L.; Kaempf, A.J.; Saraceni, M.M.; Savin, M.A.; Mitri, Z.I. Everolimus plus Exemestane Treatment in Patients with Metastatic Hormone Receptor-Positive Breast Cancer Previously Treated with Cdk4/6 Inhibitor Therapy. Oncologist 2021, 26, 101–106. [Google Scholar] [CrossRef]
- Giridhar, K.V.; Choong, G.M.; Leon-Ferre, R.A.; O’Sullivan, C.C.; Ruddy, K.J.; Haddad, T.C.; Hobday, T.J.; Peethambaram, P.P.; Moynihan, T.J.; Loprinzi, C.L.; et al. Abstract P6-18-09: Clinical Management of Metastatic Breast Cancer (MBC) after CDK 4/6 Inhibitors: A Retrospective Single-Institution Study. Cancer Res. 2019, 79, P6-18-09. [Google Scholar]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.; Yardley, D.A.; Karuturi, M.; Sanft, T.; Blau, S.; Hart, L.; et al. Phase I/II Trial of Exemestane, Ribociclib, and Everolimus in Women with HR+/HER2- Advanced Breast Cancer after Progression on CDK4/6 Inhibitors (TRINITI-1). Clin. Cancer Res. 2021, 27, 4177–4185. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. A Phase I/II, Single Arm, Open-Label Study of Ribociclib in Combination with Everolimus + Exemestane in the Treatment of Men and Postmenopausal Women with HR+, HER2− Locally Advanced or Metastatic Breast Cancer Following Progression on a CDK 4/6 Inhibitor. Available online: https://clinicaltrials.gov/ct2/show/NCT02732119 (accessed on 11 August 2021).
- Mayer, I.A.; Haley, B.B.; Abramson, V.G.; Brufsky, A.; Rexer, B.; Stringer-Reasor, E.; Jhaveri, K.L.; Sanders, M.; Ericsson-Gonzalez, P.I.; Ye, F.; et al. Abstract PD1-03: A Phase Ib Trial of Fulvestrant CDK4/6 Inhibitor (CDK4/6i) Palbociclib Pan-FGFR Tyrosine Kinase Inhibitor (TKI) Erdafitinib in FGFR-Amplified/ER/HER2-Negative Metastatic Breast Cancer (MBC). Cancer Res. 2021, 81, PD1-03. [Google Scholar]
- Pandey, K.; Park, N.; Park, K.-S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.-J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined Cdk2 and Cdk4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 2020, 12, 3566. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.R.; Bisi, J.E.; Strum, J.M. Abstract 4414: Inhibition of CDK2 Overcomes Primary and Acquired Resistance to CDK4/6 Inhibitors. Cancer Research. Available online: https://cancerres.aacrjournals.org/content/79/13_Supplement/4414 (accessed on 26 June 2021).
- Witkiewicz, A.K.; Ertel, A.; McFalls, J.; Valsecchi, M.E.; Schwartz, G.; Knudsen, E.S. RB-Pathway Disruption Is Associated with Improved Response to Neoadjuvant Chemotherapy in Breast Cancer. Clin. Cancer Res. 2012, 18, 5110–5122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rossi, L.; Biagioni, C.; McCartney, A.; Migliaccio, I.; Curigliano, G.; Sanna, G.; Moretti, E.; Minisini, A.M.; Cinieri, S.; Tondini, C.; et al. Clinical Outcomes after Palbociclib with or without Endocrine Therapy in Postmenopausal Women with Hormone Receptor Positive and HER2-Negative Metastatic Breast Cancer Enrolled in the TREnd Trial. Breast Cancer Res. 2019, 21, 71. [Google Scholar] [CrossRef]
- Clark, A.S.; McAndrew, N.P.; Troxel, A.; Feldman, M.; Lal, P.; Rosen, M.; Burrell, J.; Redlinger, C.; Gallagher, M.; Bradbury, A.R.; et al. Combination Paclitaxel and Palbociclib: Results of a Phase i Trial in Advanced Breast Cancer. Clin. Cancer Res. 2019, 25, 2072–2079. [Google Scholar] [CrossRef]
- Abramson Cancer Center of the University of Pennsylvania. A Phase II Trial of Avelumab or Hydroxychloroquine with or without Palbociclib to Eliminate Dormant Breast Cancer (PALAVY). Available online: https://clinicaltrials.gov/ct2/show/NCT04841148 (accessed on 11 August 2021).
- Mayer, I.A. BRE 17107: A Phase Ib/II Trial of Atezolizumab (an Anti-PD-L1 Monoclonal Antibody) with Cobimetinib (a MEK1/2 Inhibitor) or Idasanutlin (an MDM2 Antagonist) in Metastatic ER+ Breast Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03566485 (accessed on 26 June 2021).
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. Pd-1 and Pd-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharm. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. Cdk4/6 Inhibition Augments Antitumor Immunity by Enhancing t-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.; Wander, S.A.; Regan, M.M.; DeMichele, A.M.; Forero, A.; Rimawi, M.F.; Ma, C.X.; Cristofanilli, M.; Anders, C.K.; Huang Bartlett, C.; et al. Palbociclib after CDK and Endocrine Therapy (PACE): A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab for Endocrine Pre-Treated ER+/HER2- Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03147287 (accessed on 26 June 2021).
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 Inhibitors as Anticancer Drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A.; et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Raspé, E.; Coulonval, K.; Pita, J.M.; Paternot, S.; Rothé, F.; Twyffels, L.; Brohée, S.; Craciun, L.; Larsimont, D.; Kruys, V.; et al. CDK4 Phosphorylation Status and a Linked Gene Expression Profile Predict Sensitivity to Palbociclib. EMBO Mol. Med. 2017, 9, 1052–1066. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheidemann, E.R.; Shajahan-Haq, A.N. Resistance to CDK4/6 Inhibitors in Estrogen Receptor-Positive Breast Cancer. Int. J. Mol. Sci. 2021, 22, 12292. https://doi.org/10.3390/ijms222212292
Scheidemann ER, Shajahan-Haq AN. Resistance to CDK4/6 Inhibitors in Estrogen Receptor-Positive Breast Cancer. International Journal of Molecular Sciences. 2021; 22(22):12292. https://doi.org/10.3390/ijms222212292
Chicago/Turabian StyleScheidemann, Erin R., and Ayesha N. Shajahan-Haq. 2021. "Resistance to CDK4/6 Inhibitors in Estrogen Receptor-Positive Breast Cancer" International Journal of Molecular Sciences 22, no. 22: 12292. https://doi.org/10.3390/ijms222212292
APA StyleScheidemann, E. R., & Shajahan-Haq, A. N. (2021). Resistance to CDK4/6 Inhibitors in Estrogen Receptor-Positive Breast Cancer. International Journal of Molecular Sciences, 22(22), 12292. https://doi.org/10.3390/ijms222212292