
Sirtuins and Autophagy in Age-Associated Neurodegenerative Diseases: Lessons from the C. elegans Model

Abstract
1. Introduction
2. Neurodegeneration
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
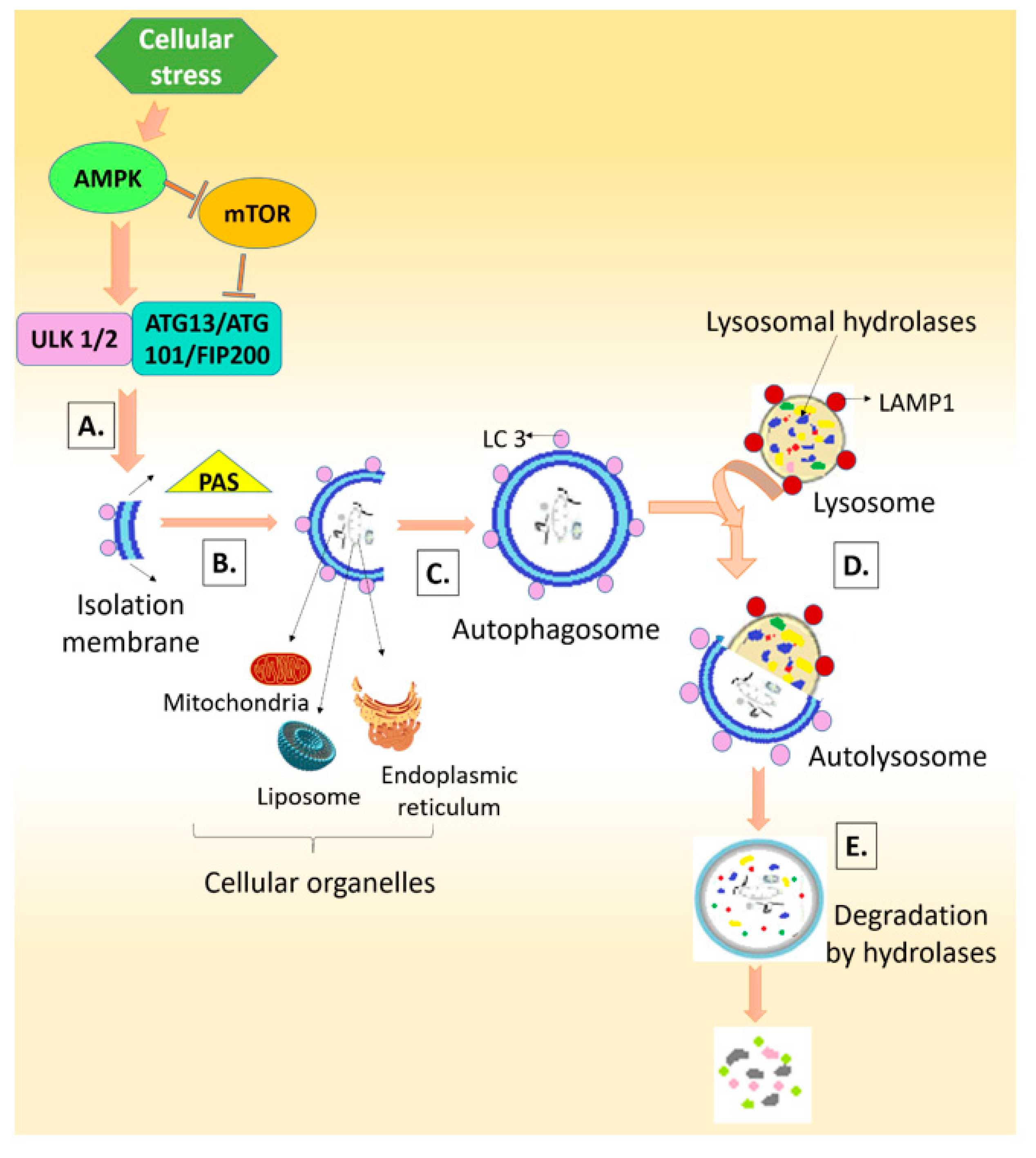
3. Autophagy: Friend or Foe
4. C. elegans: A Model for Research
5. Sirtuins: Master Regulators
6. Sirtuins and Neurodegeneration
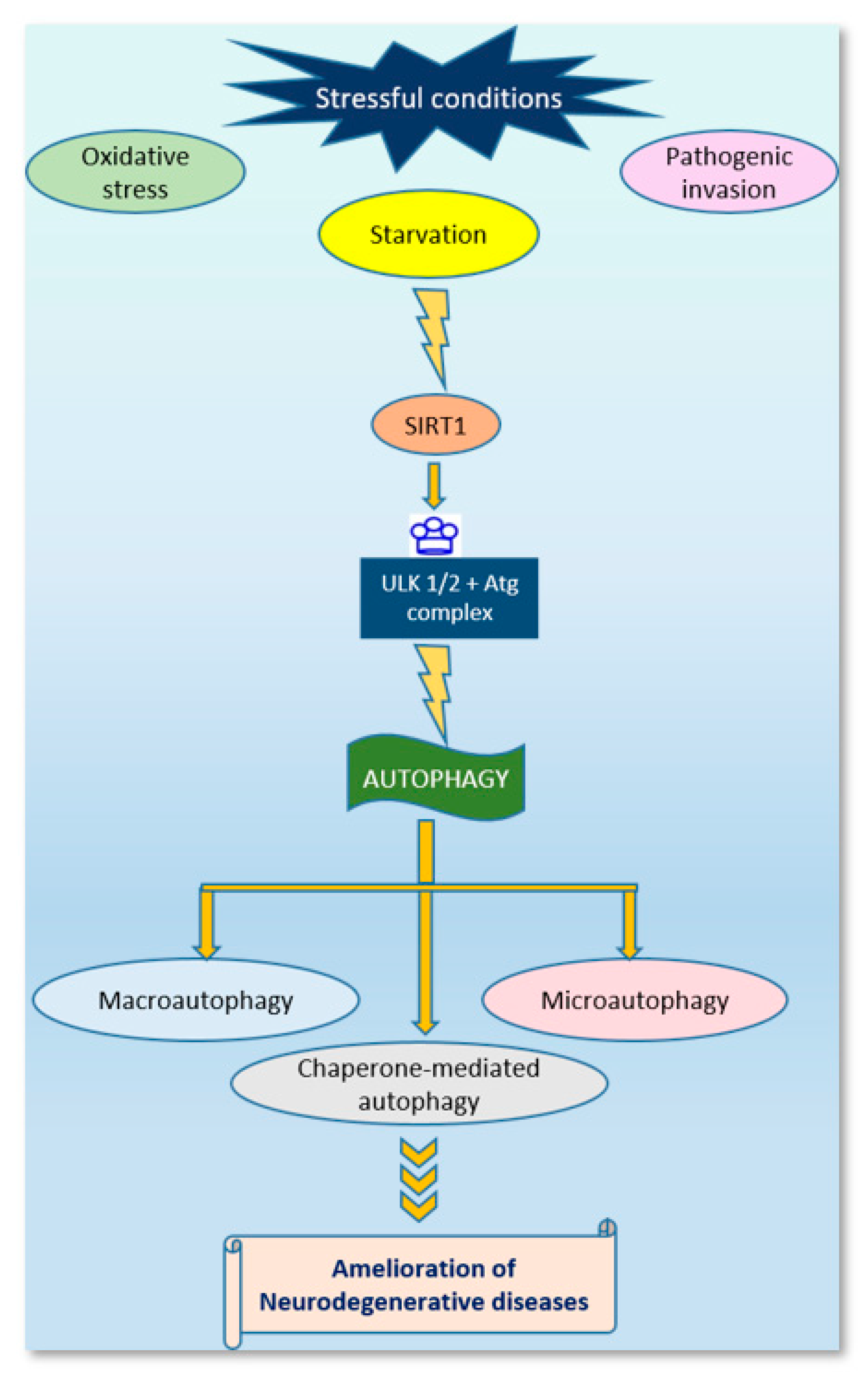
7. Sirtuins and Regulation of Autophagy in Neurodegenerative Diseases
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HDAC | Histone Deacetylase |
| WHO | World Health Organization |
| Aβ | Beta-Amyloid |
| NFTs | Neurofibrillary Tangles |
| APP | Amyloid Precursor Protein |
| AD | Alzheimer’s Disease |
| DMTs | Disease Modifying Therapeutics |
| PD | Parkinson’s Disease |
| SNCA | Synuclein Alpha |
| PINK1 | PTEN-induced Putative Kinase 1 |
| PARKN | Parkin |
| DJ-1 | Protein Deglycase |
| LRRK2 | Leucine-Rich Repeat Kinase 2 |
| LAMPs | Lysosome-Associated Membrane Proteins |
| LC3 | Light Chain 3 |
| PAS | Phagophore-Associated Site |
| YFP | Yellow fluorescent protein |
| GFP | Green fluorescent protein |
| LGG-1 | LC3, GABARAP and GATE-16 family |
| AMP | Adenosine Monophosphate |
| ATP | Adenosine Triphosphate |
| AMPK | AMP-Activated Protein Kinase |
| mTORC | Mammalian Target of Rapamycin Complex |
| ULK1/2 | UNC-51-like Autophagy-Activating Kinase 1/2 |
| ATG | Autophagy-Related |
| RNA | Ribonucleic Acid |
| FOXO | Forkhead Box transcription factor |
| HLH | Helix-Loop-Helix |
| DAF-16 | Abnormal DAuer Formation-16 |
| TFEB | Transcription Factor EB |
| Akt | A Serine/Threonine Protein Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| VATPase | Vacuolar ATPase |
| SIRT | Sirtuin |
| cAMP | Cyclic Adenosine Monophosphate |
| IRE1α | Inositol-requiring kinase 1α |
| PERK | Protein kinase (PKR)-like Endoplasmic |
| Reticulum Kinase | |
| ATF6 | Activating Transcription Factor 6 |
| CAT | Catalase |
| TLR | Toll-like Receptor |
| HIF-1 | Hypoxia-Inducible Factor 1 |
| NAD | Nicotinamide Adenine Dinucleotide |
| SOD | Superoxide Dismutase |
| ROS | Reactive Oxygen Species |
| PPARγ | Peroxisome Proliferator-Activated Receptor |
| Gamma | |
| PGC-1α | PPARγ Coactivator-1α |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| TβRI | TGF-β Receptor Protein 1 |
References
- Kumsta, C.; Chang, J.T.; Lee, R.; Tan, E.P.; Yang, Y.; Loureiro, R.; Choy, E.H.; Lim, S.H.Y.; Saez, I.; Springhorn, A.; et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kumsta, C.; Chang, J.T.; Schmalz, J.; Hansen, M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun. 2017, 8, 14337. [Google Scholar] [CrossRef] [PubMed]
- Gelino, S.; Chang, J.T.; Kumsta, C.; She, X.; Davis, A.; Nguyen, C.; Panowski, S.; Hansen, M. Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLoS Genet. 2016, 12, e1006135. [Google Scholar] [CrossRef]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Talbot, K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Khadrawyb Ya, E.H. Glutamate Excitotoxicity and Neurodegeneration. J. Mol. Genet. Med. 2014, 8, 141. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Carmichael, S.T. Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiol. Dis. 2020, 142, 104957. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Animashaun, A.; Bernardes, G. Noise promotes disengagement in dementia patients during non-invasive neurorehabilitation treatment. In Proceedings of the 4th Symposium on Occupational Safety and Health Proceedings Book, Porto, Portugal, 28–29 June 2021. [Google Scholar]
- Cognitive Vitality How Does Alzheimer’s Affect Women and Men Differently?|Cognitive Vitality|Alzheimer’s Drug Discovery Foundation. Available online: https://www.alzdiscovery.org/cognitive-vitality/blog/how-does-alzheimers-affect-women-and-men-differently (accessed on 22 October 2021).
- Beam, C.R.; Kaneshiro, C.; Jang, J.Y.; Reynolds, C.A.; Pedersen, N.L.; Gatz, M. Differences between Women and Men in Incidence Rates of Dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1077–1083. [Google Scholar] [CrossRef]
- Viña, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-β peptide. J. Alzheimer’s Dis. 2010, 20, S527–S533. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Lang, A.E.; Litvan, I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann. Neurol. 2003, 54, S15–S19. [Google Scholar] [CrossRef]
- Lima, J.A.; Hamerski, L. Alkaloids as Potential Multi-Target Drugs to Treat Alzheimer’s Disease. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Aging Parkinson’s Disease|National Institute on Aging. Available online: https://www.nia.nih.gov/health/parkinsons-disease (accessed on 22 October 2021).
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- LeWitt, P.A. Norepinephrine: The next therapeutics frontier for Parkinson’s disease. Transl. Neurodegener. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Goswami, P.; Joshi, N.; Singh, S. Neurodegenerative signaling factors and mechanisms in Parkinson’s pathology. Toxicol. Vitr. 2017, 43, 104–112. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Bar-Yosef, T.; Damri, O.; Agam, G. Dual role of autophagy in diseases of the central nervous system. Front. Cell. Neurosci. 2019, 13, 196. [Google Scholar] [CrossRef]
- Aspernig, H.; Heimbucher, T.; Qi, W.; Gangurde, D.; Curic, S.; Yan, Y.; Donner von Gromoff, E.; Baumeister, R.; Thien, A. Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 2019, 29, 1399–1409.e5. [Google Scholar] [CrossRef]
- Sampaio-Marques, B.; Ludovico, P. Sirtuins and proteolytic systems: Implications for pathogenesis of synucleinopathies. Biomolecules 2015, 5, 735–757. [Google Scholar] [CrossRef]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; Van De Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef]
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; Adams, L.M.; Hansen, M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 2017, 6, e18459. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Meléndez, A.; Levine, B. Autophagy in C. elegans. WormBook 2009, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.K.; Wightman, B.; Chalfie, M. A Transparent window into biology: A primer on Caenorhabditis elegans (18 June 2015), WormBook, ed. The C. elegans Research Community. WormBook 2015, 200, 387–407. [Google Scholar]
- Hendricks, G.; Mylonakis, E. Expanding the nematode model system: The molecular basis of inflammation and infection recovery in C. elegans. Virulence 2017, 8, 244–245. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cook, D.E.; Zdraljevic, S.; Roberts, J.P.; Andersen, E.C. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 2017, 45, D650–D657. [Google Scholar] [CrossRef]
- Jadiya, P.; Khan, A.; Sammi, S.R.; Kaur, S.; Mir, S.S.; Nazir, A. Anti-Parkinsonian effects of Bacopa monnieri: Insights from transgenic and pharmacological Caenorhabditis elegans models of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2011, 413, 605–610. [Google Scholar] [CrossRef]
- Jadiya, P.; Chatterjee, M.; Sammi, S.R.; Kaur, S.; Palit, G.; Nazir, A. Sir-2.1 modulates “calorie-restriction-mediated” prevention of neurodegeneration in Caenorhabditis elegans: Implications for Parkinson’s disease. Biochem. Biophys. Res. Commun. 2011, 413, 306–310. [Google Scholar] [CrossRef]
- Vining Smith, J.; Luo, Y. Elevation of oxidative free radicals in Alzheimer’s disease models can be attenuated by Ginkgo biloba extract EGb 761. J. Alzheimer’s Dis. 2003, 5, 287–300. [Google Scholar] [CrossRef]
- Gutierrez-Zepeda, A.; Santell, R.; Wu, Z.; Brown, M.; Wu, Y.J.; Khan, I.; Link, C.D.; Zhao, B.; Luo, Y. Soy isoflavone glycitein protects against beta amyloid-induced toxicity and oxidative stress in transgenic Caenorhabditis elegans. BMC Neurosci. 2005, 6, 54. [Google Scholar] [CrossRef]
- Alavez, S.; Vantipalli, M.C.; Zucker, D.J.S.; Klang, I.M.; Lithgow, G.J. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 2011, 472, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Keowkase, R.; Aboukhatwa, M.; Luo, Y. Fluoxetine protects against amyloid-beta toxicity, in part via daf-16 mediated cell signaling pathway, in Caenorhabditis elegans. Neuropharmacology 2010, 59, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Arya, U.; Dwivedi, H.; Subramaniam, J.R. Reserpine ameliorates Aβ toxicity in the Alzheimer’s disease model in Caenorhabditis elegans. Exp. Gerontol. 2009, 44, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef]
- Ashrafi, K.; Chang, F.Y.; Watts, J.L.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 2003, 421, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, K.; Parker, J.A. Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biol. 2019, 7, 192. [Google Scholar] [CrossRef]
- Schmeisser, K.; Parker, J.A. Nicotinamide-N-methyltransferase controls behavior, neurodegeneration and lifespan by regulating neuronal autophagy. PLoS Genet. 2018, 14, e1007561. [Google Scholar] [CrossRef] [PubMed]
- Megalou, E.V.; Tavernarakis, N. Autophagy in Caenorhabditis elegans. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 1444–1451. [Google Scholar] [CrossRef]
- Altun, Z.F.; Hall, D.H. WormAtlas Hermaphrodite Handbook-Epithelial System-Seam Cells. WormAtlas 2002. [Google Scholar] [CrossRef]
- Brabin, C.; Woollard, A. Finding a niche for seam cells? Worm 2012, 1, 107–111. [Google Scholar] [CrossRef]
- Kang, C.; You, N.J.; Avery, L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007, 21, 2161–2171. [Google Scholar] [CrossRef]
- Dall, K.B.; Færgeman, N.J. Metabolic regulation of lifespan from a C. elegans perspective. Genes Nutr. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Schmukler, E.; Pinkas-Kramarski, R. Autophagy induction in the treatment of Alzheimer’s disease. Drug Dev. Res. 2020, 81, 184–193. [Google Scholar] [CrossRef]
- Ma, J.; Yang, L.; Ren, J.; Yang, J. Autophagy, Oxidative Stress, and Redox Regulation; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 1, ISBN 9780128052532. [Google Scholar]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Xie, Y.; Xu, Y.; Zhou, H.; Xu, W.; Dong, Q. Bafilomycin A1 inhibits autophagy and induces apoptosis in MG63 osteosarcoma cells. Mol. Med. Rep. 2014, 10, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Fazlul Kabir, M.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Autophagy. In Endoplasmic Reticulum; IntechOpen: London, UK, 2019; Available online: https://www.intechopen.com/chapters/64217 (accessed on 22 October 2021).
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.H.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef]
- Safra, M.; Ben-Hamo, S.; Kenyon, C.; Henis-Korenblit, S. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J. Cell Sci. 2013, 126, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Tenor, J.L.; Aballay, A. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 2008, 9, 103–109. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin functions in health and disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef]
- Lin, H. The Enzymatic Activities of Sirtuins. In Introductory Review on Sirtuins in Biology, Aging, and Disease; Academic Press: Cambridge, MA, USA, 2018; ISBN 9780128135006. [Google Scholar]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 1–14. [Google Scholar] [CrossRef]
- Greiss, S.; Gartner, A. Europe PMC Funders Group Sirtuin/Sir2 Phylogeny, Evolutionary Considerations and Structural Conservation. Mol. Cell 2009, 28, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Satoh, A.; Imai, S.I.; Guarente, L. The brain, sirtuins, and ageing. Nat. Rev. Neurosci. 2017, 18, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Correia, S.C.; Cardoso, S.; Plácido, A.I.; Candeias, E.; Duarte, A.I.; Moreira, P.I. The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev. Neurother. 2015, 15, 867–884. [Google Scholar] [CrossRef]
- Pande, S.; Kratasyuk, V.A.; Medvedeva, N.N.; Kolenchukova, O.A.; Salmina, A.B. Nutritional biomarkers: Current view and future perspectives. Crit. Rev. Food Sci. Nutr. 2018, 58, 3055–3069. [Google Scholar] [CrossRef] [PubMed]
- Jedrusik-Bode, M.; Studencka, M.; Smolka, C.; Baumann, T.; Schmidt, H.; Kampf, J.; Paap, F.; Martin, S.; Tazi, J.; Mü ller, K.M.; et al. The sirtuin SIRT6 regulates stress granule formation in C. elegans and mammals. J. Cell Sci. 2013, 126, 5166–5177. [Google Scholar] [CrossRef]
- Wu, D.; Li, Y.; Zhu, K.S.; Wang, H.; Zhu, W.-G. Advances in Cellular Characterization of the Sirtuin Isoform, SIRT7. Front. Endocrinol. 2018, 9, 652. [Google Scholar] [CrossRef]
- Donmez, G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef]
- Silberman, D.M. Metabolism, neurodegeneration and epigenetics: Emerging role of Sirtuins. Neural Regen. Res. 2018, 13, 417–418. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Bedalov, A.; Chowdhury, S.; Simon, J.A. Biology, Chemistry, and Pharmacology of Sirtuins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Viswanathan, M.; Tissenbaum, H.A. C. elegans sirtuins. Methods Mol. Biol. 2013, 1077, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 13091–13096. [Google Scholar] [CrossRef]
- Chamoli, M.; Singh, A.; Malik, Y.; Mukhopadhyay, A. A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell 2014, 13, 641–655. [Google Scholar] [CrossRef]
- Chang, S.M.; McReynolds, M.R.; Hanna-Rose, W. Mitochondrial sirtuins sir-2.2 and sir-2.3 regulate lifespan in C. elegans. bioRxiv 2017. [Google Scholar] [CrossRef]
- Wirth, M.; Karaca, S.; Wenzel, D.; Ho, L.; Tishkoff, D.; Lombard, D.B.; Verdin, E.; Urlaub, H.; Jedrusik-Bode, M.; Fischle, W. Mitochondrial SIRT4-type proteins in Caenorhabditis elegans and mammals interact with pyruvate carboxylase and other acetylated biotin-dependent carboxylases. Mitochondrion 2013, 13, 705–720. [Google Scholar] [CrossRef]
- Simeoni, F.; Tasselli, L.; Tanaka, S.; Villanova, L.; Hayashi, M.; Kubota, K.; Isono, F.; Garcia, B.A.; Michishita-Kioi, E.; Chua, K.F. Proteomic analysis of the SIRT6 interactome: Novel links to genome maintenance and cellular stress signaling. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jedrusik-Bode, M. C. elegans sirtuin SIR-2.4 and its mammalian homolog SIRT6 in stress response. Worm 2014, 3, e29102. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Tishkoff, D.X.; Yang, B.; Wilson-Grady, J.; Yu, X.; Mazer, T.; Eckersdorff, M.; Gygi, S.P.; Lombard, D.B.; Hsu, A.L. C. elegans SIRT6/7 Homolog SIR-2.4 Promotes DAF-16 Relocalization and Function during Stress. PLoS Genet. 2012, 8, e1002948. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Nazir, A.; Jadiya, P. Sirtuin mediated neuroprotection and its association with autophagy and apoptosis: Studies employing transgenic C. elegans model. Mol. Neurodegener. 2013, 8, 65. [Google Scholar] [CrossRef][Green Version]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Lee, W.; Kim, S.H. Autophagy at synapses in neurodegenerative diseases. Arch. Pharm. Res. 2019, 42, 407–415. [Google Scholar] [CrossRef]
- Stefanoni, G.; Sala, G.; Tremolizzo, L.; Brighina, L.; Ferrarese, C. Role of autophagy in Parkinson’s disease. Autophagy Princ. Regul. Roles Dis. 2012, 26, 243–264. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Sheehan, P.; Yue, Z. Deregulation of autophagy and vesicle trafficking in Parkinson’s disease. Neurosci. Lett. 2019, 697, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shi, B.; Ma, M.; Wu, X.; Lin, X. The novel relationship between Sirt3 and autophagy in myocardial ischemia–reperfusion. J. Cell. Physiol. 2019, 234, 5488–5495. [Google Scholar] [CrossRef]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- In, H.L.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef]
- Dong, S.; Jia, C.; Zhang, S.; Fan, G.; Li, Y.; Shan, P.; Sun, L.; Xiao, W.; Li, L.; Zheng, Y.; et al. The REGγ proteasome regulates hepatic lipid metabolism through inhibition of autophagy. Cell Metab. 2013, 18, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.K. Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 2019, 10, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936–952. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Izzo, V.; Bravo-San Pedro, J.M.; Zamzami, N.; Maiuri, M.C. Mitophagy; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 9, ISBN 9780128029367. [Google Scholar]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Lombard, D.B.; Zwaans, B.M.M. SIRT3: As simple as it seems? Gerontology 2013, 60, 56–64. [Google Scholar] [CrossRef]
- Dai, S.H.; Chen, T.; Li, X.; Yue, K.Y.; Luo, P.; Yang, L.K.; Zhu, J.; Wang, Y.H.; Fei, Z.; Jiang, X.F. Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK-mTOR pathway. Free Radic. Biol. Med. 2017, 108, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Wang, K.; Yuan, Y.; Guan, Y.; Ren, X.; Li, L.; Chen, X.; Li, F.; Chen, A.F.; Zhou, J.; et al. Correction: Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget 2016, 7, 43390–43400. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, A.; Li, Y.J.; Yang, Y.; Kishimoto, Y.; Zhang, S.; Wang, Y.; Wan, R.; Raefsky, S.M.; Lu, D.; et al. SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Ramesh, S.; Govindarajulu, M.; Lynd, T.; Briggs, G.; Adamek, D.; Jones, E.; Heiner, J.; Majrashi, M.; Moore, T.; Amin, R.; et al. SIRT3 activator Honokiol attenuates β-Amyloid by modulating amyloidogenic pathway. PLoS ONE 2018, 13, e0190350. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wu, Q.; Shi, J.; Zhou, S. Regulation of SIRT3 on mitochondrial functions and oxidative stress in Parkinson’s disease. Biomed. Pharmacother. 2020, 132, 110928. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Tan, E.K. Oxidized nicotinamide adenine dinucleotide-dependent mitochondrial deacetylase sirtuin-3 as a potential therapeutic target of Parkinson’s disease. Ageing Res. Rev. 2020, 62, 101107. [Google Scholar] [CrossRef]
- Shu, M.; Zhang, W.; Jin, X.; Zeng, L.; Xiang, Y. Sirt3 gene knockout protects mice from Alzheimer’s disease through activating autophagy. Zhejiang Da Xue Xue Bao Yi Xue Ban 2020, 49, 750–757. [Google Scholar] [CrossRef]
- Yang, W.; Zou, Y.; Zhang, M.; Zhao, N.; Tian, Q.; Gu, M.; Liu, W.; Shi, R.; Lü, Y.; Yu, W. Mitochondrial Sirt3 Expression is Decreased in APP/PS1 Double Transgenic Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2015, 40, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Peritore, C.; Ginsberg, J.; Shih, J.; Arun, S.; Donmez, G. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson’s disease. Behav. Brain Res. 2015, 281, 215–221. [Google Scholar] [CrossRef]
- Araki, S.; Izumiya, Y.; Rokutanda, T.; Ianni, A.; Hanatani, S.; Kimura, Y.; Onoue, Y.; Senokuchi, T.; Yoshizawa, T.; Yasuda, O.; et al. Sirt7 Contributes to Myocardial Tissue Repair by Maintaining Transforming Growth Factor-β Signaling Pathway. Circulation 2015, 132, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 2010, 6, 186–188. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| S. No. | Pathways Regulating Autophagy | Regulatory Molecules | |
|---|---|---|---|
| Mammals | C. elegans | ||
| 1. | Nutrient/energy sensing pathway | mTORC1, Ras-cAMP-PKA, AMPK [61] | let-363, AMPK |
| 2. | Insulin/growth factor signaling pathway | PKB/Akt, Ras-MAPK | DAF-2, Ras-MAPK |
| 3. | Stress response pathway | IRE1α, PERK, ATF6α [62], ATG7, ATG8, SOD, CAT [63], HIF-1 | ire-1, pek-1, atf-6 [64], atg7, lgg-1, sod-3, ctl-1/2 |
| 4. | Pathogen-induced regulation | TLRs | TOL-1 [65] |
| 5. | Transcriptional regulation and chromatin modification | FOXO, HDAC 1,2,3 (SIRT1-7) and 6 [66] | DAF-16, HDA-1 [66], sir-2.1, sir-2.2, sir-2.3, sir-2.4 |
| S. No. | Human Sirtuin | Location | C. elegans Homolog | Enzymatic Activity | Function |
|---|---|---|---|---|---|
| 1. | SIRT1 | Nucleus (shuttles between nucleus and cytoplasm) [71] | sir-2.1 | Deacetylase activity | Transcription regulation, cell survival chromatin organization, development and differentiation, stress responses, metabolism regulation, neuroprotection, adult neurogenesis, synaptic plasticity, cognition, emotion, circadian rhythm, microglial activation [72]. |
| 2. | SIRT2 | Mainly cytoplasmic but can translocate to the nucleus as well | Deacetylase activity | DNA repair, cell cycle, mitosis, transcription regulation, adult neurogenesis, microglial activation, neuroprotection, regulation of emotions [72]. | |
| 3. | SIRT3 | Mitochondria [73] | Strong deacetylase activity | Mitochondrial functioning, metabolism regulation, ATP production, reducing oxidative stress, sleep-wake patterning, regulation of age-related hearing loss [72]. | |
| 4. | SIRT4 | Mitochondria | sir-2.2 & sir-2.3 | ADP-ribosyl transferase activity | Mitochondrial functioning, metabolism regulation |
| 5. | SIRT5 | Mitochondria | Desuccinylase and demalonylase activities; weak deacetylase activity | Fatty acid oxidation, insulin secretion [74] | |
| 6. | SIRT6 | Nucleus (translocates to cytoplasm under stress) [75] | sir-2.4 | Deacetylase activity, ADP-ribosyl transferase activity | Genome stability, DNA repair, control of circadian rythms, stress response, inflammation |
| 7. | SIRT7 | Nucleus, specifically in nucleolus | Deacetylase activity | Cell survival, rRNA regulation, cellular stress regulation [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naseer, A.; Mir, S.S.; Takacs-Vellai, K.; Nazir, A. Sirtuins and Autophagy in Age-Associated Neurodegenerative Diseases: Lessons from the C. elegans Model. Int. J. Mol. Sci. 2021, 22, 12263. https://doi.org/10.3390/ijms222212263
Naseer A, Mir SS, Takacs-Vellai K, Nazir A. Sirtuins and Autophagy in Age-Associated Neurodegenerative Diseases: Lessons from the C. elegans Model. International Journal of Molecular Sciences. 2021; 22(22):12263. https://doi.org/10.3390/ijms222212263
Chicago/Turabian StyleNaseer, Anam, Snober Shabnam Mir, Krisztina Takacs-Vellai, and Aamir Nazir. 2021. "Sirtuins and Autophagy in Age-Associated Neurodegenerative Diseases: Lessons from the C. elegans Model" International Journal of Molecular Sciences 22, no. 22: 12263. https://doi.org/10.3390/ijms222212263
APA StyleNaseer, A., Mir, S. S., Takacs-Vellai, K., & Nazir, A. (2021). Sirtuins and Autophagy in Age-Associated Neurodegenerative Diseases: Lessons from the C. elegans Model. International Journal of Molecular Sciences, 22(22), 12263. https://doi.org/10.3390/ijms222212263