
Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model

 ,
, 
 , , ,
, , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
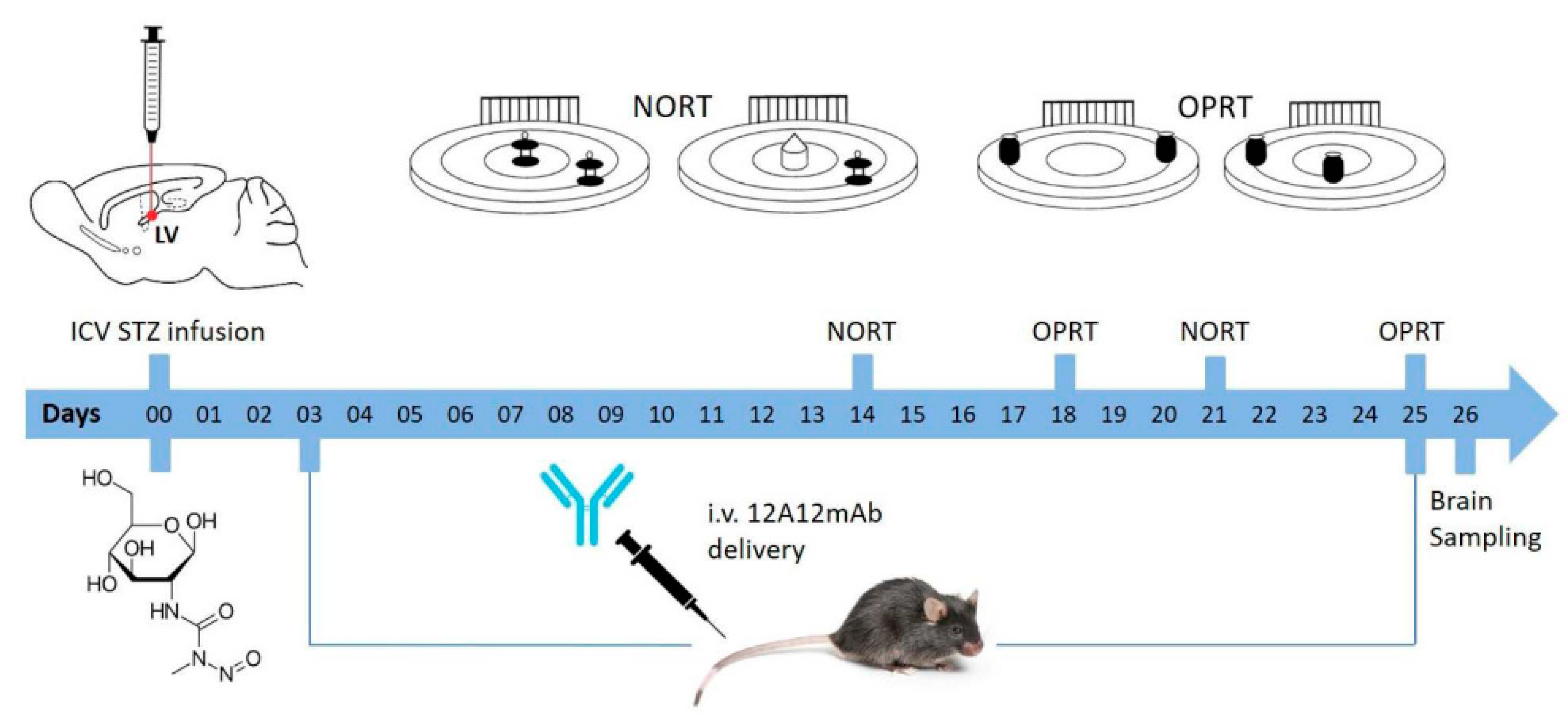
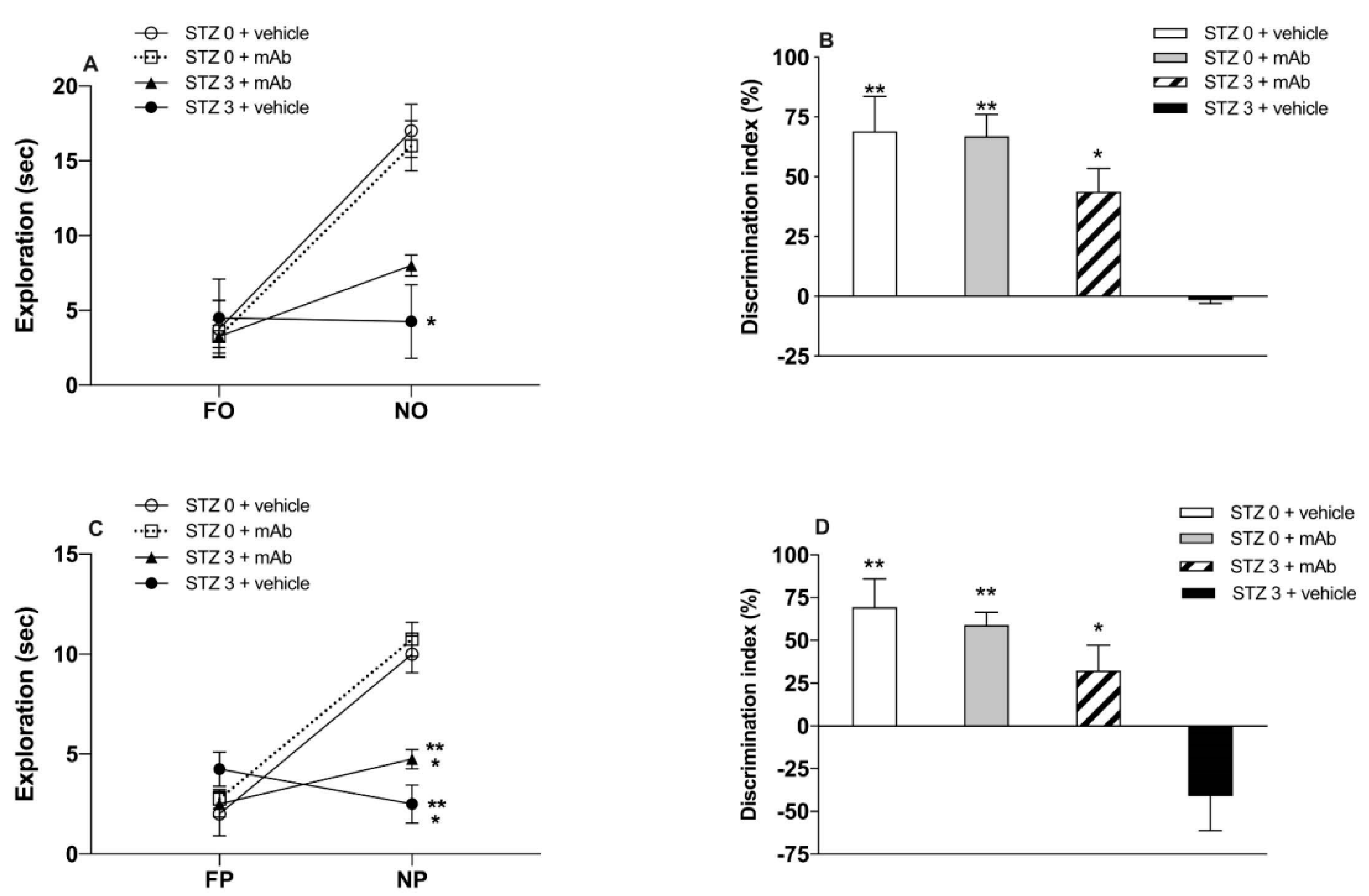
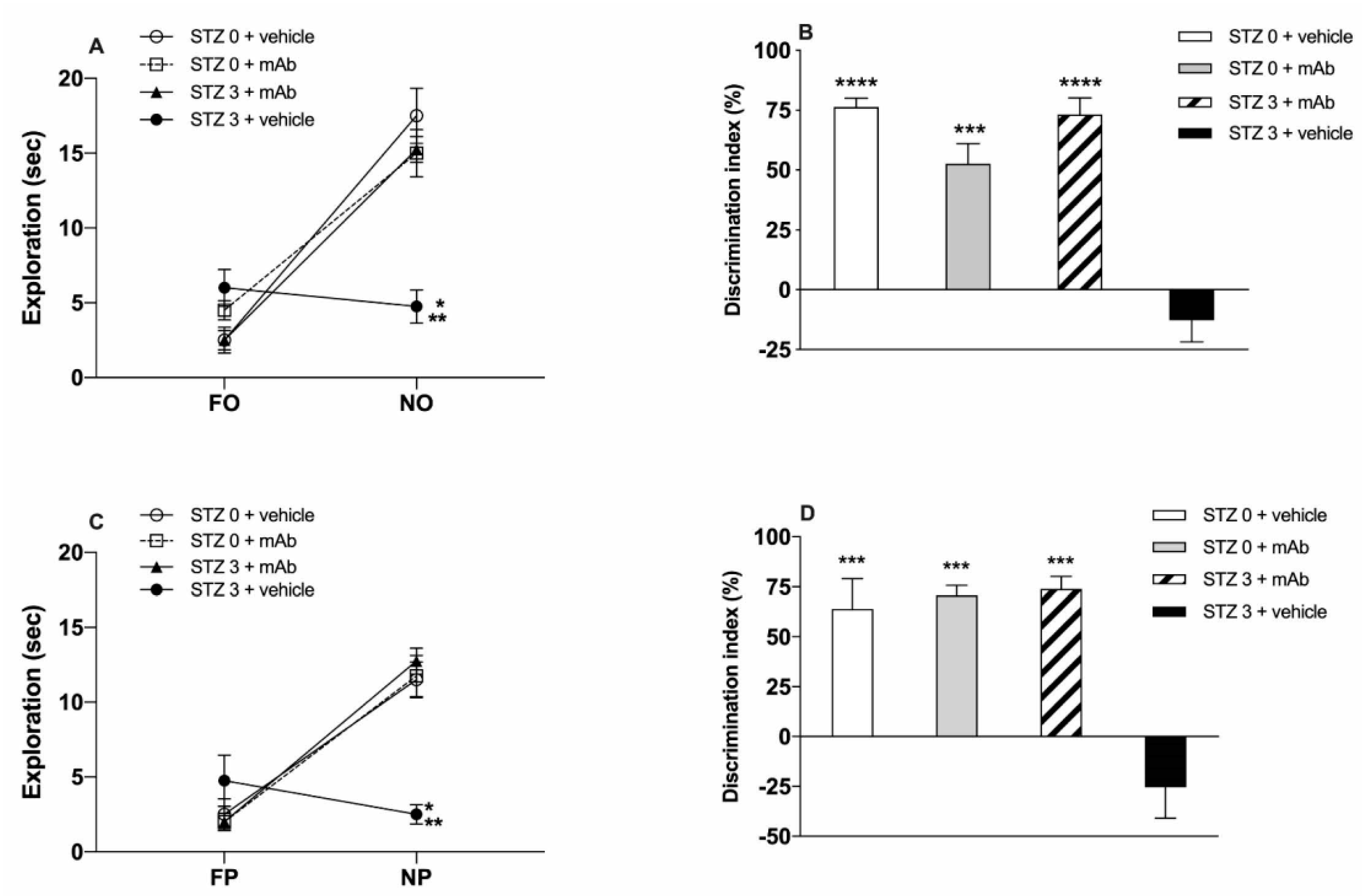
2.1. STZ-Induced sAD-Like Deficits of Memory Function Are Rescued by 12A12mAb Immunization
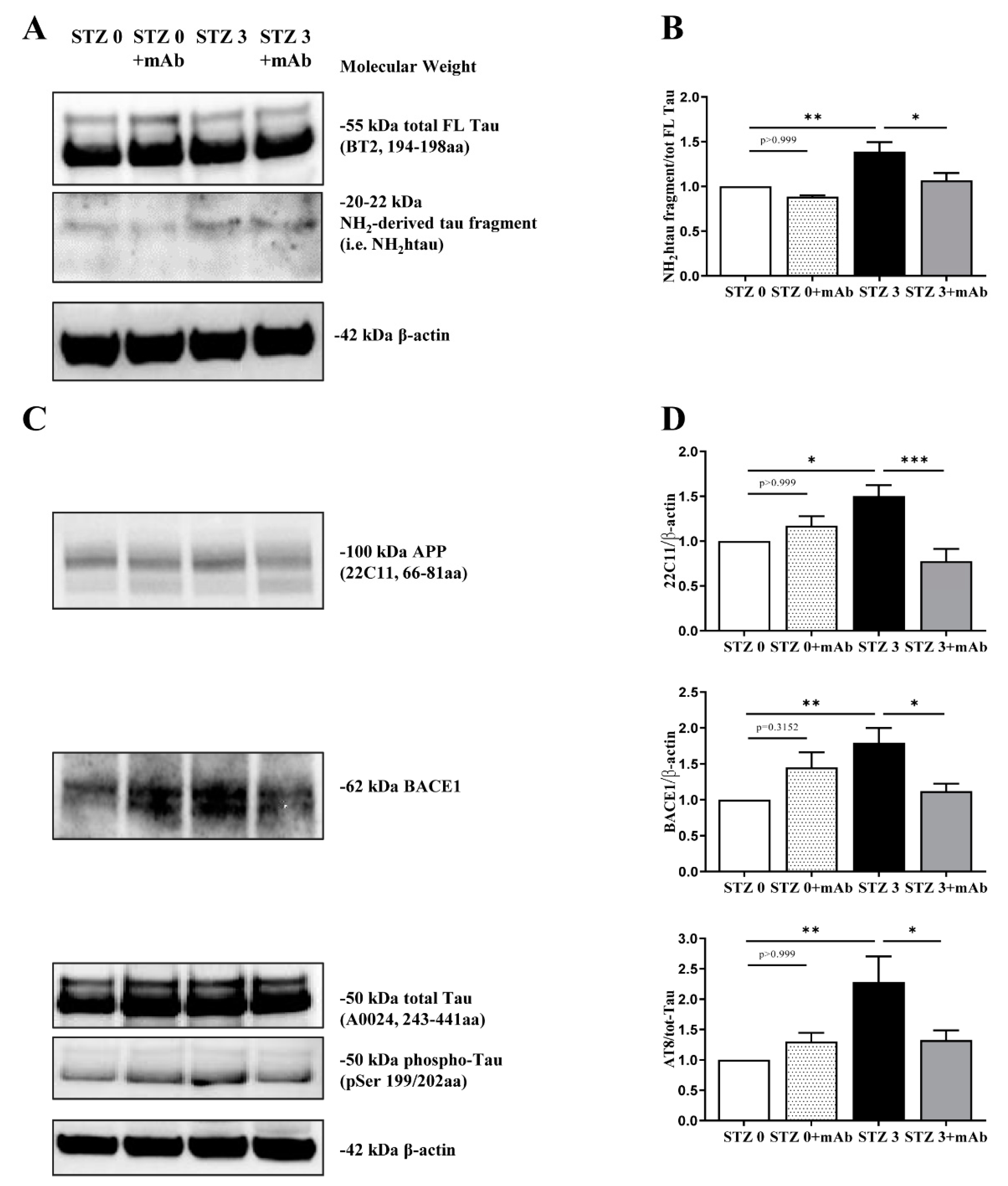
2.2. STZ-Induced sAD-like Pathological Hallmarks Are Mitigated by 12A12mAb Treatment
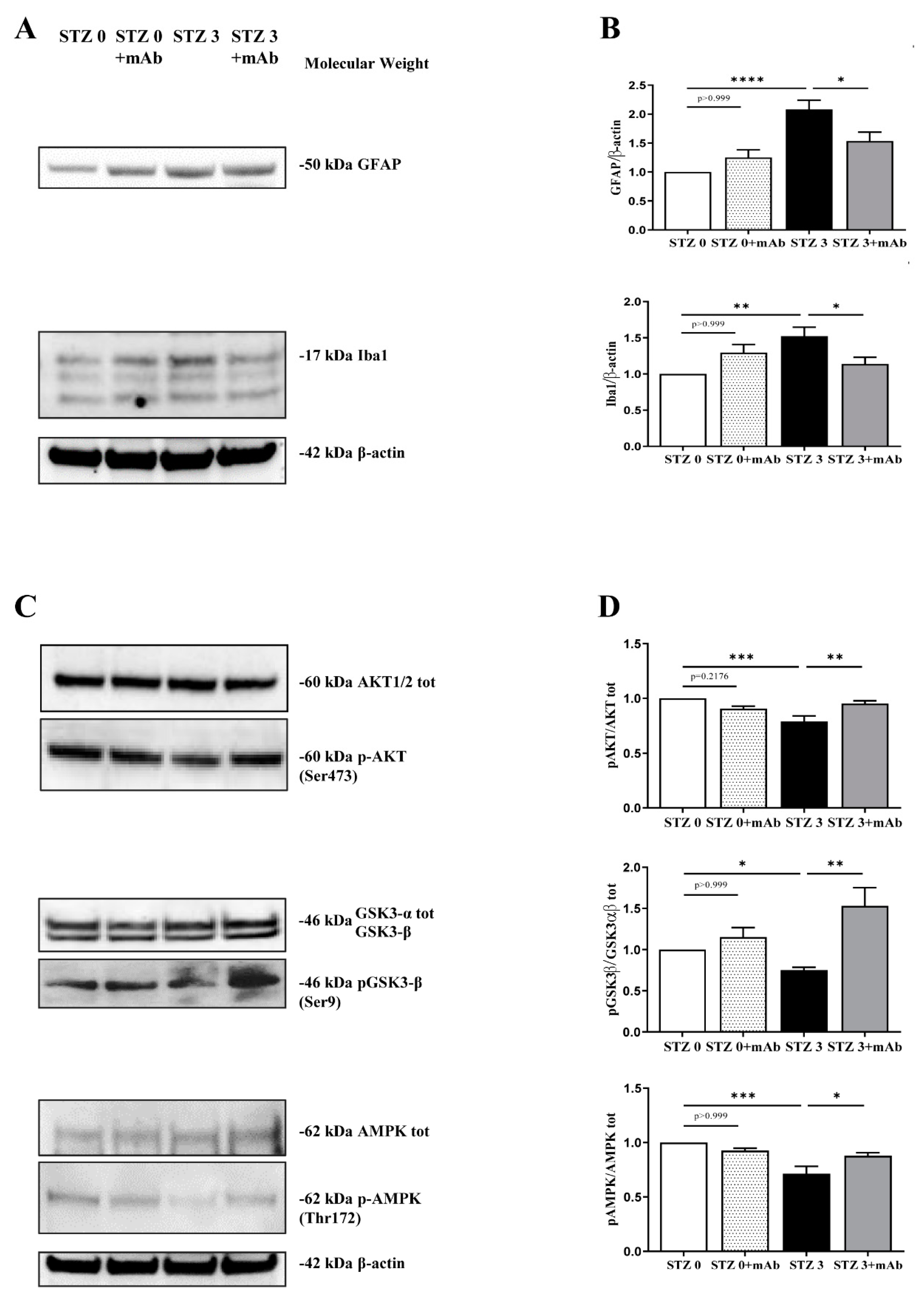
2.3. 12A12mAb Immunization Improves the STZ-Induced Neuroinflammation and Restores the PI3K/AKT/GSK3-β and AMPK Signaling Pathways
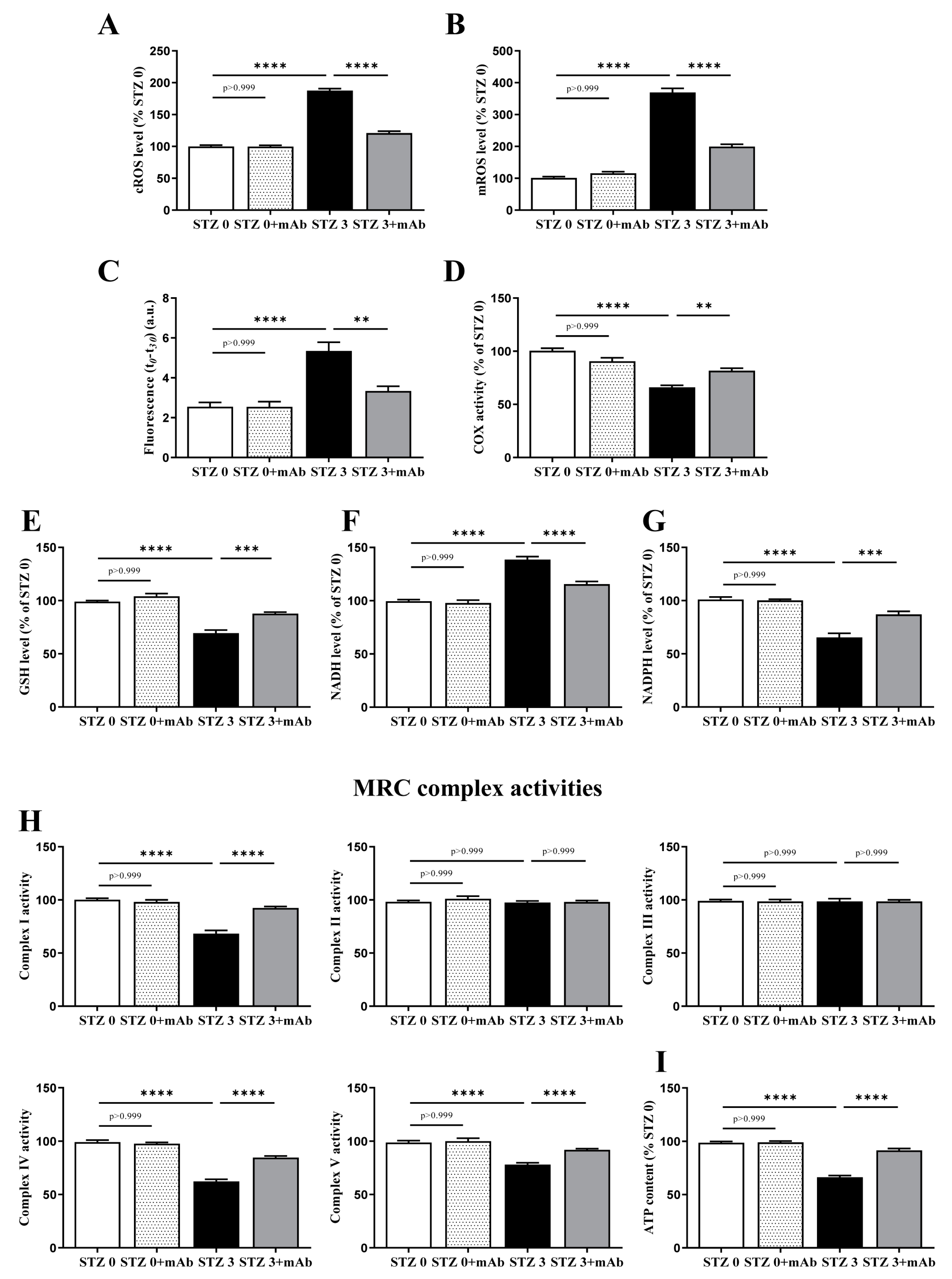
2.4. Overt Oxidative Stress and Mitochondrial Function Alterations Induced by ICV-Infusion of STZ Are Both Prevented by In Vivo 12A12mAb Delivery

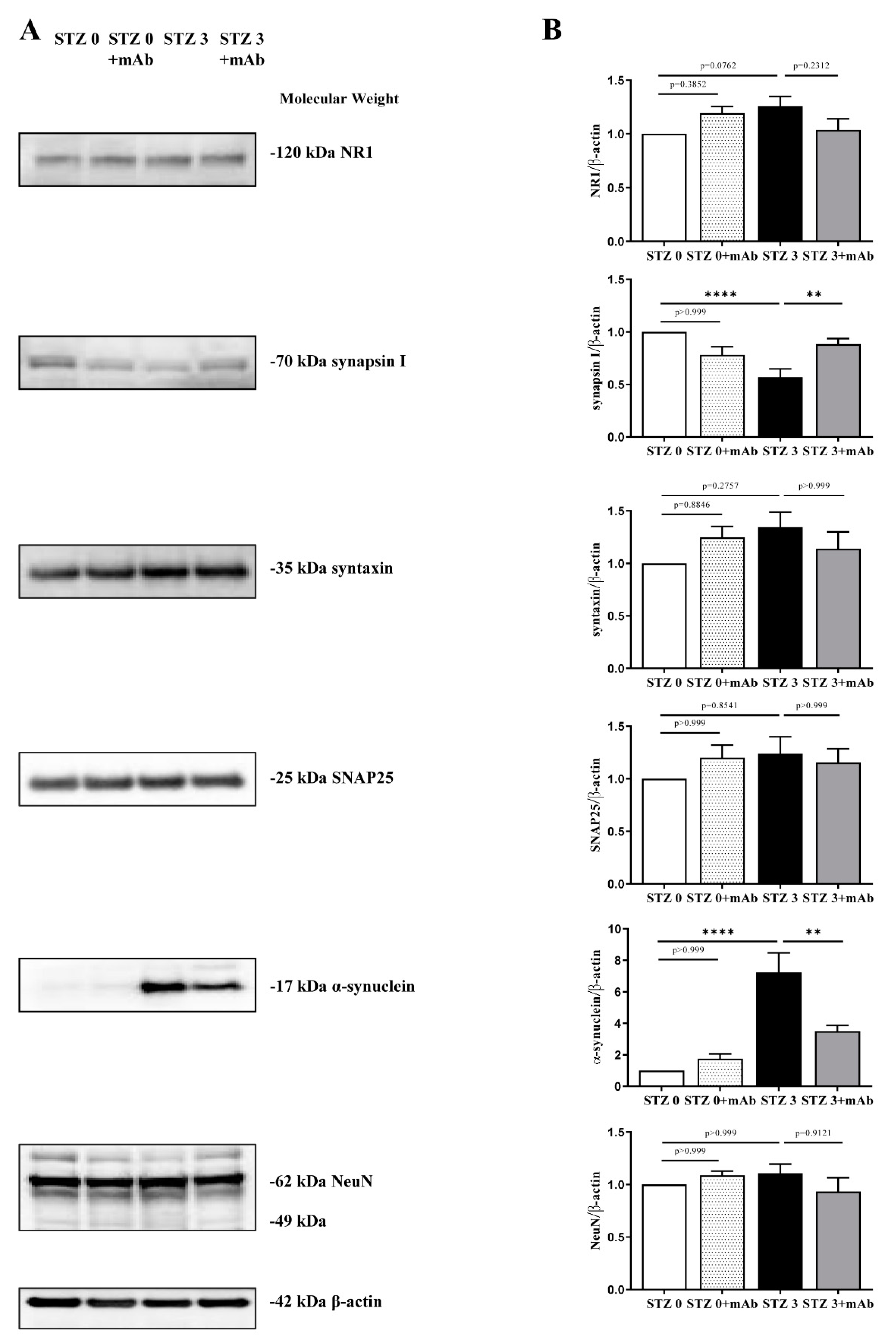
2.5. 12A12mAb Administration Normalizes the Imbalanced Expression of Key Synaptic Proteins in the Hippocampus of STZ-Lesioned Mice
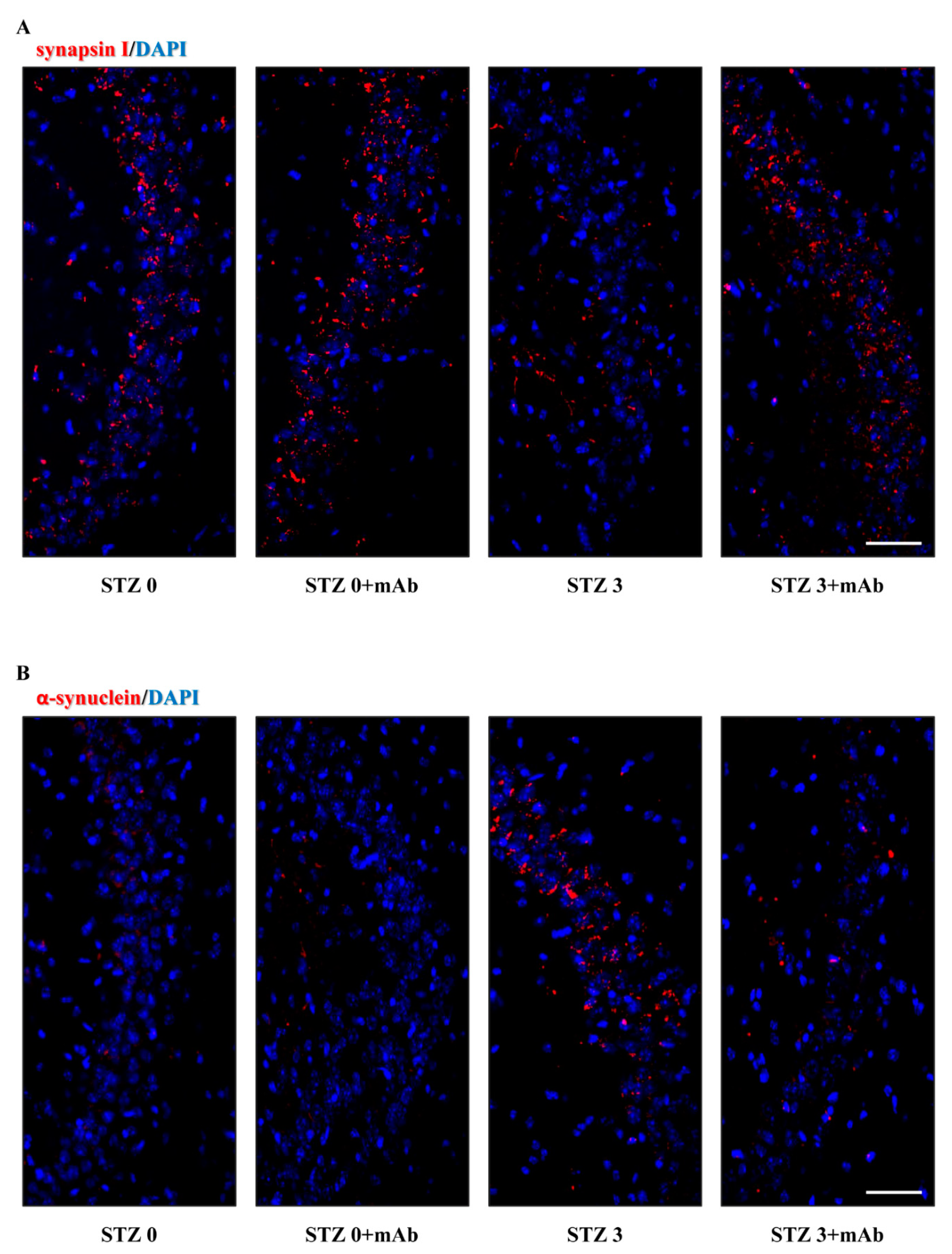
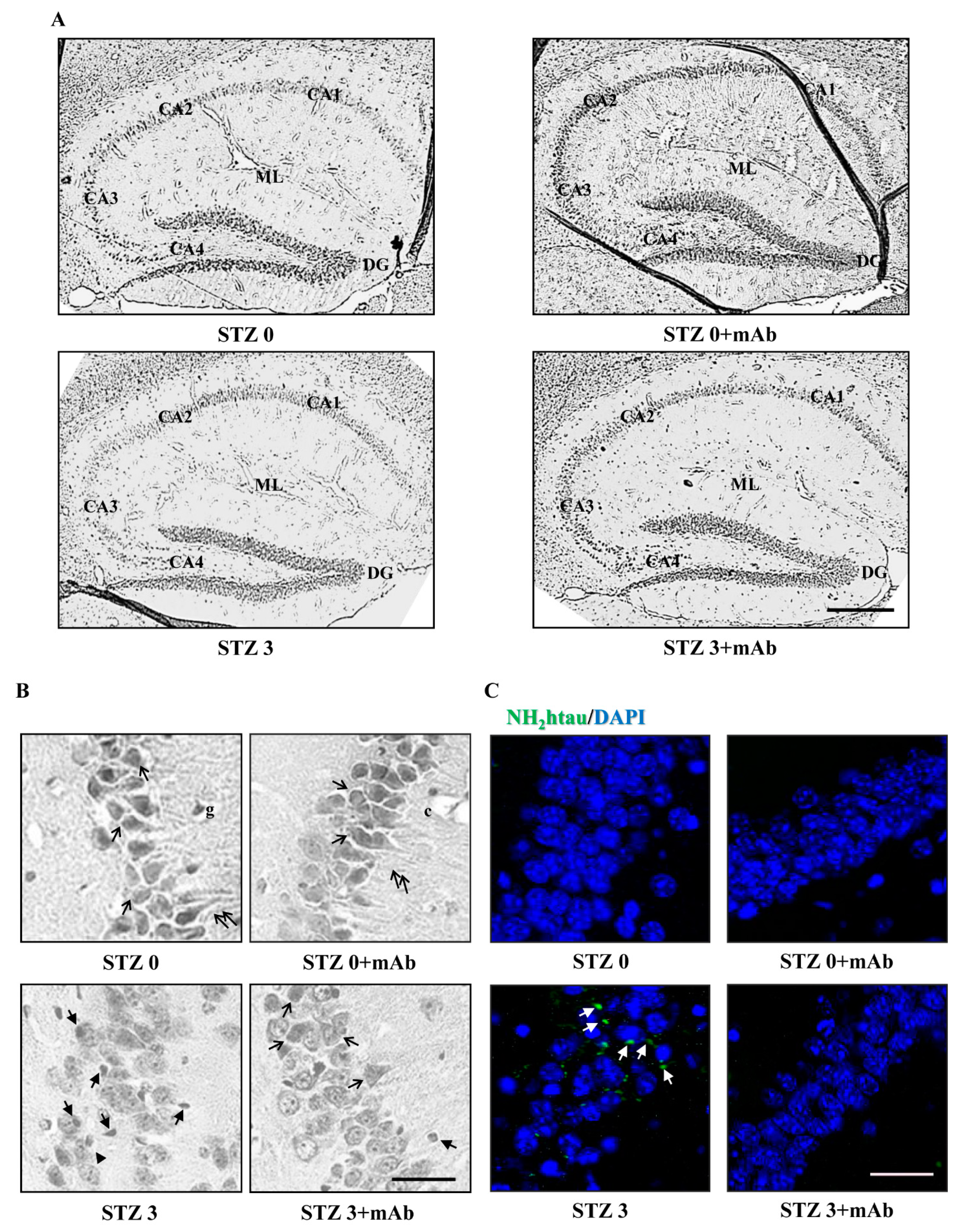
2.6. STZ-Induced Histopathologic Alterations Are Improved in the Hippocampus of Injected Mice following 12A12mAb Immunization
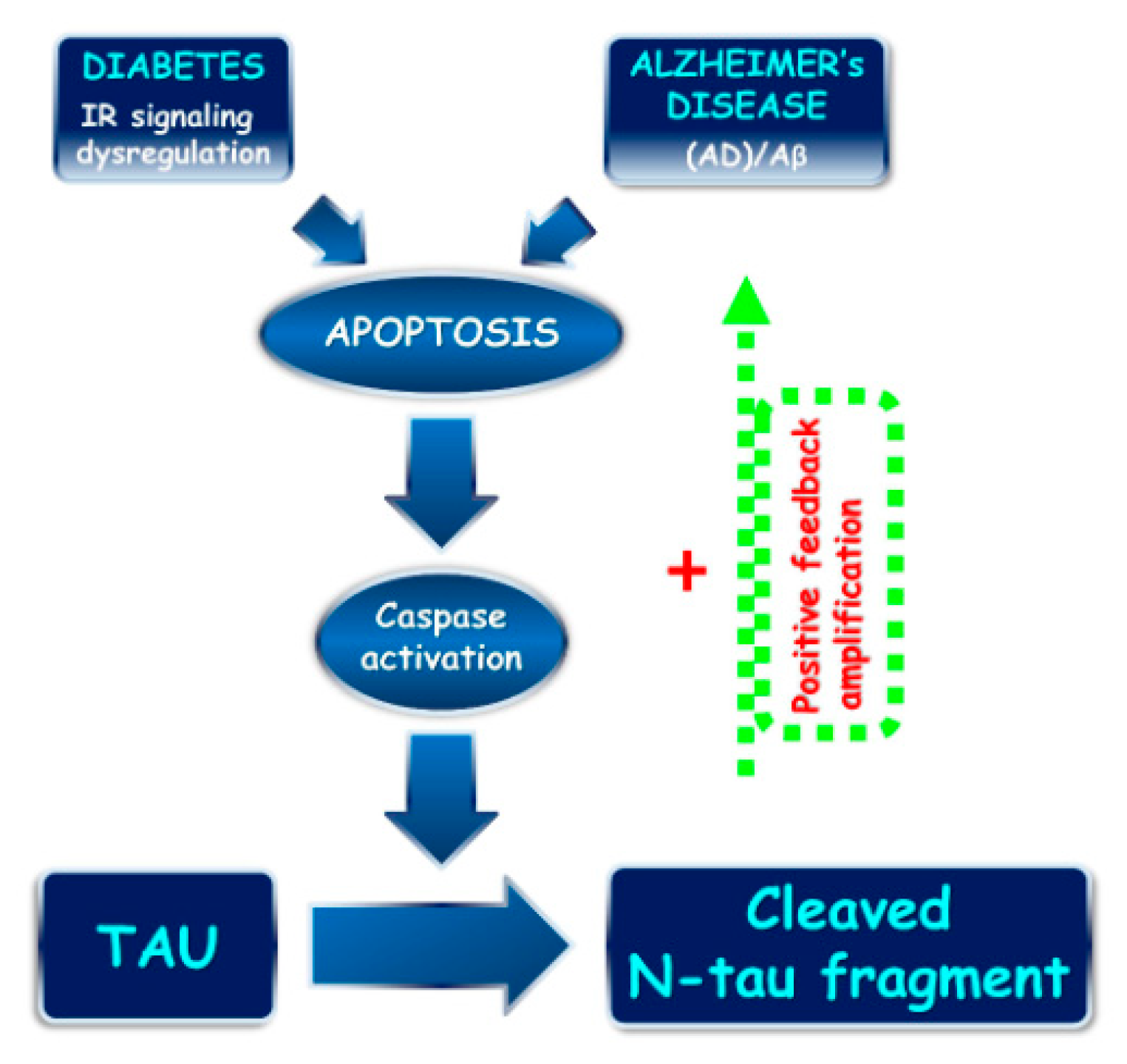
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Chemical Compounds and Antibodies
5.3. Neurosurgery and ICV-STZ Delivery
5.4. Immunization Scheme
5.5. Cognitive Assessment: Novel Object Recognition Test (NORT) and Object Place Recognition Task (OPRT)
5.6. Tissue Collection, Harvesting and Preparation
5.7. Western Blot Analysis and Semi-Quantitative Densitometry
5.8. Oxidative Stress and Mitochondrial Analysis
5.9. Immunofluorescence and Fluorescent Acquisition
5.10. Histopathological Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Competing Interests
Abbreviations
References
- Corsetti, V.; Borreca, A.; Latina, V.; Giacovazzo, G.; Pignataro, A.; Krashia, P.; Natale, F.; Cocco, S.; Rinaudo, M.; Malerba, F.; et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun. 2020, 2, fcaa039. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490, 115–125. [Google Scholar] [CrossRef]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Flores-Cuadra, J.A.; Madrid, A.; Fernández, P.L.; Pérez-Lao, A.R.; Oviedo, D.C.; Britton, G.B.; Carreira, M.B. Critical Review of the Alzheimer’s Disease Non-Transgenic Models: Can They Contribute to Disease Treatment? J. Alzheimer’s Dis. 2021, 82, S227–S250. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Velayudhan, L.; Poppe, M.; Archer, N.; Proitsi, P.; Brown, R.; Lovestone, S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br. J. Psychiatry 2010, 196, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, R.; Bolshette, N.; Gadhave, K.; Ahire, A.; Ahmed, S.; Cassano, T.; Gupta, V.B.; Lahkar, M. Insulin signaling: An opportunistic target to minify the risk of Alzheimer’s disease. Psychoneuroendocrinology 2017, 83, 159–171. [Google Scholar] [CrossRef]
- Klein, J.P.; Waxman, S.G. The brain in diabetes: Molecular changes in neurons and their implications for end-organ damage. Lancet Neurol. 2003, 2, 548–554. [Google Scholar] [CrossRef]
- Toro, P.; Schönknecht, P.; Schröder, J. Type II Diabetes in Mild Cognitive Impairment and Alzheimer’s Disease: Results from a Prospective Population-Based Study in Germany. J. Alzheimer’s Dis. 2009, 16, 687–691. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. A J. Clin. Ther. 2009, 14, 373–379. [Google Scholar]
- Knezovic, A.; Osmanovic-Barilar, J.; Curlin, M.; Hof, P.R.; Šimić, G.; Riederer, P.; Salkovic-Petrisic, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer’s disease. J. Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, Z.; Blanchard, J.; Dai, C.-L.; Sun, S.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.-X. A Non-transgenic Mouse Model (icv-STZ Mouse) of Alzheimer’s Disease: Similarities to and Differences from the Transgenic Model (3xTg-AD Mouse). Mol. Neurobiol. 2013, 47, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, K.G.; Rosário, B.; Camarini, R.; Hernandes, M.S.; Britto, L.R. Intracerebroventricular Streptozotocin as a Model of Alzheimer’s Disease: Neurochemical and Behavioral Characterization in Mice. Neurotox. Res. 2016, 31, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Gupta, Y. Intracerebroventricular injection of streptozotocin in rats produces both oxidative stress in the brain and cognitive impairment. Life Sci. 2001, 68, 1021–1029. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, Y. Effect of chronic treatment of melatonin on learning, memory and oxidative deficiencies induced by intracerebroventricular streptozotocin in rats. Pharmacol. Biochem. Behav. 2001, 70, 325–331. [Google Scholar] [CrossRef]
- Tota, S.; Awasthi, H.; Kamat, P.K.; Nath, C.; Hanif, K. Protective effect of quercetin against intracerebral streptozotocin induced reduction in cerebral blood flow and impairment of memory in mice. Behav. Brain Res. 2010, 209, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; De Jesus, D.F.; Kahraman, S.; Valdez, I.A.; Shamsi, F.; Yi, L.; Swensen, A.C.; Tseng, Y.-H.; Qian, W.-J.; Kulkarni, R.N. Insulin receptor-mediated signaling regulates pluripotency markers and lineage differentiation. Mol. Metab. 2018, 18, 153–163. [Google Scholar] [CrossRef]
- Deng, Y.; Li, B.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Dysregulation of Insulin Signaling, Glucose Transporters, O-GlcNAcylation, and Phosphorylation of Tau and Neurofilaments in the Brain: Implication for Alzheimer’s Disease. Am. J. Pathol. 2009, 175, 2089–2098. [Google Scholar] [CrossRef]
- Hoyer, S.; Riederer, P. Gene expression profile in streptozotocin rat model for sporadic Alzheimer?s disease. J. Neural Transm. 2004, 111, 367–386. [Google Scholar] [CrossRef]
- Lannert, H.; Hoyer, S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav. Neurosci. 1998, 112, 1199–1208. [Google Scholar] [CrossRef]
- Blokland, A.; Jolles, J. Spatial learning deficit and reduced hippocampal ChAT activity in rats after an ICV injection of streptozotocin. Pharmacol. Biochem. Behav. 1993, 44, 491–494. [Google Scholar] [CrossRef]
- Bassani, T.B.; Bonato, J.M.; Machado, M.M.F.; Cóppola-Segovia, V.; Moura, E.L.R.; Zanata, S.M.; Oliveira, R.M.M.W.; Vital, M.A.B.F. Decrease in Adult Neurogenesis and Neuroinflammation Are Involved in Spatial Memory Impairment in the Streptozotocin-Induced Model of Sporadic Alzheimer’s Disease in Rats. Mol. Neurobiol. 2017, 55, 4280–4296. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Kamat, P.K.; Nath, C.; Shukla, R. Glial activation and post-synaptic neurotoxicity: The key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol. Biochem. Behav. 2014, 117, 104–117. [Google Scholar] [CrossRef]
- Rajasekar, N.; Dwivedi, S.; Nath, C.; Hanif, K.; Shukla, R. Protection of streptozotocin induced insulin receptor dysfunction, neuroinflammation and amyloidogenesis in astrocytes by insulin. Neuropharmacology 2014, 86, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.D.O.; Mazucanti, C.; Xavier, G.F.; Torrão, A.D.S. Early and late neurodegeneration and memory disruption after intracerebroventricular streptozotocin. Physiol. Behav. 2012, 107, 401–413. [Google Scholar] [CrossRef]
- Santos, D.B.; Colle, D.; Moreira, E.L.G.; Peres, K.C.; Ribeiro, R.P.; Dos Santos, A.A.; De Oliveira, J.; Hort, M.A.; De Bem, A.F.; Farina, M. Probucol mitigates streptozotocin-induced cognitive and biochemical changes in mice. Neuroscience 2015, 284, 590–600. [Google Scholar] [CrossRef]
- Liu, P.; Zou, L.-B.; Wang, L.-H.; Jiao, Q.; Chi, T.-Y.; Ji, X.-F.; Jin, G. Xanthoceraside attenuates tau hyperphosphorylation and cognitive deficits in intracerebroventricular-streptozotocin injected rats. Psychopharmacology 2014, 231, 345–356. [Google Scholar] [CrossRef]
- Qi, C.-C.; Chen, X.-X.; Gao, X.-R.; Xu, J.-X.; Liu, S.; Ge, J.-F. Impaired Learning and Memory Ability Induced by a Bilaterally Hippocampal Injection of Streptozotocin in Mice: Involved With the Adaptive Changes of Synaptic Plasticity. Front. Aging Neurosci. 2021, 13, 633495. [Google Scholar] [CrossRef]
- Rajasekar, N.; Nath, C.; Hanif, K.; Shukla, R. Intranasal Insulin Administration Ameliorates Streptozotocin (ICV)-Induced Insulin Receptor Dysfunction, Neuroinflammation, Amyloidogenesis, and Memory Impairment in Rats. Mol. Neurobiol. 2017, 54, 6507–6522. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Hoyer, S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: An experimental approach. Neuropsychiatr. Disord. Integr. Approach 2007, 72, 217–233. [Google Scholar] [CrossRef]
- Du, L.-L.; Chai, D.-M.; Zhao, L.-N.; Li, X.-H.; Zhang, F.-C.; Zhang, H.-B.; Liu, L.-B.; Wu, K.; Liu, R.; Wang, J.-Z.; et al. AMPK Activation Ameliorates Alzheimer’s Disease-Like Pathology and Spatial Memory Impairment in a Streptozotocin-Induced Alzheimer’s Disease Model in Rats. J. Alzheimer’s Dis. 2014, 43, 775–784. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Knezovic, A.; Hoyer, S.; Riederer, P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm. 2012, 120, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer’s Disease: In Search of a Relevant Mechanism. Mol. Neurobiol. 2016, 53, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Tota, S.K.; Kumar, A.; Ahmad, A.S. Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: A Therapeutic Intervention for Treatment of Sporadic Alzheimer’s Disease (sAD)-Like Pathology. Mol. Neurobiol. 2016, 53, 4548–4562. [Google Scholar] [CrossRef]
- Plaschke, K.; Kopitz, J.; Siegelin, M.; Schliebs, R.; Salkovic-Petrisic, M.; Riederer, P.; Hoyer, S. Insulin-Resistant Brain State after Intracerebroventricular Streptozotocin Injection Exacerbates Alzheimer-like Changes in Tg2576 AβPP-Overexpressing Mice. J. Alzheimer’s Dis. 2010, 19, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Z.; Tian, Z.; Blanchard, J.; Dai, C.-L.; Chalbot, S.; Iqbal, K.; Liu, F.; Gong, C.-X. Intracerebroventricular Streptozotocin Exacerbates Alzheimer-Like Changes of 3xTg-AD Mice. Mol. Neurobiol. 2013, 49, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.P.; Corbett, N.J.; Kellett, K.A.B.; Hooper, N.M. Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis. 2018, 63, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole, B.B.; et al. Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef]
- Mehla, J.; Pahuja, M.; Gupta, Y.K. Streptozotocin-Induced Sporadic Alzheimer’s Disease: Selection of Appropriate Dose. J. Alzheimer’s Dis. 2013, 33, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Sil, S.; Gupta, P.; Ghosh, T. Optimization of intracerebroventricular streptozotocin dose for the induction of neuroinflammation and memory impairments in rats. Metab. Brain Dis. 2020, 35, 1279–1286. [Google Scholar] [CrossRef]
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E. Increased Tau Phosphorylation and Cleavage in Mouse Models of Type 1 and Type 2 Diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease--A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2011, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; dos Santos, R.X.C.; Perry, G.; Zhu, X.; Moreira, P.I.; Smith, M.A. Insulin-resistant brain state: The culprit in sporadic Alzheimer’s disease? Ageing Res. Rev. 2011, 10, 264–273. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, N.; Yan, F.; Jin, H.; Zhou, S.-Y.; Shi, J.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic, J.; Grünblatt, E.; Riederer, P.; Hoyer, S. Modeling Sporadic Alzheimer’s Disease: The Insulin Resistant Brain State Generates Multiple Long-Term Morphobiological Abnormalities Including Hyperphosphorylated Tau Protein and Amyloid-β. J. Alzheimer’s Dis. 2009, 18, 729–750. [Google Scholar] [CrossRef]
- Wang, L.; Li, N.; Shi, F.-X.; Xu, W.-Q.; Cao, Y.; Lei, Y.; Wang, J.-Z.; Tian, Q.; Zhou, X.-W. Upregulation of AMPK Ameliorates Alzheimer’s Disease-Like Tau Pathology and Memory Impairment. Mol. Neurobiol. 2020, 57, 3349–3361. [Google Scholar] [CrossRef]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Shi, L.; Zhong, D.; Li, Y.; Li, J.; Jin, R. AMPK: Potential Therapeutic Target for Alzheimer’s Disease. Curr. Protein Pept. Sci. 2020, 21, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 2009, 56, 779–787. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, L.; Li, J.; Du Fang, D.; Zhong, C.; Chen, J.X.; Yan, S.S. Synergistic Exacerbation of Mitochondrial and Synaptic Dysfunction and Resultant Learning and Memory Deficit in a Mouse Model of Diabetic Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, F.; Gong, C.; Shi, X.; Wang, G. Protective effect of daidzein against streptozotocin-induced Alzheimer’s disease via improving cognitive dysfunction and oxidative stress in rat model. J. Biochem. Mol. Toxicol. 2019, 33, e22319. [Google Scholar] [CrossRef]
- Soni, N.D.; Ramesh, A.; Roy, D.; Patel, A.B. Brain energy metabolism in intracerebroventricularly administered streptozotocin mouse model of Alzheimer’s disease: A 1H-[13C]-NMR study. Br. J. Pharmacol. 2021, 41, 2344–2355. [Google Scholar] [CrossRef]
- Xu, P.; Ning, J.; Jiang, Q.; Li, C.; Yan, J.; Zhao, L.; Gao, H.; Zheng, H. Region-specific metabolic characterization of the type 1 diabetic brain in mice with and without cognitive impairment. Neurochem. Int. 2021, 143, 104941. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Djordjevic, J.; Thomson, E.; Smith, D.R.; Albensi, B.C.; Fernyhough, P. Depressed mitochondrial function and electron transport Complex II-mediated H2O2 production in the cortex of type 1 diabetic rodents. Mol. Cell. Neurosci. 2018, 90, 49–59. [Google Scholar] [CrossRef]
- Javed, H.; Khan, M.M.; Ahmad, A.; Vaibhav, K.; Ahmad, M.; Khan, A.; Ashafaq, M.; Islam, F.; Siddiqui, M.; Safhi, M. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience 2012, 210, 340–352. [Google Scholar] [CrossRef]
- Zhou, S.; Yu, G.; Chi, L.; Zhu, J.; Zhang, W.; Zhang, Y.; Zhang, L. Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. NeuroToxicology 2013, 38, 136–145. [Google Scholar] [CrossRef]
- Verma, V.; Singh, D.; Kh, R. Sinapic Acid Alleviates Oxidative Stress and Neuro-Inflammatory Changes in Sporadic Model of Alzheimer’s Disease in Rats. Brain Sci. 2020, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Kirkinezos, I.G.; Moraes, C.T. Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging. J. Signal Transduct. 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Mirò, O.; Alonso, J.-R.; Jarreta, D.; Casademont, J.; Urbano-Màrquez, A.; Cardellach, F. Smoking disturbs mitochondrial respiratory chain function and enhances lipid peroxidation on human circulating lymphocytes. Carcinog. Carcinog. 1999, 20, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zimbron, L.F.; Luna-Muñoz, J.; Mena, R.; Vazquez-Ramirez, R.; Kubli-Garfias, C.; Cribbs, D.H.; Manoutcharian, K.; Gevorkian, G. Amyloid-β Peptide Binds to Cytochrome C Oxidase Subunit 1. PLoS ONE 2012, 7, e42344. [Google Scholar] [CrossRef] [PubMed]
- Bobba, A.; Amadoro, G.; Valenti, D.; Corsetti, V.; Lassandro, R.; Atlante, A. Mitochondrial respiratory chain Complexes I and IV are impaired by β-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 2013, 13, 298–311. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- De Bari, L.; Favia, M.; Bobba, A.; Lassandro, R.; Guerra, L.; Atlante, A. Aberrant GSH reductase and NOX activities concur with defective CFTR to pro-oxidative imbalance in cystic fibrosis airways. J. Bioenerg. Biomembr. 2018, 50, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Blacker, T.; Duchen, M. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free. Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef]
- Young, T.A.; Cunningham, C.C.; Bailey, S.M. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: Studies using myxothiazol. Arch. Biochem. Biophys. 2002, 405, 65–72. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of Reactive Oxygen Species in Brain Mitochondria: Contribution by Electron Transport Chain and Non–Electron Transport Chain Sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef]
- Arendt, T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Yan, W.; Pang, M.; Yu, Y.; Gou, X.; Si, P.; Zhawatibai, A.; Zhang, Y.; Zhang, M.; Guo, T.; Yi, X.; et al. The neuroprotection of liraglutide on diabetic cognitive deficits is associated with improved hippocampal synapses and inhibited neuronal apoptosis. Life Sci. 2019, 231, 116566. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z.; Liang, Z.; Sun, S.; Dai, C.-L.; Lee, M.H.; LaFerla, F.M.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; et al. Brain Gene Expression of a Sporadic (icv-STZ Mouse) and a Familial Mouse Model (3xTg-AD Mouse) of Alzheimer’s Disease. PLoS ONE 2012, 7, e51432. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, L.; Yang, Q.; Du, X.; Li, M.; Yuan, M.; Zhou, Z. Changes in hippocampal synapses and learning-memory abilities in a streptozotocin-treated rat model and intervention by using fasudil hydrochloride. Neuroscience 2012, 200, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Angelova, H.; Pechlivanova, D.; Krumova, E.; Miteva-Staleva, J.; Kostadinova, N.; Dzhambazova, E.; Landzhov, B. Moderate protective effect of Kyotorphin against the late consequences of intracerebroventricular streptozotocin model of Alzheimer’s disease. Amino Acids 2019, 51, 1501–1513. [Google Scholar] [CrossRef]
- Rostami, F.; Javan, M.; Moghimi, A.; Haddad-Mashadrizeh, A.; Fereidoni, M. Streptozotocin-induced hippocampal astrogliosis and insulin signaling malfunction as experimental scales for subclinical sporadic Alzheimer model. Life Sci. 2017, 188, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Amadoro, G.; Gentile, A.; Capsoni, S.; Ciotti, M.; Cencioni, M.; Atlante, A.; Canu, N.; Rohn, T.; Cattaneo, A.; et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol. Cell. Neurosci. 2008, 38, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Rissman, R.A.; Davis, M.; Kim, Y.E.; Cotman, C.W.; Headb, E. Caspase-9 Activation and Caspase Cleavage of tau in the Alzheimer’s Disease Brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Dhaliwal, J.; Sah, S.P. 7,8-Dihydroxyflavone improves cognitive functions in ICV-STZ rat model of sporadic Alzheimer’s disease by reversing oxidative stress, mitochondrial dysfunction, and insulin resistance. Psychopharmacolog 2021, 238, 1991–2009. [Google Scholar] [CrossRef]
- Ghoneum, M.H.; El Sayed, N.S. Protective Effect of Biobran/MGN-3 against Sporadic Alzheimer’s Disease Mouse Model: Possible Role of Oxidative Stress and Apoptotic Pathways. Oxidative Med. Cell. Longev. 2021, 2021, 8845064. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimer’s Dement. 2010, 6, 420–424. [Google Scholar] [CrossRef]
- Sonkusare, S.; Srinivasan, K.; Kaul, C.; Ramarao, P. Effect of donepezil and lercanidipine on memory impairment induced by intracerebroventricular streptozotocin in rats. Life Sci. 2005, 77, 1–14. [Google Scholar] [CrossRef]
- Saxena, G.; Singh, S.P.; Agrawal, R.; Nath, C. Effect of donepezil and tacrine on oxidative stress in intracerebral streptozotocin-induced model of dementia in mice. Eur. J. Pharmacol. 2008, 581, 283–289. [Google Scholar] [CrossRef]
- Ponce-Lopez, T.; Liy-Salmeron, G.; Hong, E.; Meneses, A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3β decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res. 2011, 1426, 73–85. [Google Scholar] [CrossRef]
- Martinisi, A.; Flach, M.; Sprenger, F.; Frank, S.; Tolnay, M.; Winkler, D.T. Severe oligomeric tau toxicity can be reversed without long-term sequelae. Brain 2021, 144, 963–974. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.-G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Frolich, L.; Blum-Degen, D.; Riederer, P.; Hoyer, S. A Disturbance in the Neuronal Insulin Receptor Signal Transduction in Sporadic Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1999, 893, 290–293. [Google Scholar] [CrossRef]
- Shingo, A.S.; Kanabayashi, T.; Murase, T.; Kito, S. Cognitive decline in STZ-3V rats is largely due to dysfunctional insulin signalling through the dentate gyrus. Behav. Brain Res. 2012, 229, 378–383. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic-Barilar, J.; Brückner, M.K.; Hoyer, S.; Arendt, T.; Riederer, P. Cerebral amyloid angiopathy in streptozotocin rat model of sporadic Alzheimer’s disease: A long-term follow up study. J. Neural Transm. 2011, 118, 765–772. [Google Scholar] [CrossRef]
- Grünblatt, E.; Salkovic-Petrisic, M.; Osmanovic, J.; Riederer, P.; Hoyer, S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Santos, M.S.; Casadesus, G.; LaManna, J.C.; Perry, G.; Smith, M.A.; Moreira, P.I. Mitochondrial Abnormalities in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 406–419. [Google Scholar] [CrossRef]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.M.D.; Madhunapantula, S.; Nataraj, S.K.M.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Nataraj, S.K.M.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1773–1784. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Phiel, C.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nat. Cell Biol. 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Qureshi, H.Y.; Cafferty, P.W.; Sobue, K.; Agarwal-Mawal, A.; Neufield, K.D.; Paudel, H.K. Glycogen Synthase Kinase-3β Is Complexed with Tau Protein in Brain Microtubules. J. Biol. Chem. 2002, 277, 11933–11940. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Xu, Q.-Q.; Xian, Y.-F.; Lin, Z.-X. Sulforaphene Ameliorates Neuroinflammation and Hyperphosphorylated Tau Protein via Regulating the PI3K/Akt/GSK-3βPathway in Experimental Models of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2020, 2020, 475419. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef]
- Guo, T.; Dakkak, D.; Rodriguez-Martin, T.; Noble, W.; Hanger, D.P. A pathogenic tau fragment compromises microtubules, disrupts insulin signaling and induces the unfolded protein response. Acta Neuropathol. Commun. 2019, 7, 34. [Google Scholar] [CrossRef]
- Ishrat, T.; Hoda, N.; Khan, M.M.; Yousuf, S.; Ahmad, M.; Ahmad, A.; Islam, F. Amelioration of cognitive deficits and neurodegeneration by curcumin in rat model of sporadic dementia of Alzheimer’s type (SDAT). Eur. Neuropsychopharmacol. 2009, 19, 636–647. [Google Scholar] [CrossRef]
- Reeta, K.H.; Singh, D.; Gupta, Y.K. Edaravone attenuates intracerebroventricular streptozotocin-induced cognitive impairment in rats. Eur. J. Neurosci. 2017, 45, 987–997. [Google Scholar] [CrossRef]
- Reeta, K.; Singh, D.; Gupta, Y. Chronic treatment with taurine after intracerebroventricular streptozotocin injection improves cognitive dysfunction in rats by modulating oxidative stress, cholinergic functions and neuroinflammation. Neurochem. Int. 2017, 108, 146–156. [Google Scholar] [CrossRef]
- Fattah, S.A.; Waly, H.; El-Enein, A.A.; Kamel, A.; Labib, H. Mesenchymal stem cells versus curcumin in enhancing the alterations in the cerebellar cortex of streptozocin-induced diabetic albino rats. The role of GFAP, PLC and α-synuclein. J. Chem. Neuroanat. 2020, 109, 101842. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Kim, B.; Backus, C.; Oh, S.; Feldman, E.L. Hyperglycemia-Induced Tau Cleavage in vitro and in vivo: A Possible Link between Diabetes and Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 34, 727–739. [Google Scholar] [CrossRef]
- Li, D.-X.; Wang, C.-N.; Wang, Y.; Ye, C.-L.; Jiang, L.; Zhu, X.-Y.; Liu, Y.-J. NLRP3 inflammasome-dependent pyroptosis and apoptosis in hippocampus neurons mediates depressive-like behavior in diabetic mice. Behav. Brain Res. 2020, 391, 112684. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Nakamura, M.; Wolpert, E.B.; Reiter, C.E.N.; Seigel, G.M.; Antonetti, D.A.; Gardner, T. Insulin Rescues Retinal Neurons from Apoptosis by a Phosphatidylinositol 3-Kinase/Akt-mediated Mechanism That Reduces the Activation of Caspase-3. J. Biol. Chem. 2001, 276, 32814–32821. [Google Scholar] [CrossRef]
- Calissano, P.; Matrone, C.; Amadoro, G. Apoptosis and in vitro Alzheimer’s disease neuronal models. Commun. Integr. Biol. 2009, 2, 163–169. [Google Scholar] [CrossRef]
- Amadoro, G.; Latina, V.; Calissano, P. A long story for a short peptide: Therapeutic efficacy of a cleavage-specific tau antibody. Neural Regen. Res. 2021, 16, 2417–2419. [Google Scholar] [CrossRef]
- Klann, I.P.; Martini, F.; Rosa, S.G.; Nogueira, C.W. Ebselen reversed peripheral oxidative stress induced by a mouse model of sporadic Alzheimer’s disease. Mol. Biol. Rep. 2020, 47, 2205–2215. [Google Scholar] [CrossRef]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Sciacca, M.F.M.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017, 8, 64745–64778. [Google Scholar] [CrossRef]
- Coccurello, R.; Adriani, W.; Oliverio, A.; Mele, A. Effect of intra-accumbens dopamine receptor agents on reactivity to spatial and non-spatial changes in mice. Psychopharmacology 2000, 152, 189–199. [Google Scholar] [CrossRef]
- Coccurello, R.; Oliverio, A.; Mele, A. Dopamine–Glutamate Interplay in the Ventral Striatum Modulates Spatial Learning in a Receptor Subtype-Dependent Manner. Neuropsychopharmacology 2012, 37, 1122–1133. [Google Scholar] [CrossRef]
- Barbato, C.; Giacovazzo, G.; Albiero, F.; Scardigli, R.; Scopa, C.; Ciotti, M.T.; Strimpakos, G.; Coccurello, R.; Ruberti, F. Cognitive Decline and Modulation of Alzheimer’s Disease-Related Genes After Inhibition of MicroRNA-101 in Mouse Hippocampal Neurons. Mol. Neurobiol. 2020, 57, 3183–3194. [Google Scholar] [CrossRef]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Squire, L.R.; Wixted, J.; Clark, R.E. Recognition memory and the medial temporal lobe: A new perspective. Nat. Rev. Neurosci. 2007, 8, 872–883. [Google Scholar] [CrossRef]
- Planel, E.; Richter, K.E.; Nolan, C.E.; Finley, J.E.; Liu, L.; Wen, Y.; Krishnamurthy, P.; Herman, M.; Wang, L.; Schachter, J.B.; et al. Anesthesia Leads to Tau Hyperphosphorylation through Inhibition of Phosphatase Activity by Hypothermia. J. Neurosci. 2007, 27, 3090–3097. [Google Scholar] [CrossRef]
- Waddell, W.-J.; Hill, C. A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med. 1956, 48, 311–314. [Google Scholar]
- Favia, M.; De Bari, L.; Lassandro, R.; Atlante, A. Modulation of glucose-related metabolic pathways controls glucose level in airway surface liquid and fight oxidative stress in cystic fibrosis cells. J. Bioenerg. Biomembr. 2019, 51, 203–218. [Google Scholar] [CrossRef]
- Favia, M.; Atlante, A. Cellular Redox State Acts as Switch to Determine the Direction of NNT-Catalyzed Reaction in Cystic Fibrosis Cells. Int. J. Mol. Sci. 2021, 22, 967. [Google Scholar] [CrossRef]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef]
- Vergani, L.; Floreani, M.; Russell, A.; Ceccon, M.; Napoli, E.; Cabrelle, A.; Valente, L.; Bragantini, F.; Léger, B.; Dabbeni-Sala, F. Antioxidant defences and homeostasis of reactive oxygen species in different human mitochondrial DNA-depleted cell lines. JBIC J. Biol. Inorg. Chem. 2004, 271, 3646–3656. [Google Scholar] [CrossRef]
- Gibon, Y.; Larher, F. Cycling Assay for Nicotinamide Adenine Dinucleotides: NaCl Precipitation and Ethanol Solubilization of the Reduced Tetrazolium. Anal. Biochem. 1997, 251, 153–157. [Google Scholar] [CrossRef]
- Benit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rotig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Cooper, C. Interaction of Peroxynitrite with Mitochondrial Cytochrome Oxidase: Catalytic production of nitric oxide and irreversible inhibition of enzyme activity. J. Biol. Chem. 1998, 273, 30961–30972. [Google Scholar] [CrossRef]
- Khan, H.A. Bioluminometric assay of ATP in mouse brain: Determinant factors for enhanced test sensitivity. J. Biosci. 2003, 28, 379–382. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latina, V.; Giacovazzo, G.; Calissano, P.; Atlante, A.; La Regina, F.; Malerba, F.; Dell’Aquila, M.; Stigliano, E.; Balzamino, B.O.; Micera, A.; et al. Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model. Int. J. Mol. Sci. 2021, 22, 12158. https://doi.org/10.3390/ijms222212158
Latina V, Giacovazzo G, Calissano P, Atlante A, La Regina F, Malerba F, Dell’Aquila M, Stigliano E, Balzamino BO, Micera A, et al. Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model. International Journal of Molecular Sciences. 2021; 22(22):12158. https://doi.org/10.3390/ijms222212158
Chicago/Turabian StyleLatina, Valentina, Giacomo Giacovazzo, Pietro Calissano, Anna Atlante, Federico La Regina, Francesca Malerba, Marco Dell’Aquila, Egidio Stigliano, Bijorn Omar Balzamino, Alessandra Micera, and et al. 2021. "Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model" International Journal of Molecular Sciences 22, no. 22: 12158. https://doi.org/10.3390/ijms222212158
APA StyleLatina, V., Giacovazzo, G., Calissano, P., Atlante, A., La Regina, F., Malerba, F., Dell’Aquila, M., Stigliano, E., Balzamino, B. O., Micera, A., Coccurello, R., & Amadoro, G. (2021). Tau Cleavage Contributes to Cognitive Dysfunction in Strepto-Zotocin-Induced Sporadic Alzheimer’s Disease (sAD) Mouse Model. International Journal of Molecular Sciences, 22(22), 12158. https://doi.org/10.3390/ijms222212158