
Loss of Function of Mutant IDS Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type II


Abstract
:1. Background of Mucopolysaccharidosis Type II (MPS II)
2. Current Therapy for MPS II and Its Difficulties
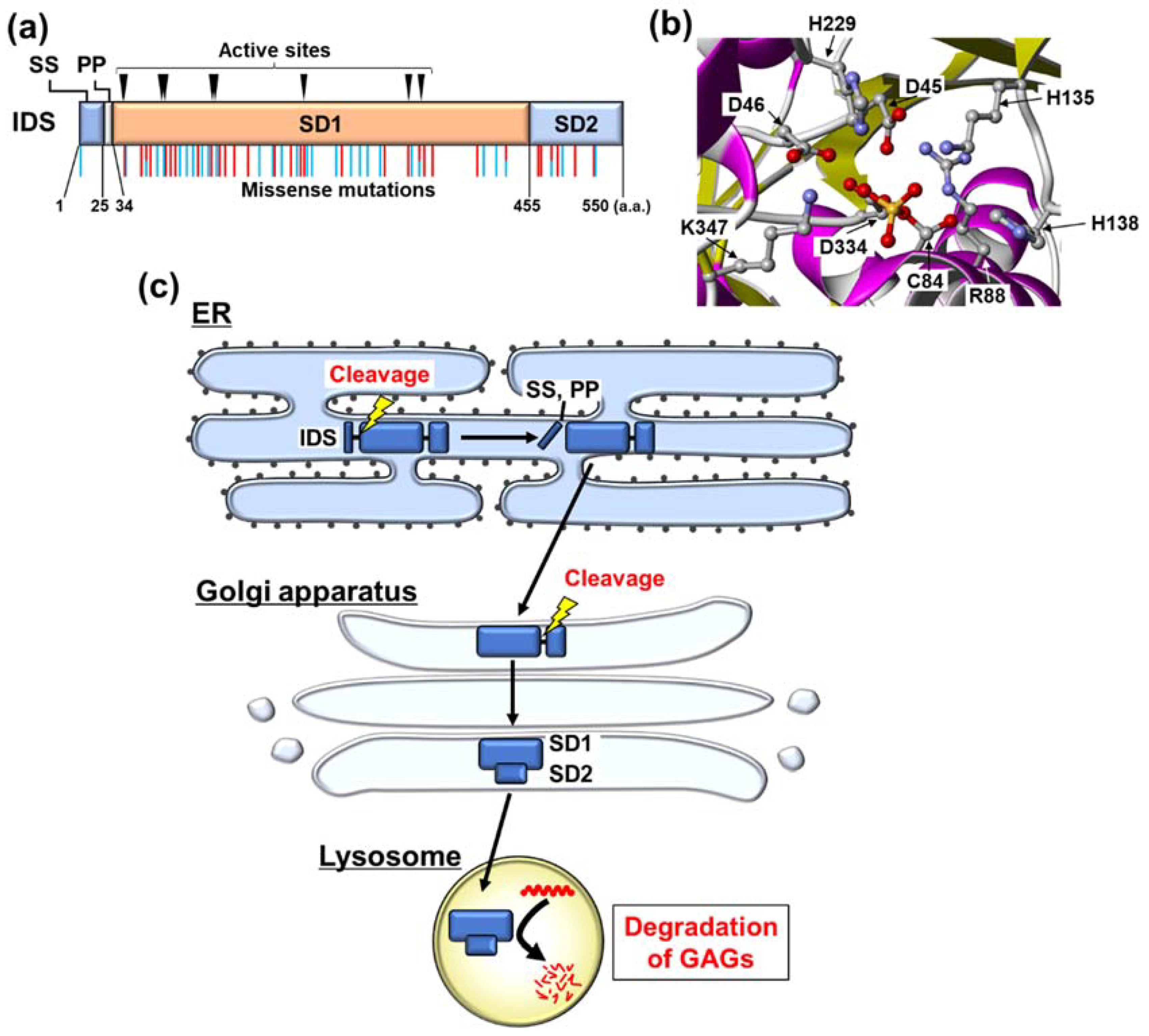
3. Biochemical Characteristics of IDS
4. Misfolding of IDS Caused by Its Mutation
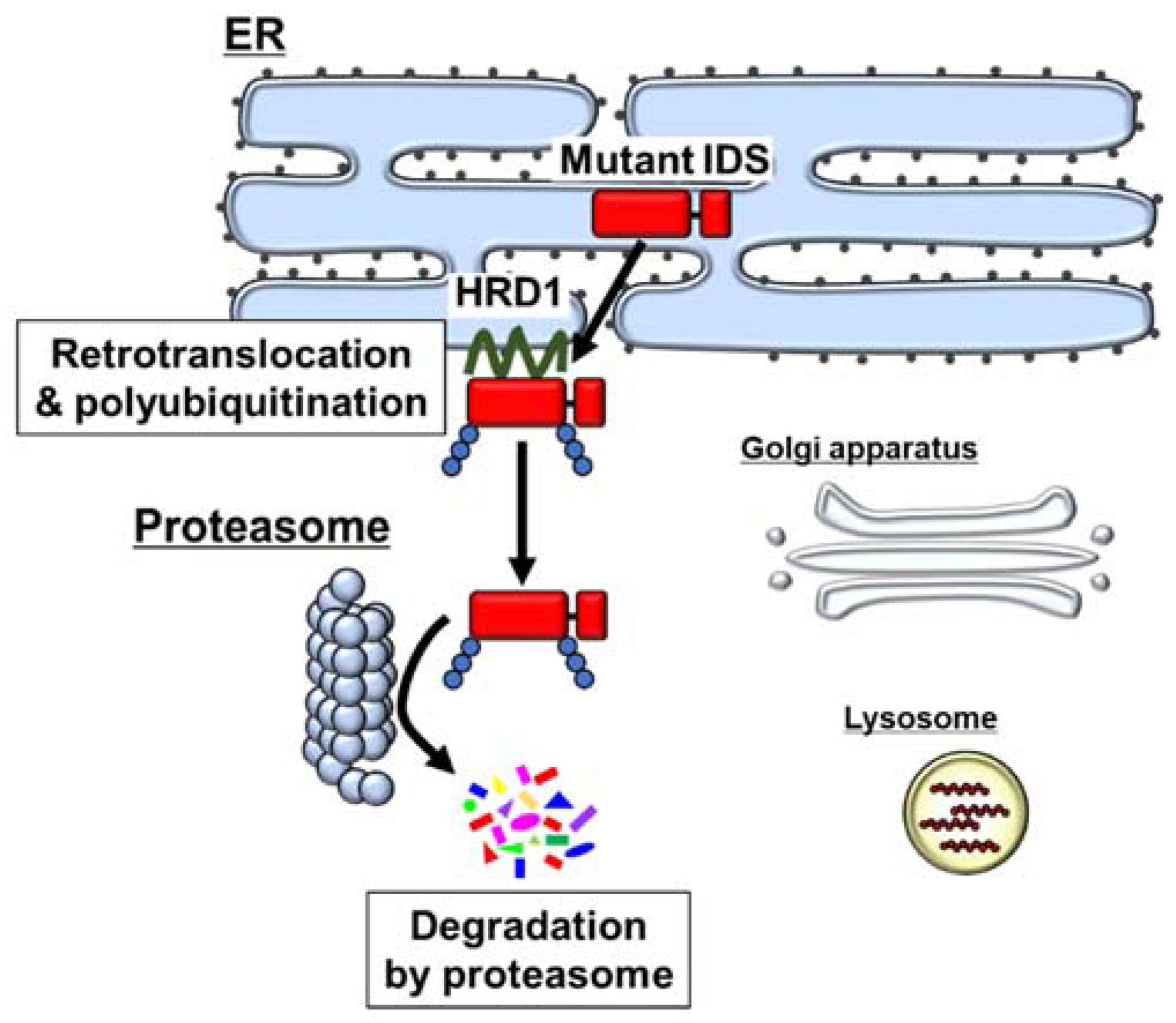
5. Degradation of Mutant IDS by the ER-Associated Degradation (ERAD) System
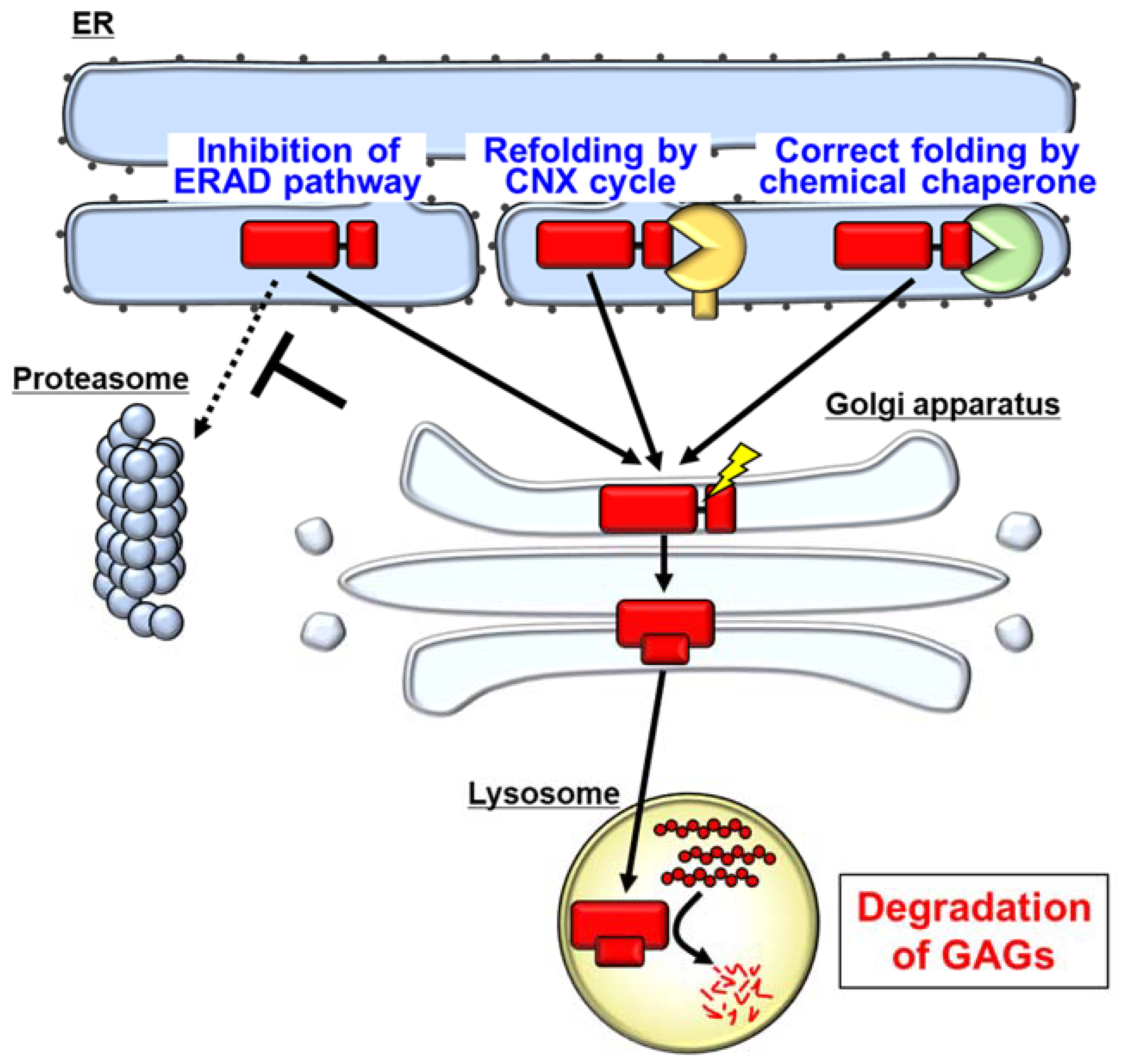
6. Pharmacological Chaperones as a Possible Therapeutic Strategy for MPS II
7. Rare Diseases and ERAD
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCA4 | ATP binding cassette subfamily A member 4 |
| α-Gal A | α-galactosidase A |
| AQP2 | aquaporin 2 |
| BBB | Blood–brain barrier |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CNS | Central nervous system |
| CNX | Calnexin |
| D2S0 | Δ-unsaturated 2-sulfouronic acid-N-sulfoglucosamine |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum-associated degradation |
| ERT | Enzyme replacement therapy |
| GAG | Glycosaminoglycan |
| GCase | Glucocerebrosidase |
| HexA | Β-hexosaminidase A |
| HGMD | Human Gene Mutation Database |
| HRD1 | Hydroxymethyl glutaryl-coenzyme A reductase degradation protein 1 |
| HSC | Hematopoietic stem cell |
| IDS | Iduronate-2-sulfatase |
| IDUA | α-L-iduronidase |
| MPS | Mucopolysaccharidosis |
| MPS I | Mucopolysaccharidosis type I |
| MPS II | Mucopolysaccharidosis type II |
| NKCC2 | Sodium-potassium-chloride cotransporter 2 |
| PMP22 | Peripheral myelin protein 22 |
| POMC | Pro-opiomelanocortin |
| PP | Propeptide |
| SD1 | Subdomain 1 |
| SD2 | Subdomain 2 |
| SS | Signal peptide |
| XBP1 | X-box binding protein 1 |
References
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Bellows, A.B.; Thompson, J.N. Molecular basis of iduronate-2-sulphatase gene mutations in patients with mucopolysaccharidosis type II (Hunter syndrome). J. Med Genet. 1999, 36, 21–27. [Google Scholar]
- Bach, G.; Eisenberg, F.; Cantz, M.; Neufeld, E.F. The Defect in the Hunter Syndrome: Deficiency of Sulfoiduronate Sulfatase. Proc. Natl. Acad. Sci. USA 1973, 70, 2134–2138. [Google Scholar] [CrossRef] [Green Version]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bodamer, O.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampe, C.; Yund, B.; Orchard, P.; Lund, T.; Wesley, J.; McIvor, R. Differences in MPS I and MPS II Disease Manifestations. Int. J. Mol. Sci. 2021, 22, 7888. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Ellaway, C.; Nicholls, K.; Reardon, K.; Szer, J. Treatable lysosomal storage diseases in the advent of disease-specific therapy. Intern. Med. J. 2020, 50, 5–27. [Google Scholar] [CrossRef]
- Zapolnik, P.; Pyrkosz, A. Gene Therapy for Mucopolysaccharidosis Type II—A Review of the Current Possibilities. Int. J. Mol. Sci. 2021, 22, 5490. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, J.B.; Poe, M.D.; Escolar, M.L. Natural Progression of Neurological Disease in Mucopolysaccharidosis Type II. Pediatr. 2011, 127, e1258–e1265. [Google Scholar] [CrossRef] [PubMed]
- D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demydchuk, M.; Hill, C.H.; Zhou, A.; Bunkóczi, G.; Stein, P.E.; Marchesan, D.; Deane, J.E.; Read, R.J. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat. Commun. 2017, 8, 15786. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ohno, K.; Okuyama, T.; Sakuraba, H. Structural Basis of Mucopolysaccharidosis Type II and Construction of a Database of Mutant Iduronate 2-Sulfatases. PLoS ONE 2016, 11, e0163964. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter Syndromes: Mutual Correction of the Defect in Cultured Fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef]
- Hasilik, A.; Neufeld, E. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J. Biol. Chem. 1980, 255, 4946–4950. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Cho, S.Y.; Park, S.W.; Kim, S.J.; Ko, A.-R.; Kwon, E.-K.; Han, S.J.; Jin, D.-K. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome). Orphanet J. Rare Dis. 2013, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J.; Wraith, J.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Muenzer, J.; Beck, M.; Giugliani, R.; Suzuki, Y.; Tylki-Szymanska, A.; Valayannopoulos, V.; Vellodi, A.; Wraith, J. Idursulfase treatment of Hunter syndrome in children younger than 6 years: Results from the Hunter Outcome Survey. Genet. Med. 2011, 13, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parini, R.; Rigoldi, M.; Tedesco, L.; Boffi, L.; Brambilla, A.; Bertoletti, S.; Boncimino, A.; Del Longo, A.; De Lorenzo, P.; Gaini, R.; et al. Enzymatic replacement therapy for Hunter disease: Up to 9years experience with 17 patients. Mol. Genet. Metab. Rep. 2015, 3, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lagler, F.B. Current and Emerging Therapies for Mucopolysaccharidoses. Physiol. Pharmacol. Bone 2019, 261, 39–56. [Google Scholar] [CrossRef]
- Warkentin, P.; Dixon, M.S.; Schafer, I.; Strandjord, S.; Coccia, P.F. Bone marrow transplantation in Hunter syndrome: A preliminary report. Birth Defects Orig. Artic. Ser. 1986, 22, 31–39. [Google Scholar] [PubMed]
- Mullen, C.; Thompson, J.N.; Richard, L.; Chan, K.W. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia. Bone Marrow Transplant. 2000, 25, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.K.; Kurtzberg, J. Cord blood and bone marrow transplantation in inherited metabolic diseases: Scientific basis, current status and future directions. Br. J. Haematol. 2010, 148, 356–372. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; Van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic Cell Transplantation for Mucopolysaccharidosis Patients Is Safe and Effective: Results after Implementation of International Guidelines. Biol. Blood Marrow Transplant. 2015, 21, 1106–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Kurtzberg, J. Early HSCT corrects the skeleton in MPS. Blood 2015, 125, 1518–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielicki, J.; Freeman, C.; Clements, P.R.; Hopwood, J.J. Human liver iduronate-2-sulphatase. Purification, characterization and catalytic properties. Biochem. J. 1990, 271, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.J.; Morris, P.; Anson, D.S.; Occhiodoro, T.; Bielicki, J.; Clements, P.R.; Hopwood, J.J. Hunter syndrome: Isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 8531–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millat, G.; Froissart, R.; Maire, I.; Bozon, M. Characterization of iduronate sulphatase mutants affecting N-glycosylation sites and the cysteine-84 residue. Biochem. J. 1997, 326, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Selmer, T.; Ingendoh, A.; von Figurat, K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 1995, 82, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Froissart, R.; Millat, G.; Mathieu, M.; Bozon, D.; Maire, I. Processing of iduronate 2-sulphatase in human fibroblasts. Biochem. J. 1995, 309, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukegawa-Hayasaka, K.; Kato, Z.; Nakamura, H.; Tomatsu, S.; Fukao, T.; Kuwata, K.; Orii, T.; Kondo, N. Effect of Hunter disease (mucopolysaccharidosis type II) mutations on molecular phenotypes of iduronate-2-sulfatase: Enzymatic activity, protein processing and structural analysis. J. Inherit. Metab. Dis. 2006, 29, 755–761. [Google Scholar] [CrossRef]
- Cardona, C.; Benincore, E.; Pimentel, N.; Reyes, L.H.; Patarroyo, C.; Rodríguez-López, A.; Martin-Rufian, M.; Barrera, L.A.; Alméciga-Díaz, C.J. Identification of the iduronate-2-sulfatase proteome in wild-type mouse brain. Heliyon 2019, 5, e01667. [Google Scholar] [CrossRef] [Green Version]
- Kobolák, J.; Molnar, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Giudice, M.L.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the neuropathology of lysosomal storage disorders through disease-specific human induced pluripotent stem cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef] [Green Version]
- Osaki, Y.; Saito, A.; Kanemoto, S.; Kaneko, M.; Matsuhisa, K.; Asada, R.; Masaki, T.; Orii, K.; Fukao, T.; Tomatsu, S.; et al. Shutdown of ER-associated degradation pathway rescues functions of mutant iduronate 2-sulfatase linked to mucopolysaccharidosis type II. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, Y.; Matsuhisa, K.; Che, W.; Kaneko, M.; Asada, R.; Masaki, T.; Imaizumi, K.; Saito, A. Calnexin promotes the folding of mutant iduronate 2-sulfatase related to mucopolysaccharidosis type II. Biochem. Biophys. Res. Commun. 2019, 514, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Marazza, A.; Galli, C.; Fasana, E.; Sgrignani, J.; Burda, P.; Fassi, E.M.A.; Baumgartner, M.R.; Cavalli, A.; Molinari, M. Endoplasmic Reticulum and Lysosomal Quality Control of Four Nonsense Mutants of Iduronate 2-Sulfatase Linked to Hunter’s Syndrome. DNA Cell Biol. 2020, 39, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Lamriben, L.; Graham, J.B.; Adams, B.M.; Hebert, D.N. N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle. Traffic 2016, 17, 308–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Määttänen, P.; Gehring, K.; Bergeron, J.J.; Thomas, D.Y. Protein quality control in the ER: The recognition of misfolded proteins. Semin. Cell Dev. Biol. 2010, 21, 500–511. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Johnson, J.D.; Arvan, P.; Han, J.; Kaufman, R.J. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat. Rev. Endocrinol. 2021, 17, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losada Díaz, J.; Cepeda Del, C.J.; Rodriguez-López, E.; Alméciga-Díaz, C. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2019, 21, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2018, 123, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Endocrinology, T.L.D. & Spotlight on rare diseases. Lancet Diabetes Endocrinol. 2019, 7, 75. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Kinch, L.; Otwinowski, Z.; Grishin, N.V. Mutation severity spectrum of rare alleles in the human genome is predictive of disease type. PLoS Comput. Biol. 2020, 16, e1007775. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Genereux, J.C.; Pankow, S.; Aerts, J.M.; Yates, J.R.; Kelly, J.W. ERdj3 Is an Endoplasmic Reticulum Degradation Factor for Mutant Glucocerebrosidase Variants Linked to Gaucher’s Disease. Chem. Biol. 2014, 21, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef]
- Fog, C.; Zago, P.; Malini, E.; Solanko, L.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.; Bembi, B.; et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Chang, H.; Kawasaki, K.; Yasuda, K.; Wu, H.; Garman, S.; Fan, J. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Nicholls, K.; Shankar, S.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Dersh, D.; Iwamoto, Y.; Argon, Y. Tay–Sachs disease mutations in HEXA target the α chain of hexosaminidase A to endoplasmic reticulum–associated degradation. Mol. Biol. Cell 2016, 27, 3813–3827. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Zeitlin, P. Sodium 4-phenylbutyrate downregulates Hsc70: Implications for intracellular trafficking of ΔF508-CFTR. Am. J. Physiol. Physiol. 2000, 278, C259–C267. [Google Scholar] [CrossRef] [PubMed]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.L.; Omura, S.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis ofCFTR mutations?correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef]
- Marr, N.; Bichet, D.-G.; Hoefs, S.; Savelkoul, P.J.M.; Konings, I.B.M.; de Mattia, F.; Graat, M.P.J.; Arthus, M.-F.; Lonergan, M.; Fujiwara, T.M.; et al. Cell-Biologic and Functional Analyses of Five NewAquaporin-2Missense Mutations that Cause Recessive Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2002, 13, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Chiang, W.-C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, D.; Matthes, M.T.; Coppinger, J.; Palczewski, K.; Lavail, M.M.; Lin, J.H. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol. Neurobiol. 2015, 52, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Aron, L.; Piccoli, G.; Ueffing, M. Clearance of RhodopsinP23H aggregates requires the ERAD effector VCP. Biochim. Biophys. Acta (BBA) -Bioenerg. 2010, 1803, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Tayo, B.O.; Bandyopadhyay, A.; Wang, H.; Feng, T.; Franceschini, N.; Tang, H.; Gao, J.; Sung, Y.J.; Elston, R.C.; et al. The Association of the Vanin-1 N131S Variant with Blood Pressure Is Mediated by Endoplasmic Reticulum-Associated Degradation and Loss of Function. PLoS Genet. 2014, 10, e1004641. [Google Scholar] [CrossRef]
- Sabirzhanova, I.; Lopes-Pacheco, M.; Rapino, D.; Grover, R.; Handa, J.T.; Guggino, W.B.; Cebotaru, L. Rescuing Trafficking Mutants of the ATP-binding Cassette Protein, ABCA4, with Small Molecule Correctors as a Treatment for Stargardt Eye Disease. J. Biol. Chem. 2015, 290, 19743–19755. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Hashimoto, Y.; Akuzawa, T.; Hirai, R.; Kobayashi, H.; Sato, K. Rer1 and calnexin regulate endoplasmic reticulum retention of a peripheral myelin protein 22 mutant that causes type 1A Charcot-Marie-Tooth disease. Sci. Rep. 2014, 4, 6992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seaayfan, E.; Defontaine, N.; Demaretz, S.; Zaarour, N.; Laghmani, K. OS9 Protein Interacts with Na-K-2Cl Co-transporter (NKCC2) and Targets Its Immature Form for the Endoplasmic Reticulum-associated Degradation Pathway. J. Biol. Chem. 2016, 291, 4487–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.H.; Shi, G.; Somlo, D.R.; Haataja, L.; Song, S.; Long, Q.; Nillni, E.A.; Low, M.J.; Arvan, P.; Myers, M.G.; et al. Hypothalamic ER–associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Investig. 2018, 128, 1125–1140. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Disease | ERAD Substrate | References |
|---|---|---|
| Gaucher disease | GCase | [55,56,57,58] |
| Fabry disease | α-Gal A | [59,60] |
| Tay-Sachs disease | HexA (α subunit) | [61] |
| Cystic fibrosis | CFTR | [62,63,64,65] |
| diabetes insipidus | AQP2 | [66] |
| retinitis pigmentosa | rhodopsin | [67,68] |
| High blood pressure | vanin-1 | [69] |
| Stargardt disease | ABCA4 | [70] |
| Charcot-Marie-Tooth disease | PMP22 | [71] |
| Type I Bartter syndrome | NKCC2 | [72] |
| Type II Bartter syndrome | POMC | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuhisa, K.; Imaizumi, K. Loss of Function of Mutant IDS Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type II. Int. J. Mol. Sci. 2021, 22, 12227. https://doi.org/10.3390/ijms222212227
Matsuhisa K, Imaizumi K. Loss of Function of Mutant IDS Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type II. International Journal of Molecular Sciences. 2021; 22(22):12227. https://doi.org/10.3390/ijms222212227
Chicago/Turabian StyleMatsuhisa, Koji, and Kazunori Imaizumi. 2021. "Loss of Function of Mutant IDS Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type II" International Journal of Molecular Sciences 22, no. 22: 12227. https://doi.org/10.3390/ijms222212227