
Patient-Specific iPSC-Derived Neural Differentiated and Hepatocyte-like Cells, Carrying the Compound Heterozygous Mutation p.V1023Sfs*15/p.G992R, Present the “Variant” Biochemical Phenotype of Niemann-Pick Type C1 Disease


 and
and
Abstract
:1. Introduction
2. Results
2.1. Generation of iPSC-Derived Neural Differentiated Cells
2.2. Generation of iPSC-Derived Hepatocyte-like Cells
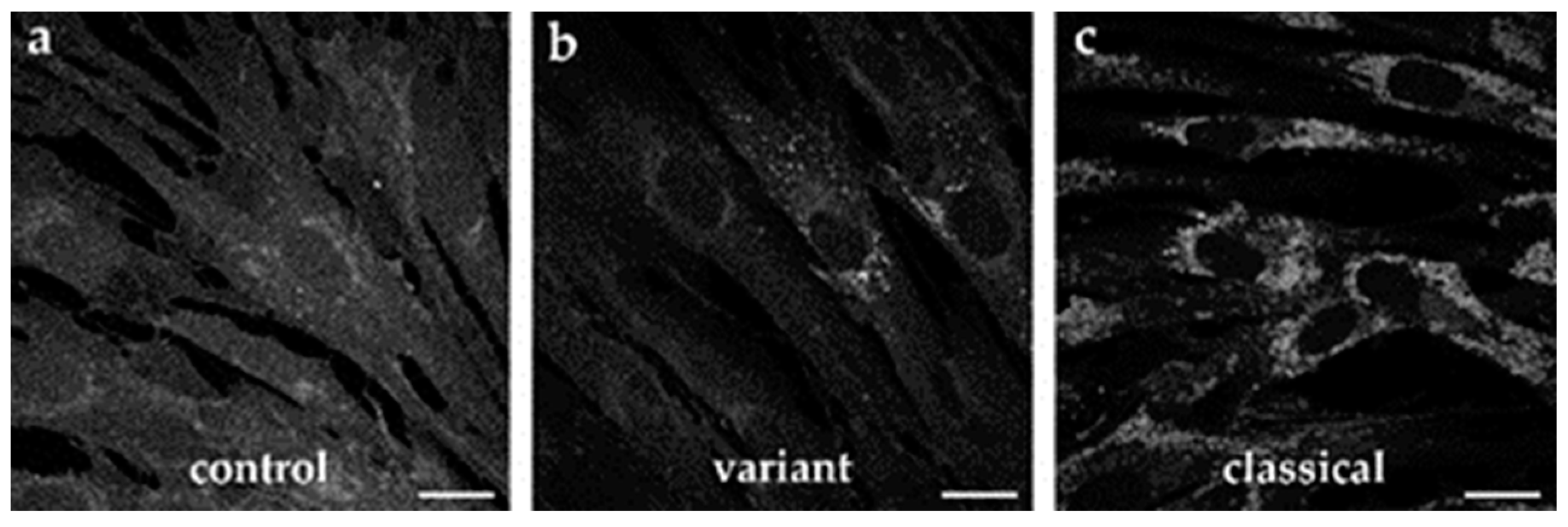
2.3. Cholesterol Accumulation in Patient-Specific Fibroblasts and iPSC-Derived Neural Differentiated and Hepatocyte-like Cells
2.4. NPC1 Expression in Patient-Specific Fibroblasts and iPSC-Derived Neural Differentiated and Hepatocyte-like Cells
2.5. Clinical Representation
3. Discussion
3.1. In Vitro Analysis of Patient-Specific Fibroblasts Revealed a “Variant” Biochemical Phenotype
3.2. iPSC-Derived Neural Differentiated Cell, Hepatocyte-like Cells and Fibroblasts Show a Comparable Phenotype
3.3. Cinical Presentation of a Patient with a “Variant” Biochemical Phenotype
3.4. Therapeutic Options for Patients with a “Variant” Biochemical Phenotype
4. Materials and Methods
4.1. Cell Culture of Human Dermal Fibroblasts
4.2. Neural Differentiation
4.3. Hepatic Differentiation
4.4. Periodic Acid-Schiff Assay
4.5. Cellular Uptake and Release of Indocyanine Green
4.6. Uptake of Low-Density Lipoprotein (LDL)
4.7. Filipin Staining
4.8. Immunocytochemistry
4.9. Colocalization Analysis
4.10. Western Blot Analysis
4.11. Endoglycosidase H Assay
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| αFP | Alpha-Fetoprotein |
| ALB | Albumin |
| BSA | Bovine Serum Albumin |
| ChT | Chitotriosidase |
| DE | Definitive Endoderm |
| DMSO | Dimethyl sulfoxide |
| Endo H | Endoglycosidase H |
| ER | Endoplasmic reticulum |
| GCS | Glucosylceramide synthase |
| GFAP | Glial fibrillary acidic protein |
| HDL | High Density Lipoprotein |
| HLCs | Hepatocyte-like cells |
| HNF4α | Hepatocyte nuclear factor 4 alpha |
| HPCs | Hepatic progenitor cells |
| ICG | Indocyanine green |
| iPSCs | Induced pluripotent stem cells |
| LDL | Low Density Lipoprotein |
| LSM | Laser Scanning Microscope |
| LSO | Lysosome-like storage organelle |
| NDCs | Neural differentiated cells |
| NPCs | Neural progenitor cells |
| NPC1 | Niemann-Pick type C1 protein |
| NP-C1 | Niemann-Pick disease type C1 |
| PAS | Periodic acid-Schiff |
| PFA | Paraformaldehyde |
| PSA-NCAM | Polysialylated-neural cell adhesion molecule |
References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulman, L.M.; David, N.J.; Weiner, W.J. Psychosis as the initial manifestation of adult-onset Niemann-Pick disease type C. Neurology 1995, 45, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Campo, J.V.; Stowe, R.; Slomka, G.; Byler, D.; Gracious, B. Psychosis as a presentation of physical disease in adolescence: A case of Niemann-Pick disease, type C. Dev. Med. Child Neurol. 1998, 40, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterfang, M.; Fietz, M.; Fahey, M.; Sullivan, D.; Leane, P.; Lubman, D.I.; Velakoulis, D. The Neuropsychiatry of Niemann-Pick Type C Disease in Adulthood. JNP 2006, 18, 158–170. [Google Scholar] [CrossRef]
- Muller, C.P.; Stephany, D.A.; Winkler, D.F.; Hoeg, J.M.; Demosky, S.J.; Wunderlich, J.R. Filipin as a flow microfluorometry probe for cellular cholesterol. Cytometry 1984, 5, 42–54. [Google Scholar] [CrossRef]
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Gazzah, N.; Juge, M.-C.; Pentchev, P.G.; Revol, A.; Louisot, P. Type C Niemann-Pick disease: Spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1991, 1096, 328–337. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 Disease: Correlations between NPC1 Mutations, Levels of NPC1 Protein, and Phenotypes Emphasize the Functional Significance of the Putative Sterol-Sensing Domain and of the Cysteine-Rich Luminal Loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Pagano, R.E.; et al. Niemann-Pick C Variant Detection by Altered Sphingolipid Trafficking and Correlation with Mutations within a Specific Domain of NPC1. Am. J. Hum. Genet. 2001, 68, 1361–1372. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, I.; Marcão, A.; Amaral, O.; Sá Miranda, M.; Vanier, M.T.; Millat, G. Niemann-Pick type C disease: NPC1 mutations associated with severe and mild cellular cholesterol trafficking alterations. Hum. Genet. 2001, 109, 24–32. [Google Scholar] [CrossRef]
- Peter, F.; Trilck, M.; Rabenstein, M.; Rolfs, A.; Frech, M.J. Dataset in support of the generation of Niemann-Pick disease Type C1 patient-specific iPS cell lines carrying the novel NPC1 mutation c.1180T>C or the prevalent c.3182T>C mutation—Analysis of pluripotency and neuronal differentiation. Data Brief 2017, 12, 123–131. [Google Scholar] [CrossRef]
- Wang, Y.; Alhaque, S.; Cameron, K.; Meseguer-Ripolles, J.; Lucendo-Villarin, B.; Rashidi, H.; Hay, D.C. Defined and Scalable Generation of Hepatocyte-like Cells from Human Pluripotent Stem Cells. JoVE 2017, 121, 55355. [Google Scholar] [CrossRef] [Green Version]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Vanier, M.T.; Wenger, D.A.; Patel, S.; Brady, R.O. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. USA 1985, 82, 8247–8251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipalia, N.H.; Huang, A.; Ralph, H.; Rujoi, M.; Maxfield, F.R. Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. J. Lipid Res. 2006, 47, 284–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argoff, C.E.; Comly, M.E.; Blanchette-Mackie, J.; Kruth, H.S.; Pye, H.T.; Goldin, E.; Kaneski, C.; Vanier, M.T.; Brady, R.O.; Pentchev, P.G. Type C Niemann-Pick disease: Cellular uncoupling of cholesterol homeostasis is linked to the severity of disruption in the intracellular transport of exogenously derived cholesterol. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1991, 1096, 319–327. [Google Scholar] [CrossRef]
- Greer, W.L.; Dobson, M.J.; Girouard, G.S.; Byers, D.M.; Riddell, D.C.; Neumann, P.E. Mutations in NPC1 Highlight a Conserved NPC1-Specific Cysteine-Rich Domain. Am. J. Hum. Genet. 1999, 65, 1252–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millat, G.; Baïlo, N.; Molinero, S.; Rodriguez, C.; Chikh, K.; Vanier, M.T. Niemann–Pick C disease: Use of denaturing high performance liquid chromatography for the detection of NPC1 and NPC2 genetic variations and impact on management of patients and families. Mol. Genet. Metab. 2005, 86, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Tängemo, C.; Weber, D.; Theiss, S.; Mengel, E.; Runz, H. Niemann-Pick Type C disease: Characterizing lipid levels in patients with variant lysosomal cholesterol storage. J. Lipid Res. 2011, 52, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shammas, H.; Kuech, E.-M.; Rizk, S.; Das, A.M.; Naim, H.Y. Different Niemann-Pick C1 Genotypes Generate Protein Phenotypes that Vary in their Intracellular Processing, Trafficking and Localization. Sci. Rep. 2019, 9, 5292. [Google Scholar] [CrossRef] [Green Version]
- Kulinski, A.; Vance, J.E. Lipid Homeostasis and Lipoprotein Secretion in Niemann-Pick C1-deficient Hepatocytes. J. Biol. Chem. 2007, 282, 1627–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, A.I.; Maxfield, F.R. Niemann-Pick type C disease: Molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011, 116, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Völkner, C.; Liedtke, M.; Petters, J.; Lukas, J.; Escobar, H.M.; Knuebel, G.; Bullerdiek, J.; Holzmann, C.; Hermann, A.; Frech, M.J. Generation of an iPSC line (AKOSi004-A) from fibroblasts of a female adult NPC1 patient, carrying the compound heterozygous mutation p.Val1023Serfs*15/p.Gly992Arg and of an iPSC line (AKOSi005-A) from a female adult control individual. Stem Cell Res. 2021, 50, 102127. [Google Scholar] [CrossRef] [PubMed]
- Trilck, M.; Peter, F.; Zheng, C.; Frank, M.; Dobrenis, K.; Mascher, H.; Rolfs, A.; Frech, M.J. Diversity of glycosphingolipid GM2 and cholesterol accumulation in NPC1 patient-specific iPSC-derived neurons. Brain Res. 2017, 1657, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Liedtke, M.; Hermann, A.; Frech, M.J. Pluripotent Stem Cells for Disease Modeling and DrugDiscovery in Niemann-Pick Type C1. Int. J. Mol. Sci. 2021, 22, 710. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and Chemical Correction of Cholesterol Accumulation and Impaired Autophagy in Hepatic and Neural Cells Derived from Niemann-Pick Type C Patient-Specific iPS Cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [Green Version]
- Soga, M.; Ishitsuka, Y.; Hamasaki, M.; Yoneda, K.; Furuya, H.; Matsuo, M.; Ihn, H.; Fusaki, N.; Nakamura, K.; Nakagata, N.; et al. HPGCD Outperforms HPBCD as a Potential Treatment for Niemann-Pick Disease Type C During Disease Modeling with iPS Cells. Stem Cells 2015, 33, 1075–1088. [Google Scholar] [CrossRef]
- Choi, H.Y.; Karten, B.; Chan, T.; Vance, J.E.; Greer, W.L.; Heidenreich, R.A.; Garver, W.S.; Francis, G.A. Impaired ABCA1-dependent Lipid Efflux and Hypoalphalipoproteinemia in Human Niemann-Pick type C Disease. J. Biol. Chem. 2003, 278, 32569–32577. [Google Scholar] [CrossRef] [Green Version]
- Ries, M.; Schaefer, E.; Lührs, T.; Mani, L.; Kuhn, J.; Vanier, M.T.; Krummenauer, F.; Gal, A.; Beck, M.; Mengel, E. Critical assessment of chitotriosidase analysis in the rational laboratory diagnosis of children with Gaucher disease and Niemann–Pick disease type A/B and C. J. Inherit. Metab. Dis. 2006, 29, 647–652. [Google Scholar] [CrossRef]
- McKay Bounford, K.; Gissen, P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J. Neurol. 2014, 261, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Wajner, A.; Michelin, K.; Burin, M.G.; Pires, R.F.; Pereira, M.L.S.; Giugliani, R.; Coelho, J.C. Biochemical characterization of chitotriosidase enzyme: Comparison between normal individuals and patients with Gaucher and with Niemann–Pick diseases. Clin. Biochem. 2004, 37, 893–897. [Google Scholar] [CrossRef]
- Ribas, G.S.; Souza, H.M.; de Mari, J.; Deon, M.; Mescka, C.; Saraiva-Pereira, M.L.; Kessler, R.; Trapp, F.; Michelin, K.; Burin, M.; et al. Selective screening of Niemann–Pick type C Brazilian patients by cholestane-3β,5α,6β-triol and chitotriosidase measurements followed by filipin staining and NPC1/NPC2 gene analysis. Clin. Chim. Acta 2016, 459, 57–62. [Google Scholar] [CrossRef]
- Pineda, M.; Perez-Poyato, M.S.; O’Callaghan, M.; Vilaseca, M.A.; Pocovi, M.; Domingo, R.; Ruiz Portal, L.; Verdú Pérez, A.; Temudo, T.; Gaspar, A.; et al. Clinical experience with miglustat therapy in pediatric patients with Niemann–Pick disease type C: A case series. Mol. Genet. Metab. 2010, 99, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Piña-Aguilar, R.E.; Vera-Loaiza, A.; Chacón-Camacho, O.F.; Zenteno, J.C.; Nuñez-Orozco, L.; Santillán-Hernández, Y. Clinical and Genetic Characteristics of Mexican Patients with Juvenile Presentation of Niemann-Pick Type C Disease. Case Rep. Neurol. Med. 2014, 2014, 785890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, F.; Dubbioso, R.; Iodice, R.; Topa, A.; Dardis, A.; Russo, C.V.; Ruggiero, L.; Tozza, S.; Filla, A.; Santoro, L. Central cholinergic dysfunction in the adult form of Niemann Pick disease type C: A further link with Alzheimer’s disease? J. Neurol. 2014, 261, 804–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain 2006, 130, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterfang, M.; Fietz, M.; Abel, L.; Bowman, E.; Mocellin, R.; Velakoulis, D. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2009, 32, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Macías-Vidal, J.; Rodríguez-Pascau, L.; Sánchez-Ollé, G.; Lluch, M.; Vilageliu, L.; Grinberg, D.; Coll, M.J. Molecular analysis of 30 Niemann-Pick type C patients from Spain. Clin. Genet. 2011, 80, 39–49. [Google Scholar] [CrossRef]
- Abel, L.A.; Walterfang, M.; Fietz, M.; Bowman, E.A.; Velakoulis, D. Saccades in adult Niemann-Pick disease type C reflect frontal, brainstem, and biochemical deficits. Neurology 2009, 72, 1083–1086. [Google Scholar] [CrossRef]
- Jahnova, H.; Dvorakova, L.; Vlaskova, H.; Hulkova, H.; Poupetova, H.; Hrebicek, M.; Jesina, P. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: A surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J. Rare Dis. 2014, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Watari, H.; Blanchette-Mackie, E.J.; Dwyer, N.K.; Glick, J.M.; Patel, S.; Neufeld, E.B.; Brady, R.O.; Pentchev, P.G.; Strauss, J.F. Niemann-Pick C1 protein: Obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc. Natl. Acad. Sci. USA 1999, 96, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Platt, F.M. Lipids on Trial: The Search for the Offending Metabolite in Niemann-Pick type C Disease. Traffic 2010, 11, 419–428. [Google Scholar] [CrossRef]
- Vanier, M.T. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef]
- Yanagimoto, C.; Harada, M.; Kumemura, H.; Abe, M.; Koga, H.; Sakata, M.; Kawaguchi, T.; Terada, K.; Hanada, S.; Taniguchi, E.; et al. Copper incorporation into ceruloplasmin is regulated by Niemann-Pick C1 protein. Hepatol. Res. 2011, 41, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Parker, R.; Davidson, C.; Yanjanin, N.; Kelley, T.J.; Corey, D.; Atkinson, J.; Porter, F.; Arai, H.; Walkley, S.U.; et al. Altered vitamin E status in Niemann-Pick type C disease. J. Lipid Res. 2011, 52, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.-T.; Tu, X.; Yang, K.; Wu, J.; Repa, J.J.; Yan, N. Tonic prime-boost of STING signalling mediates Niemann–Pick disease type C. Nature 2021, 596, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Miglustat: A Review of Its Use in Niemann-Pick Disease Type C. Drugs 2014, 74, 61–74. [Google Scholar] [CrossRef]
- Jamrozik, Z.; Szczudlik, P.; Ługowska, A.; Weiß, S.; Rolfs, A.; Czartoryska, B.; Kwieciński, H. A case report of ‘variant’ biochemical phenotype of Niemann-Pick C disease and a discussion of therapeutic options. Neurol. Neurochir. Pol. 2013, 47, 86–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesús, S.; Cáceres-Redondo, M.T.; Carrillo, F.; Cordones, I.; Escudero, M.; Macías-Vidal, J.; Coll, M.J.; Bautista, J.; Mir, P. Adult form of Niemann-Pick type C with the variant biochemical phenotype on treatment with Miglustat. Parkinsonism Relat. Disord. 2013, 19, 916–917. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | Patient | References | |
|---|---|---|---|
| creatinkinase | U/l | 17 | 30–179 |
| triglyceride | mg/dL | 152 | <150 |
| cholesterol (total) | mg/dL | 216 | <200 |
| HDL-cholesterol | mg/dL | 40 | >40 |
| LDL/HDL | 3.6 | <3 | |
| iron | µg/dL | 39 | 50–170 |
| transferrin | % | 8.7 | 15–45 |
| erythrocytes | /pl | 4.91 | 3.7–4.8 |
| chitotriosidase | nmol/mL/h | 203 | 20–100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Völkner, C.; Liedtke, M.; Untucht, R.; Hermann, A.; Frech, M.J. Patient-Specific iPSC-Derived Neural Differentiated and Hepatocyte-like Cells, Carrying the Compound Heterozygous Mutation p.V1023Sfs*15/p.G992R, Present the “Variant” Biochemical Phenotype of Niemann-Pick Type C1 Disease. Int. J. Mol. Sci. 2021, 22, 12184. https://doi.org/10.3390/ijms222212184
Völkner C, Liedtke M, Untucht R, Hermann A, Frech MJ. Patient-Specific iPSC-Derived Neural Differentiated and Hepatocyte-like Cells, Carrying the Compound Heterozygous Mutation p.V1023Sfs*15/p.G992R, Present the “Variant” Biochemical Phenotype of Niemann-Pick Type C1 Disease. International Journal of Molecular Sciences. 2021; 22(22):12184. https://doi.org/10.3390/ijms222212184
Chicago/Turabian StyleVölkner, Christin, Maik Liedtke, Robert Untucht, Andreas Hermann, and Moritz J. Frech. 2021. "Patient-Specific iPSC-Derived Neural Differentiated and Hepatocyte-like Cells, Carrying the Compound Heterozygous Mutation p.V1023Sfs*15/p.G992R, Present the “Variant” Biochemical Phenotype of Niemann-Pick Type C1 Disease" International Journal of Molecular Sciences 22, no. 22: 12184. https://doi.org/10.3390/ijms222212184