
Insertion of Calcium-Permeable AMPA Receptors during Epileptiform Activity In Vitro Modulates Excitability of Principal Neurons in the Rat Entorhinal Cortex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
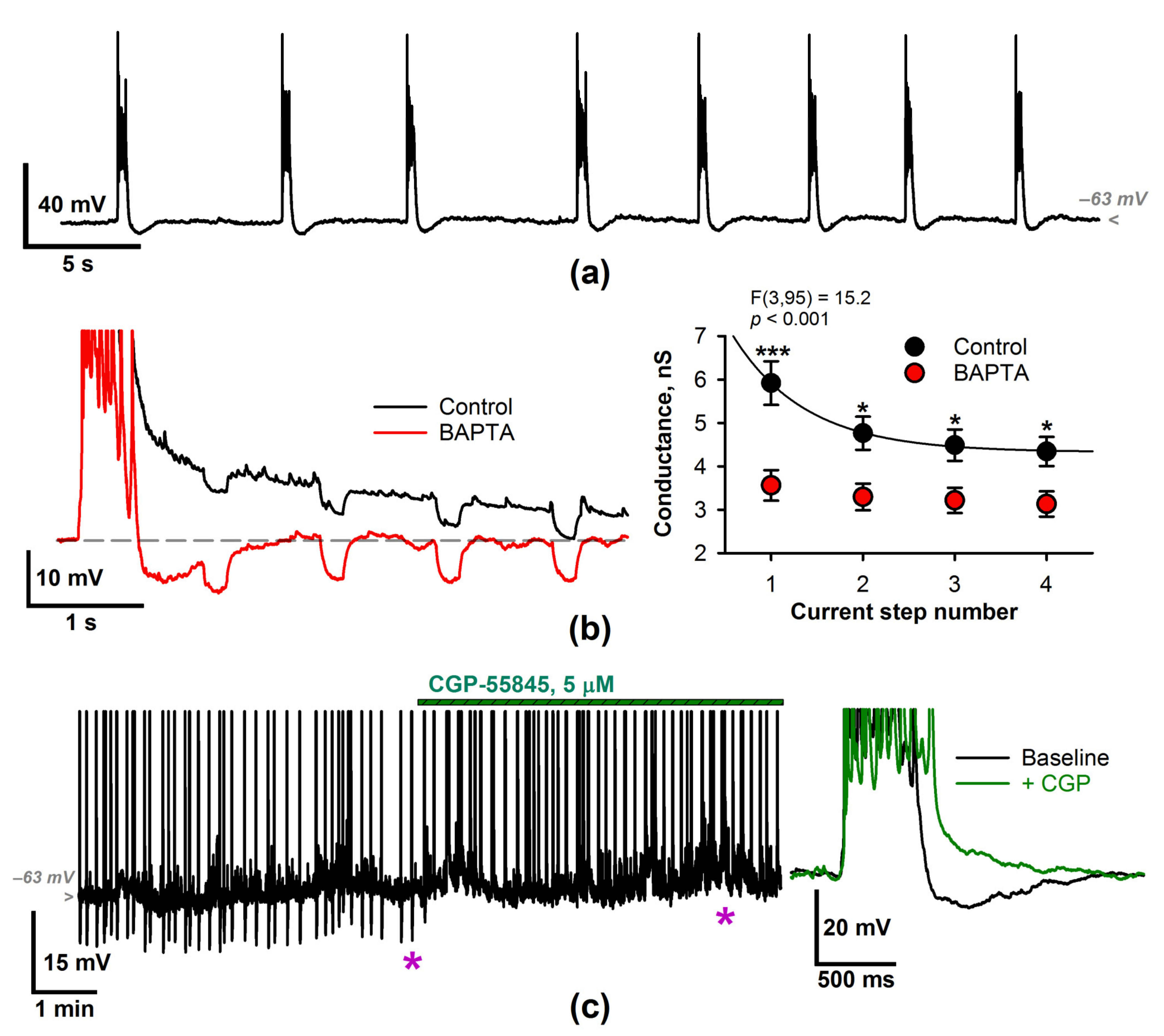
2.1. Epileptiform Activity Increases the Membrane Input Conductance of the Entorhinal Neurons
2.2. The Effect of a CP-AMPAR Blockade on the Generation of SSDs
2.3. The Input Conductance Decrease following IEM-1460 Application Results in an Increased Probability of Discharges
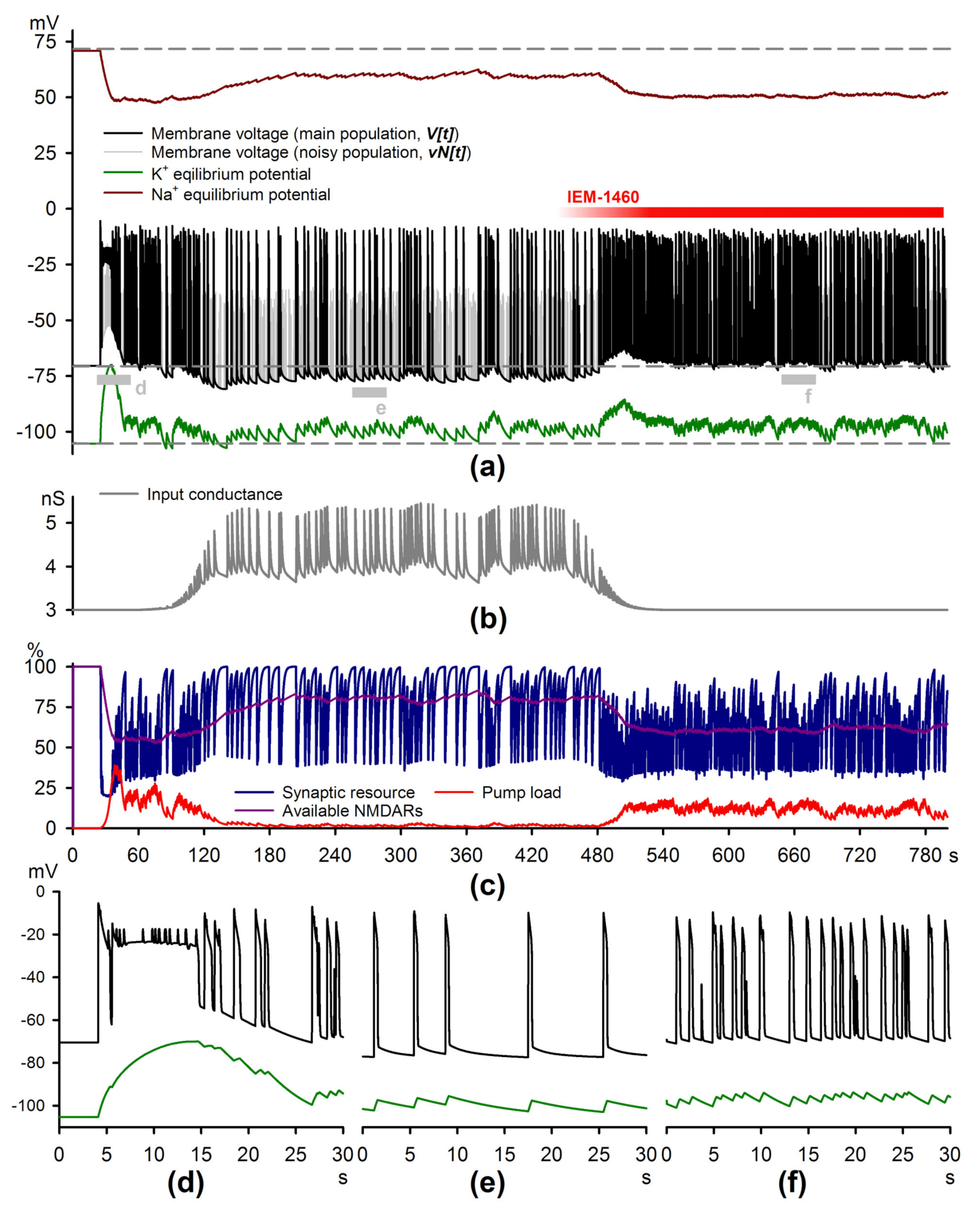
2.4. Simulations of the Epileptiform Activity
3. Discussion
3.1. Membrane Conductance as a Factor of Seizure Generation
3.2. The Physiological Role of Abnormal Expression of CP-AMPARs during Seizures
4. Materials and Methods
4.1. Animals
4.2. Brain Slice Preparation
4.3. In Vitro Model of Epileptiform Activity
4.4. The Whole-Cell Patch-Clamp Recordings
4.5. Statistics
4.6. The Mathematical Model of Epileptiform Activity
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Ma, Y.-Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circ. 2021, 11, 711564. [Google Scholar] [CrossRef]
- Isaac, J.T.R.; Ashby, M.C.; McBain, C.J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 2007, 54, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Lalanne, T.; Oyrer, J.; Farrant, M.; Sjöström, P.J. Synapse Type-Dependent Expression of Calcium-Permeable AMPA Receptors. Front. Synaptic Neurosci. 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Buldakova, S.L.; Kim, K.K.; Tikhonov, D.B.; Magazanik, L.G. Selective blockade of Ca2+ permeable AMPA receptors in CA1 area of rat hippocampus. Neuroscience 2007, 144, 88–99. [Google Scholar] [CrossRef]
- Burman, R.J.; Selfe, J.S.; Lee, J.H.; van den Berg, M.; Calin, A.; Codadu, N.K.; Wright, R.; Newey, S.E.; Parrish, R.R.; Katz, A.A.; et al. Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain 2019, 142, 3482–3501. [Google Scholar] [CrossRef] [Green Version]
- Goodkin, H.P.; Yeh, J.-L.; Kapur, J. Status Epilepticus Increases the Intracellular Accumulation of GABAA Receptors. J. Neurosci. 2005, 25, 5511–5520. [Google Scholar] [CrossRef] [Green Version]
- Goodkin, H.P.; Joshi, S.; Mtchedlishvili, Z.; Brar, J.; Kapur, J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J. Neurosci. 2008, 28, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Meletti, S.; Lucchi, C.; Monti, G.; Giovannini, G.; Bedin, R.; Trenti, T.; Rustichelli, C.; Biagini, G. Decreased allopregnanolone levels in cerebrospinal fluid obtained during status epilepticus. Epilepsia 2017, 58, e16–e20. [Google Scholar] [CrossRef]
- Meletti, S.; Lucchi, C.; Monti, G.; Giovannini, G.; Bedin, R.; Trenti, T.; Rustichelli, C.; Biagini, G. Low levels of progesterone and derivatives in cerebrospinal fluid of patients affected by status epilepticus. J. Neurochem. 2018, 147, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchi, C.; Costa, A.M.; Senn, L.; Messina, S.; Rustichelli, C.; Biagini, G. Augmentation of endogenous neurosteroid synthesis alters experimental status epilepticus dynamics. Epilepsia 2020, 61, e129–e134. [Google Scholar] [CrossRef] [PubMed]
- Abegg, M.H.; Savic, N.; Ehrengruber, M.U.; McKinney, R.A.; Gähwiler, B.H. Epileptiform activity in rat hippocampus strengthens excitatory synapses. J. Physiol. 2004, 554, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debanne, D.; Thompson, S.M.; Gähwiler, B.H. A brief period of epileptiform activity strengthens excitatory synapses in the rat hippocampus in vitro. Epilepsia 2006, 47, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Rajasekaran, K.; Sun, H.; Williamson, J.; Kapur, J. Enhanced AMPA receptor-mediated neurotransmission on CA1 pyramidal neurons during status epilepticus. Neurobiol. Dis. 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Rajasekaran, K.; Todorovic, M.; Kapur, J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann. Neurol. 2012, 72, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ergina, J.L.; Amakhin, D.V.; Postnikova, T.Y.; Soboleva, E.B.; Zaitsev, A.V. Short-Term Epileptiform Activity Potentiates Excitatory Synapses but Does Not Affect Intrinsic Membrane Properties of Pyramidal Neurons in the Rat Hippocampus In Vitro. Biomedicines 2021, 9, 1374. [Google Scholar] [CrossRef] [PubMed]
- Amakhin, D.V.; Soboleva, E.B.; Ergina, J.L.; Malkin, S.L.; Chizhov, A.V.; Zaitsev, A.V. Seizure-Induced Potentiation of AMPA Receptor-Mediated Synaptic Transmission in the Entorhinal Cortex. Front. Cell. Neurosci. 2018, 12, 486. [Google Scholar] [CrossRef]
- Rajasekaran, K.; Joshi, S.; Kozhemyakin, M.; Todorovic, M.S.; Kowalski, S.; Balint, C.; Kapur, J. Receptor trafficking hypothesis revisited: Plasticity of AMPA receptors during established status epilepticus. Epilepsia 2013, 54, 14–16. [Google Scholar] [CrossRef]
- Joshi, S.; Kapur, J. Mechanisms of status epilepticus: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor hypothesis. Epilepsia 2018, 59, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Szczurowska, E.; Mares, P. An antagonist of calcium permeable AMPA receptors, IEM1460: Anticonvulsant action in immature rats? Epilepsy Res. 2015, 109, 106–113. [Google Scholar] [CrossRef]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adotevi, N.; Lewczuk, E.; Sun, H.; Joshi, S.; Dabrowska, N.; Shan, S.; Williamson, J.; Kapur, J. α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor Plasticity Sustains Severe, Fatal Status Epilepticus. Ann. Neurol. 2020, 87, 84–96. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Zaitsev, A.V. Calcium-permeable AMPA receptors are essential to the synaptic plasticity induced by epileptiform activity in rat hippocampal slices. Biochem. Biophys. Res. Commun. 2020, 529, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, J.V.; Burman, R.J.; Katz, A.A.; Akerman, C.J. Ion dynamics during seizures. Front. Cell. Neurosci. 2015, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevelyan, A.J.; Sussillo, D.; Yuste, R. Feedforward Inhibition Contributes to the Control of Epileptiform Propagation Speed. J. Neurosci. 2007, 27, 3383–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Qiao, Z.; Liu, N.; Gao, L.; Wei, L.; Liu, A.; Ma, Z.; Wang, F.; Hou, S.; Li, J.; et al. Stereotypical patterns of epileptiform calcium signal in hippocampal CA1, CA3, dentate gyrus and entorhinal cortex in freely moving mice. Sci. Rep. 2019, 9, 4518. [Google Scholar] [CrossRef] [PubMed]
- Nagarkatti, N.; Deshpande, L.S.; DeLorenzo, R.J. Development of the calcium plateau following status epilepticus: Role of calcium in epileptogenesis. Expert Rev. Neurother. 2009, 9, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badea, T.; Goldberg, J.; Mao, B.; Yuste, R. Calcium imaging of epileptiform events with single-cell resolution. J. Neurobiol. 2001, 48, 215–227. [Google Scholar] [CrossRef]
- Pisani, A.; Bonsi, P.; Martella, G.; De Persis, C.; Costa, C.; Pisani, F.; Bernardi, G.; Calabresi, P. Intracellular calcium increase in epileptiform activity: Modulation by levetiracetam and lamotrigine. Epilepsia 2004, 45, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avoli, M.; D’Antuono, M.; Louvel, J.; Köhling, R.; Biagini, G.; Pumain, R.; D’Arcangelo, G.; Tancredi, V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 2002, 68, 167–201. [Google Scholar] [CrossRef]
- Vismer, M.S.; Forcelli, P.A.; Skopin, M.D.; Gale, K.; Koubeissi, M.Z. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circ. 2015, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Chizhov, A.V.; Zefirov, A.V.; Amakhin, D.V.; Smirnova, E.Y.; Zaitsev, A.V. Minimal model of interictal and ictal discharges “Epileptor-2”. PLoS Comput. Biol. 2018, 14. [Google Scholar] [CrossRef]
- Zhang, C.L.; Dreier, G.P.; Heinemann, U. Paroxysmal epileptiform discharges in temporal lobe slices after prolonged exposure to low magnesium are resistant to clinically used anticonvulsants. Epilepsy Res. 1995, 20, 105–111. [Google Scholar] [CrossRef]
- Empson, R.M.; Jefferys, J.G.R. Ca2+ entry through L-type Ca2+ channels helps terminate epileptiform activity by activation of a Ca2+ dependent afterhyperpolarization in hippocampal CA3. Neuroscience 2001, 102, 297–306. [Google Scholar] [CrossRef]
- De Sevilla, D.F.; Garduño, J.; Galván, E.; Buño, W. Calcium-Activated Afterhyperpolarizations Regulate Synchronization and Timing of Epileptiform Bursts in Hippocampal CA3 Pyramidal Neurons. J. Neurophysiol. 2006, 96, 3028–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.S. Epileptiform events induced by GABA-antagonists in entorhinal cortical cells in vitro are partly mediated by N-methyl-D-aspartate receptors. Brain Res. 1988, 457, 113–121. [Google Scholar] [CrossRef]
- Zorumski, C.F.; Thio, L.L.; Clark, G.D.; Clifford, D.B. Calcium influx through N-methyl-d-aspartate channels activates a potassium current in postnatal rat hippocampal neurons. Neurosci. Lett. 1989, 99, 293–299. [Google Scholar] [CrossRef]
- Mahanty, N.K.; Sah, P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 1998, 394, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Lalanne, T.; Oyrer, J.; Mancino, A.; Gregor, E.; Chung, A.; Huynh, L.; Burwell, S.; Maheux, J.; Farrant, M.; Sjöström, P.J. Synapse-specific expression of calcium-permeable AMPA receptors in neocortical layer 5. J. Physiol. 2016, 594, 837–861. [Google Scholar] [CrossRef]
- Beck, H.; Yaari, Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat. Rev. Neurosci. 2008, 9, 357–369. [Google Scholar] [CrossRef]
- Amarillo, Y.; Zagha, E.; Mato, G.; Rudy, B.; Nadal, M.S. The interplay of seven subthreshold conductances controls the resting membrane potential and the oscillatory behavior of thalamocortical neurons. J. Neurophysiol. 2014, 112, 393–410. [Google Scholar] [CrossRef]
- Naylor, D.E.; Liu, H.; Niquet, J.; Wasterlain, C.G. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 2013, 54, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaila, K.; Ruusuvuori, E.; Seja, P.; Voipio, J.; Puskarjov, M. GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 2014, 26, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Arnold, E.C.; McMurray, C.; Gray, R.; Johnston, D. Epilepsy-induced reduction in HCN channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal CA1 neurons. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halabisky, B.; Parada, I.; Buckmaster, P.S.; Prince, D.A. Excitatory input onto hilar somatostatin interneurons is increased in a chronic model of epilepsy. J. Neurophysiol. 2010, 104, 2214–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Thamattoor, A.K.; Leroy, C.; Buckmaster, P.S. Surviving mossy cells enlarge and receive more excitatory synaptic input in a mouse model of temporal lobe epilepsy. Hippocampus 2015, 25, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-induced Status Epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef]
- Smirnova, E.Y.; Amakhin, D.V.; Malkin, S.L.; Chizhov, A.V.; Zaitsev, A.V. Acute Changes in Electrophysiological Properties of Cortical Regular-Spiking Cells Following Seizures in a Rat Lithium–Pilocarpine Model. Neuroscience 2018, 379, 202–215. [Google Scholar] [CrossRef]
- Alger, B.E.; Williamson, A. A transient calcium-dependent potassium component of the epileptiform burst after-hyperpolarization in rat hippocampus. J. Physiol. 1988, 399, 191–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, M.E.; Yoshida, M.; Fransén, E.A. Low-frequency summation of synaptically activated transient receptor potential channel-mediated depolarizations. Eur. J. Neurosci. 2011, 34, 578–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Reboreda, A.; Alonso, A.; Barker, P.A.; Séguéla, P. TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 2011, 21, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Haj-Dahmane, S.; Andrade, R. Ionic Mechanism of the Slow Afterdepolarization Induced by Muscarinic Receptor Activation in Rat Prefrontal Cortex. J. Neurophysiol. 1998, 80, 1197–1210. [Google Scholar] [CrossRef]
- Riquelme, D.A.; Peralta, F.A.; Navarro, F.D.; Moreno, C.; Leiva-Salcedo, E. Ican (Trpm4) contributes to the intrinsic excitability of prefrontal cortex layer 2/3 pyramidal neurons. Int. J. Mol. Sci. 2021, 22, 5268. [Google Scholar] [CrossRef]
- Neymotin, S.A.; McDougal, R.A.; Bulanova, A.S.; Zeki, M.; Lakatos, P.; Terman, D.; Hines, M.L.; Lytton, W.W. Calcium regulation of HCN channels supports persistent activity in a multiscale model of neocortex. Neuroscience 2016, 316, 344–366. [Google Scholar] [CrossRef] [Green Version]
- Albertson, A.J.; Williams, S.B.; Hablitz, J.J. Regulation of epileptiform discharges in rat neocortex by HCN channels. J. Neurophysiol. 2013, 110, 1733–1743. [Google Scholar] [CrossRef] [Green Version]
- Gu, N.; Vervaeke, K.; Hu, H.; Storm, J.F. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J. Physiol. 2005, 566, 689–715. [Google Scholar] [CrossRef]
- MartÍnez-François, J.R.; Fernández-Agüera, M.C.; Nathwani, N.; Lahmann, C.; Burnham, V.L.; Danial, N.N.; Yellen, G. BAD and KATP channels regulate neuron excitability and epileptiform activity. Elife 2018, 7, e32721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemak, M.S.; Voloshanenko, O.; Draguhn, A.; Egorov, A.V. KATP channels modulate intrinsic firing activity of immature entorhinal cortex layer III neurons. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Igelström, K.M. Is slack an intrinsic seizure terminator? Neuroscientist 2013, 19, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, M.N.; Mohan, S.; Biala, Y.; Yaari, Y. Differential contributions of Ca2+-activated K+ channels and Na+/K+-ATPases to the generation of the slow afterhyperpolarization in CA1 pyramidal cells. Hippocampus 2018, 28, 338–357. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Sherathiya, V.N.; Matthew Oh, M.; Disterhoft, J.F. Persistent firing in lec iii neurons is differentially modulated by learning and aging. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.; Foehring, R.C.; Tzingounis, A. V The calcium-activated slow AHP: Cutting through the Gordian knot. Front. Cell. Neurosci. 2012, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.C.; Combe, C.L.; Gasparini, S. Differential contribution of Ca2+-dependent mechanisms to hyperexcitability in layer V neurons of the medial entorhinal cortex. Front. Cell. Neurosci. 2017, 11, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grooms, S.Y.; Opitz, T.; Bennett, M.V.; Zukin, R.S. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proc. Natl. Acad. Sci. USA 2000, 97, 3631–3636. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Roth, S.U.; Kiessling, M. Kainate-induced epilepsy alters protein expression of AMPA receptor subunits GluR1, GluR2 and AMPA receptor binding protein in the rat hippocampus. Acta Neuropathol. 2001, 101, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.M.; Koh, S.; Rio, C.; Wang, C.; Lamperti, E.D.; Sharma, D.; Corfas, G.; Jensen, F.E. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J. Neurosci. 2001, 21, 8154–8163. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.K. Selective reduction of GluR2 protein in adult hippocampal CA3 neurons following status epilepticus but prior to cell loss. Hippocampus 1998, 8, 511–525. [Google Scholar] [CrossRef]
- Malkin, S.L.; Amakhin, D.V.; Veniaminova, E.A.; Kim, K.K.; Zubareva, O.E.; Magazanik, L.G.; Zaitsev, A.V. Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar] [CrossRef]
- Lippman-Bell, J.J.; Zhou, C.; Sun, H.; Feske, J.S.; Jensen, F.E. Early-life seizures alter synaptic calcium-permeable AMPA receptor function and plasticity. Mol. Cell. Neurosci. 2016, 76, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Benke, T. O brother, wherefore are thou? Calcium-permeable AMPA receptors make an appearance in adult status epilepticus. Epilepsy Curr. 2013, 13, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Borowicz, K.; Banach, M. Effect of IEM 1460—a selective antagonist of GluR2-lacking AMPA receptors—on the action of conventional antiepileptic drugs against maximal electroshock in mice. J. Pre-Clin. Clin. Res. 2007, 1, 39–40. [Google Scholar]
- Amakhin, D.V.; Ergina, J.L.; Chizhov, A.V.; Zaitsev, A.V. Synaptic conductances during interictal discharges in pyramidal neurons of rat entorhinal cortex. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Chizhov, A.V.; Amakhin, D.V.; Zaitsev, A.V. Spatial propagation of interictal discharges along the cortex. Biochem. Biophys. Res. Commun. 2019, 508, 1245–1251. [Google Scholar] [CrossRef]
- Jahr, C.; Stevens, C. Voltage dependence of NMDA-activated macroscopic conductances predicted by single-channel kinetics. J. Neurosci. 1990, 10, 3178–3182. [Google Scholar] [CrossRef] [PubMed]
- Tsodyks, M.; Pawelzik, K.; Markram, H. Neural Networks with Dynamic Synapses. Neural Comput. 1998, 10, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Kager, H.; Wadman, W.J.; Somjen, G.G. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J. Neurophysiol. 2000, 84, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Cressman, J.R.; Ullah, G.; Ziburkus, J.; Schiff, S.J.; Barreto, E.; Barreto, E. The influence of sodium and potassium dynamics on excitability, seizures, and the stability of persistent states: I. Single neuron dynamics. J. Comput. Neurosci. 2009, 26, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Antonov, S.M.; Johnson, J.W.; Lukomskaya, N.Y.; Potapyeva, N.N.; Gmiro, V.E.; Magazanik, L.G. Novel adamantane derivatives act as blockers of open ligand-gated channels and as anticonvulsants. Mol. Pharmacol. 1995, 47, 558–567. [Google Scholar] [PubMed]
- Wollmuth, L.P.; Sakmann, B. Different mechanisms of Ca2+ transport in NMDA and Ca2+-permeable AMPA glutamate receptor channels. J. Gen. Physiol. 1998, 112, 623–636. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amakhin, D.V.; Soboleva, E.B.; Chizhov, A.V.; Zaitsev, A.V. Insertion of Calcium-Permeable AMPA Receptors during Epileptiform Activity In Vitro Modulates Excitability of Principal Neurons in the Rat Entorhinal Cortex. Int. J. Mol. Sci. 2021, 22, 12174. https://doi.org/10.3390/ijms222212174
Amakhin DV, Soboleva EB, Chizhov AV, Zaitsev AV. Insertion of Calcium-Permeable AMPA Receptors during Epileptiform Activity In Vitro Modulates Excitability of Principal Neurons in the Rat Entorhinal Cortex. International Journal of Molecular Sciences. 2021; 22(22):12174. https://doi.org/10.3390/ijms222212174
Chicago/Turabian StyleAmakhin, Dmitry V., Elena B. Soboleva, Anton V. Chizhov, and Aleksey V. Zaitsev. 2021. "Insertion of Calcium-Permeable AMPA Receptors during Epileptiform Activity In Vitro Modulates Excitability of Principal Neurons in the Rat Entorhinal Cortex" International Journal of Molecular Sciences 22, no. 22: 12174. https://doi.org/10.3390/ijms222212174
APA StyleAmakhin, D. V., Soboleva, E. B., Chizhov, A. V., & Zaitsev, A. V. (2021). Insertion of Calcium-Permeable AMPA Receptors during Epileptiform Activity In Vitro Modulates Excitability of Principal Neurons in the Rat Entorhinal Cortex. International Journal of Molecular Sciences, 22(22), 12174. https://doi.org/10.3390/ijms222212174