
Emerging Approaches for Solid Tumor Treatment Using CAR-T Cell Therapy




Abstract
:1. Introduction
2. Tumor-Specific CAR-T Cell Immunotherapy
2.1. Hematologic Malignancies
2.2. Solid Tumors
2.2.1. Lung Cancer
2.2.2. Brain Tumor
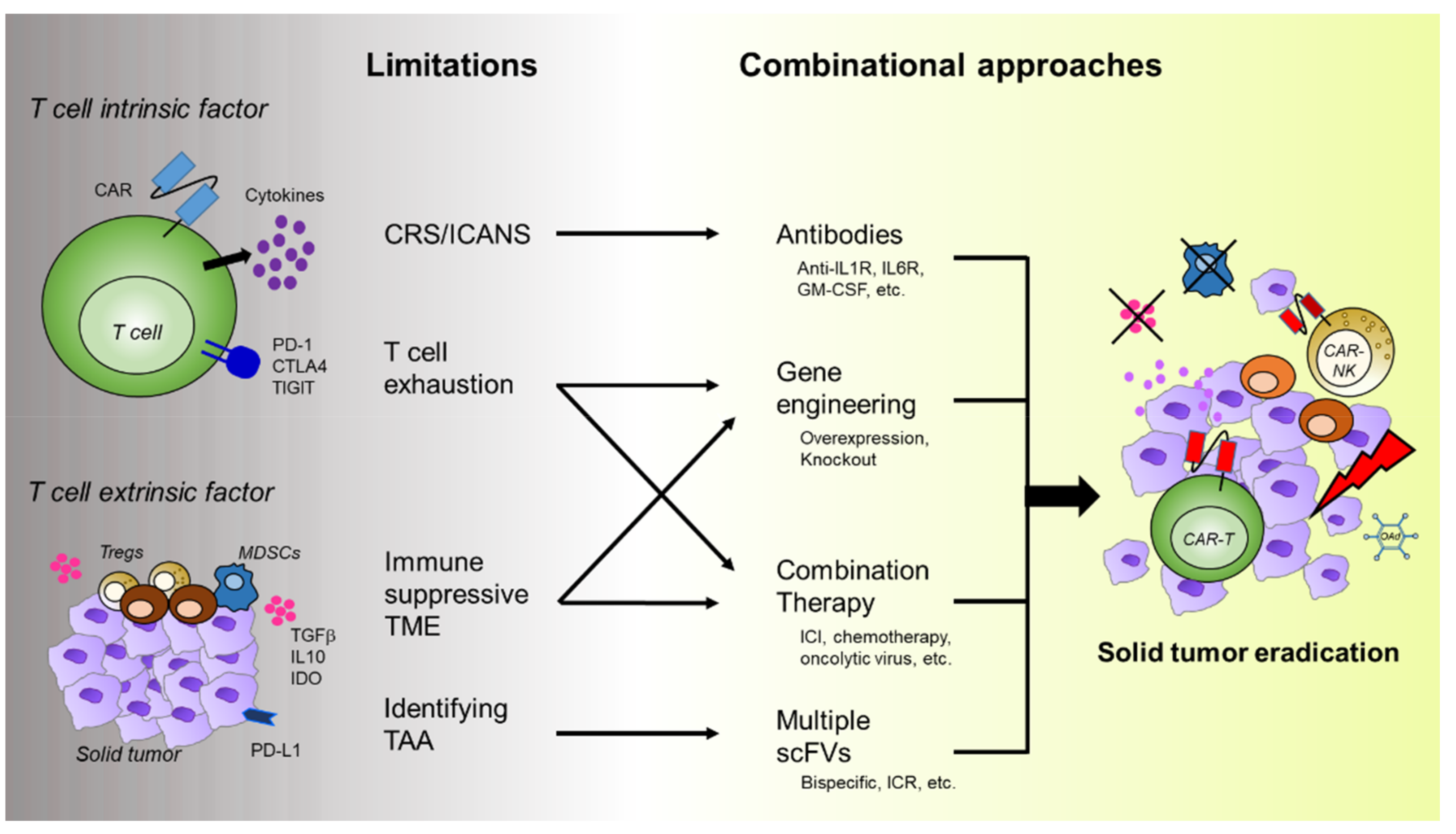
3. Challenges of CAR-T Cell Therapy for Solid Tumors
3.1. T Cell Intrinsic Factors
3.1.1. T Cell Exhaustion
3.1.2. Toxicity of CAR-T Cells
3.2. T Cell Extrinsic Factors
3.2.1. Immunosuppressive Tumor Microenvironments (TMEs)
3.2.2. Tumor-Associated Antigens
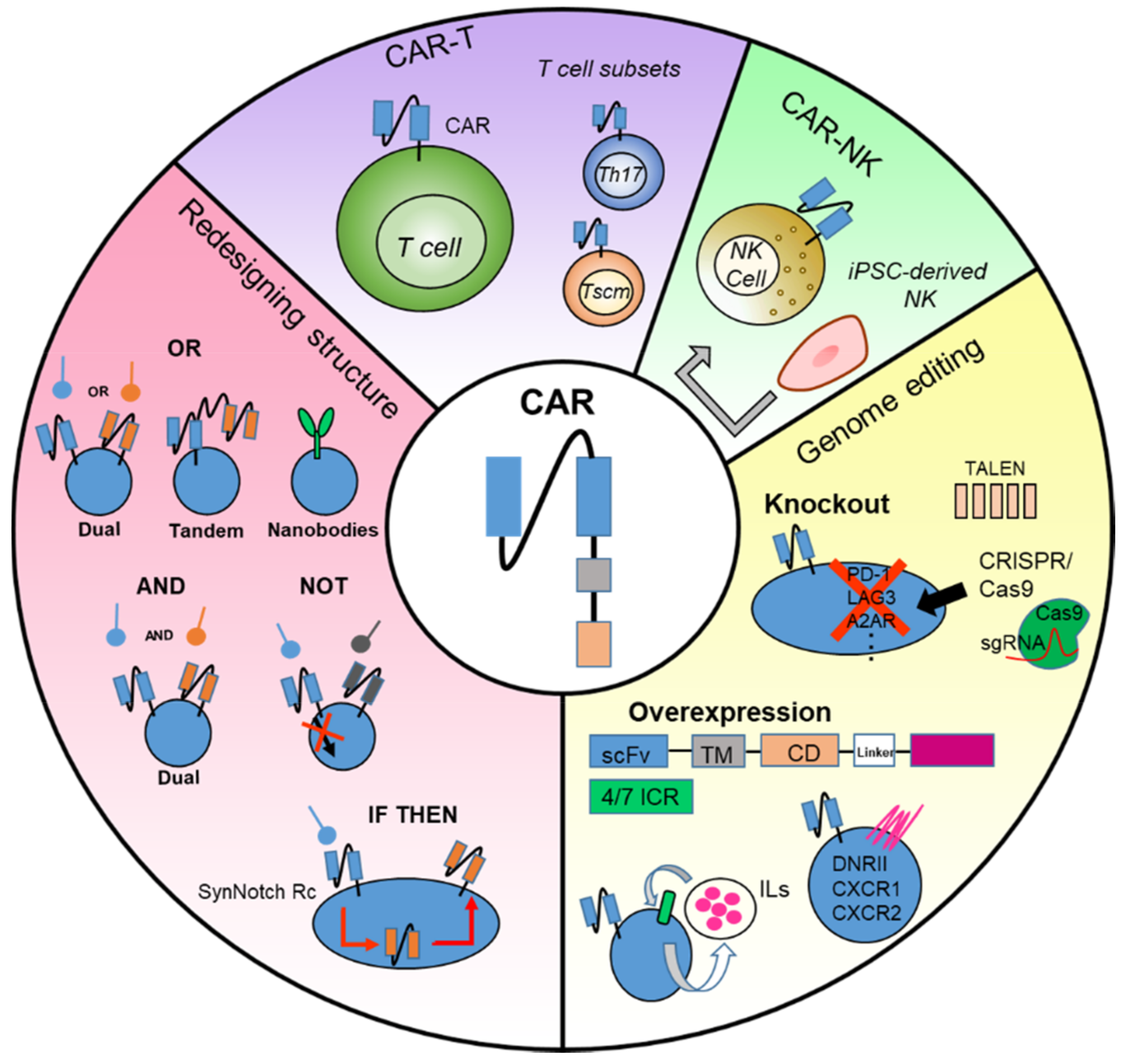
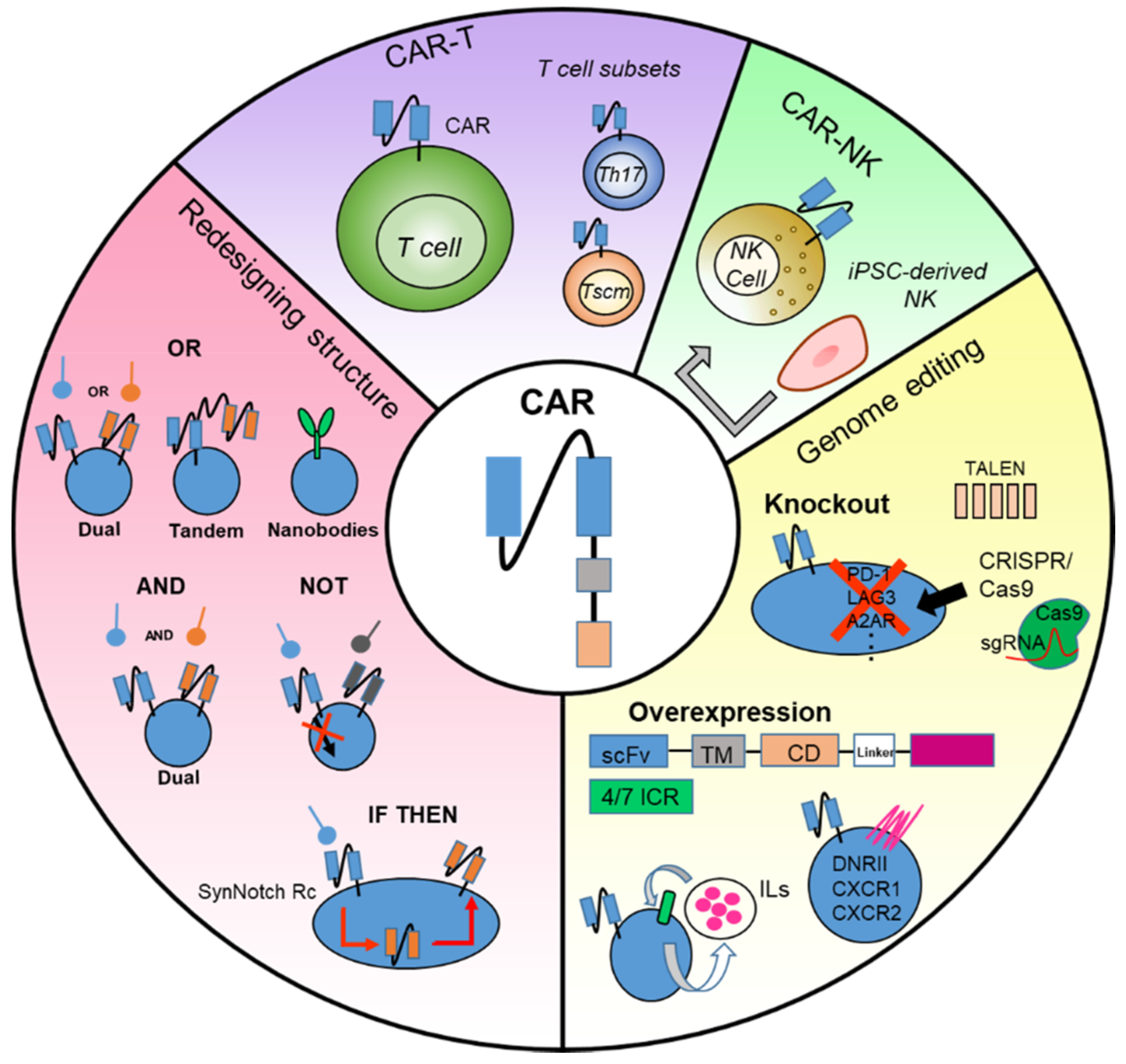
4. Emerging Approaches for the Improvement of CAR-T Cell Therapy
4.1. Redesigning CAR Structure
4.1.1. Multiple CARs with “OR” Strategy
4.1.2. Multiple CARs with “AND” or “NOT” Strategy
4.1.3. Multiple CARs with “IF/THEN” Strategy
4.2. Genome Editing of CAR-T Cell
4.2.1. Knockout of Inhibitory Molecules
4.2.2. Overexpression of T Cell Stimulus Genes
4.2.3. Promoting Inflammatory and Proliferative Activities of CAR-T Cells
4.3. Combination Therapy
4.3.1. Chemotherapy or Other Strategies
4.3.2. Immune Checkpoint Blockade
4.4. Development of CAR-NK Cell Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non–small-cell lung cancer: Role of the immune system and potential for immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zeng, G. Cancer and innate immune system interactions: Translational potentials for cancer immunotherapy. J. Immunother. 2012, 35, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T-cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2230–2239. [Google Scholar] [CrossRef]
- Noh, J.-Y.; Seo, H.; Lee, J.; Jung, H. Immunotherapy in Hematologic Malignancies: Emerging Therapies and Novel Approaches. Int. J. Mol. Sci. 2020, 21, 8000. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Berdeja, J.G.; Madduri, D.; Jakubowiak, A.J.; Agha, M.E.; Cohen, A.D.; Hari, P.; Yeh, T.-M.; Olyslager, Y.; Banerjee, A. Ciltacabtagene autoleucel, a B-cell maturation antigen (BCMA)-directed chimeric antigen receptor T-cell (CAR-T) therapy, in relapsed/refractory multiple myeloma (R/R MM): Updated results from CARTITUDE-1. J. Clin. Oncol. 2021, 39 (Suppl. S15), 8005. [Google Scholar] [CrossRef]
- Liu, G.; Rui, W.; Zhao, X.; Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol. Immunol. 2021, 18, 1085–1095. [Google Scholar] [CrossRef]
- Bolton, K.L.; Gillis, N.K.; Coombs, C.C.; Takahashi, K.; Zehir, A.; Bejar, R.; Garcia-Manero, G.; Futreal, A.; Jensen, B.C.; Diaz Jr, L.A. Managing clonal hematopoiesis in patients with solid tumors. J. Clin. Oncol. 2019, 37, 7–11. [Google Scholar] [CrossRef]
- Inamura, K. Lung cancer: Understanding its molecular pathology and the 2015 WHO classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Qu, J.; Mei, Q.; Chen, L.; Zhou, J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): Current status and future perspectives. Cancer Immunol. Immunother. 2020, 70, 619–631. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecek, M.; Lupo-Stanghellini, M.-T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, A.; Goodpaster, T.; Randolph-Habecker, J.; Hoffstrom, B.G.; Jalikis, F.G.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D. Analysis of ROR1 protein expression in human cancer and normal tissues. Clin. Cancer Res. 2017, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Yanagisawa, K.; Sugiyama, R.; Hosono, Y.; Shimada, Y.; Arima, C.; Kato, S.; Tomida, S.; Suzuki, M.; Osada, H. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 2012, 21, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of targeting ROR1 in primates with chimeric antigen receptor–modified T cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric antigen receptor T-cell therapy in glioblastoma: Charging the T cells to fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. Immunother. Cancer 2015, 3, O11. [Google Scholar] [CrossRef] [Green Version]
- Schmidts, A.; Maus, M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Karschnia, P.; Blobner, J.; Teske, N.; Schöberl, F.; Fitzinger, E.; Dreyling, M.; Tonn, J.-C.; Thon, N.; Subklewe, M.; von Baumgarten, L. CAR T-Cells for CNS Lymphoma: Driving into New Terrain? Cancers 2021, 13, 2503. [Google Scholar] [CrossRef] [PubMed]
- Mulazzani, M.; Fräßle, S.P.; von Mücke-Heim, I.; Langer, S.; Zhou, X.; Ishikawa-Ankerhold, H.; Leube, J.; Zhang, W.; Dötsch, S.; Svec, M. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 24275–24284. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Wijewarnasuriya, D.; Bebernitz, C.; Lopez, A.V.; Rafiq, S.; Brentjens, R.J. Excessive costimulation leads to dysfunction of adoptively transferred T cells. Cancer Immunol. Res. 2020, 8, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey Jr, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell–mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.; Cross, R.; Watson, K.; Liao, Y.; Shi, W.; Prince, H.; Beavis, P.; Trapani, J.; Kershaw, M.; Ritchie, D. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.-M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [Green Version]
- Baymon, D.E.; Boyer, E.W. Chimeric antigen receptor T-cell toxicity. Curr. Opin. Pediatr. 2019, 31, 251–255. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-β-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, F.; Cao, J.; Neelalpu, S.S. Versatile CAR T-cells for cancer immunotherapy. Contemp. Oncol. 2018, 22, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; Delpozzo, F.; Desantis, G.; Papalini, F.; Simonato, F.; Sonda, N.; Zilio, S.; Bronte, V. Therapeutic targeting of myeloid-derived suppressor cells. Contemp. Oncol. 2009, 9, 470–481. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznansky, M.C.; Olszak, I.T.; Foxall, R.; Evans, R.H.; Luster, A.D.; Scadden, D.T. Active movement of T cells away from a chemokine. Nat. Med. 2000, 6, 543–548. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Mellor, A.L.; Keskin, D.B.; Johnson, T.; Chandler, P.; Munn, D.H. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J. Immunol. 2002, 168, 3771–3776. [Google Scholar] [CrossRef] [Green Version]
- Kosti, P.; Maher, J.; Arnold, J.N. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front. Immunol. 2018, 9, 1104. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Straten, T. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365–73 wild type peptide loaded on dendritic cells in vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand. J. Immunol. 2000, 51, 128–133. [Google Scholar]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, S.; Yang, J.C. Landscape of tumor antigens in T cell immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Van Belzen, I.A.; Kesmir, C. Immune biomarkers for predicting response to adoptive cell transfer as cancer treatment. Immunogenetics 2019, 71, 71–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic viruses and immune checkpoint inhibition: The best of both worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelderman, S.; Kvistborg, P. Tumor antigens in human cancer control. Biochim. Biophys. Acta 2016, 1865, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on tumor neoantigens and their utility: Why it is good to be different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, C.; Sleijfer, S.; Vulto, A.G.; Kruit, W.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The principles of engineering immune cells to treat cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Brentjens, R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther. 2017, 178, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric antigen receptor-modified T cell therapy in multiple myeloma: Beyond B cell maturation antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.; El Chaer, F.; Koka, R.; Singh, Z.; Hutnick, E.; Ruehle, K.; Lee, S.T.; Kocoglu, M.H.; Shanholtz, C.; Badros, A. Rapid relapse of large B-cell lymphoma after CD19 directed CAR-T-cell therapy due to CD-19 antigen loss. Am. J. Hematol. 2019, 94, E273–E275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Heuser, C.; Gerken, M.; Fischer, B.; Lewalter, K.; Diehl, V.; Pohl, C.; Abken, H. T cell activation by recombinant FcεRI γ-chain immune receptors: An extracellular spacer domain impairs antigen-dependent T cell activation but not antigen recognition. Gene Ther. 2000, 7, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Lai, Y.; Zhao, R.; Wei, X.; Weng, J.; Lai, P.; Li, B.; Lin, S.; Wang, S.; Wu, Q. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J. Hematol. Oncol. 2017, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef]
- Lindner, S.; Johnson, S.; Brown, C.; Wang, L. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Wang, Z.; Wang, Y.; Liu, Y.; Dai, H.; Tong, C.; Guo, Y.; Guo, B.; Ti, D.; Han, X. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Osborne, W.; Marzolini, M.; Tholouli, E.; Ramakrishnan, A.; Bachier, C.R.; McSweeney, P.A.; Irvine, D.; Zhang, M.; Al-Hajj, M.A.; Pule, M. Phase I Alexander study of AUTO3, the first CD19/22 dual targeting CAR T cell therapy, with pembrolizumab in patients with relapsed/refractory (r/r) DLBCL. J. Clin. Oncol. 2020, 38, 8001. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhong, J.F.; Zhang, X.; Zhang, C. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J. Hematol. Oncol. 2017, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.a.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Muyldermans, S.; Lauwereys, M. Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J. Mol. Recognit. 1999, 12, 131–140. [Google Scholar] [CrossRef]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody based dual specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1–and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 2019, 35, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, D.M.; Foster, J.; Van Der Stegen, S.J.; Parente-Pereira, A.C.; Chiapero-Stanke, L.; Delinassios, G.J.; Burbridge, S.E.; Kao, V.; Liu, Z.; Bosshard-Carter, L. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 565–576. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Chun, J.Y.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef]
- Odagiu, L.; May, J.; Boulet, S.; Baldwin, T.A.; Labrecque, N. Role of the orphan nuclear receptor NR4A family in T-cell biology. Front. Endocrinol 2020, 11, 624122. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Wiede, F.; Lu, K.H.; Du, X.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Goh, P.K.; Kearney, C.; Meyran, D.; Beavis, P.A. PTPN 2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020, 39, e103637. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.-Y.; Kim, Y.-Y.; Yu, H.-S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.; Restifo, N.; Rosenberg, S.; Morgan, R. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W. CXCR1-or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Miao, L.; Ren, Z.; Tang, F.; Li, Y. Gene-Edited Interleukin CAR-T Cells Therapy in the Treatment of Malignancies: Present and Future. Front. Immunol. 2021, 12, 718686. [Google Scholar] [CrossRef]
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 2018, 8, 972–987. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 inverted cytokine receptor improving CAR-T cell potency in immunosuppressive solid-tumor microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2020, 218, e20200844. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S. Glypican-3–specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Lange, S.; Sand, L.G.; Bell, M.; Patil, S.L.; Langfitt, D.; Gottschalk, S. A chimeric GM-CSF/IL18 receptor to sustain CAR T-cell function. Cancer Discov. 2021, 11, 1661–1671. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.; Todd, K.L.; Petley, E.V.; Chan, J.D. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Mayol, K.; Biajoux, V.; Marvel, J.; Balabanian, K.; Walzer, T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood 2011, 118, 4863–4871. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Chou, C.K.; Schietinger, A.; Liggitt, H.D.; Tan, X.; Funk, S.; Freeman, G.J.; Ratliff, T.L.; Greenberg, N.M.; Greenberg, P.D. Cell-intrinsic abrogation of TGF-β signaling delays but does not prevent dysfunction of self/tumor-specific CD8 T cells in a murine model of autochthonous prostate cancer. J. Immunol. 2012, 189, 3936–3946. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, Q.; Risu, N.; Fu, J.; Zou, Y.; Tang, J.; Li, L.; Liu, H.; Zhou, G.; Zhu, X. Galunisertib enhances chimeric antigen receptor-modified T cell function. Eur. J. Histochem. 2020, 64, 3122. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Cheng, F.; Yu, B.; Nefedova, Y.; Sotomayor, E.; Lush, R.; Gabrilovich, D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003, 63, 4441–4449. [Google Scholar] [PubMed]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.K.; Englisch, A.; Brenner, B.; Smith, T.; Hoyos, V.; Suzuki, M.; Brenner, M.K. Mesenchymal stromal cell delivery of oncolytic immunotherapy improves CAR-T cell antitumor activity. Mol. Ther. 2021, 29, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Puzanov, I.; Hecht, J.R.; Hodi, F.S.; Szabo, Z.; Murugappan, S.; Kaufman, H.L. Clinical development of talimogene laherparepvec (T-VEC): A modified herpes simplex virus type-1–derived oncolytic immunotherapy. Expert Rev. Anticancer Ther. 2015, 15, 1389–1403. [Google Scholar] [CrossRef]
- Hadryś, A.; Sochanik, A.; McFadden, G.; Jazowiecka-Rakus, J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020, 874, 172991. [Google Scholar] [CrossRef]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.-T.; Liu, J.-H.; Song, T.-T.; Ma, B.; Amidula, N.; Bai, C. EGLIF-CAR-T Cells Secreting PD-1 Blocking Antibodies Significantly Mediate the Elimination of Gastric Cancer. Cancer Manag. Res. 2020, 12, 8893–8902. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Sajeve, T.S.; Evidio, D.-M.; Pavlick, A.C.; Whitman, E.D. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): Combination or built-in CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 1–10. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Ngai, D.A.; McGee, E.; Chintala, N.K.; Messinger, J.C.; Vincent, A. Abstract CT036: A phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: Safety and efficacy. Cancer Res. 2019, 79, CT036. [Google Scholar]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, S.; Wu, C.; Li, J.; Li, Z.; Wen, C.; Hu, S.; An, G.; Meng, H.; Zhang, X. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy 2018, 10, 935–949. [Google Scholar] [CrossRef]
- Wang, J.; Lupo, K.B.; Chambers, A.M.; Matosevic, S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J. Immunother. Cancer 2018, 6, 136. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Tay, J.C.; Wang, S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol. Ther. Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woll, P.S.; Martin, C.H.; Miller, J.S.; Kaufman, D.S. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J. Immunol. 2005, 175, 5095–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, K.; Capitini, C.M.; Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res. Ther. 2020, 11, 234. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “off-the-shelf” natural killer cells from peripheral blood cell-derived induced pluripotent stem cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A. Markers and function of human NK cells in normal and pathological conditions. Cytom. B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Target | CAR-T Cell Mode † | Combination (Strategy) | Malignancy | Preclinical Study | Clinical Trial | Clinical Trial Register | Ref |
|---|---|---|---|---|---|---|---|
| CD19/CD22 | 41BB | Bispecific CAR-T cell | Leukemia/Lymphoma | - | - | NCT03233854 | [89] |
| CD19/CD20 | 41BB | Bispecific CAR-T cell | Leukemia/Lymphoma | - | - | NCT03019055 | [97] |
| HER2/IL3Ra2 | CD28 | Pooled CAR-T cells | Glioblastoma model | - | [92] | ||
| CD19/CD123 | 41BB | Pooled CAR-T cell | Leukemia model | - | [93] | ||
| PSMA/CD19 | PD1/CD28 | Bispecific CAR-T cell | Leukemia model | - | [107] | ||
| CD22/CD19 | 41BB | Tandem CAR-T cell | B-ALL | - | - | [94] | |
| CD19/MSLN | synNotch/41BB | Bispecific CAR-T cell (synNotch CAR) | Leukemia model | - | [108] | ||
| CD19/CD22 | OX40/41BB | Bispecific CAR-T & Pembrolizumab | DLBCL | - | - | NCT03287817 | [95] |
| CD20/HER2 | CD28 | Nanobody based tandem CAR-T cell | Leukemia cell | * | [104] | ||
| EPHA2 or HER2/GM-CSF | CD28/IL18R | IL18R (MYD88 signaling) | Ewing sarcoma model Osteosarcoma model | - | [137] | ||
| GD2 | 41BB/IL7Ra | C7R (STAT5 signaling) | GD2-expressing brain tumors, RR neuroblastoma, other GD2-expressing cancers | - | - | NCT04099797 NCT03635632 | [135] |
| PSCA/TGF-β /IL4 | CD28/41BB/IL7Ra | 4/7 ICR | Pancreatic adenocarcinoma model | - | [131] | ||
| GPC3/IL4 | CD28/IL21R | 4/21 ICR | IL-4-expressing hepatocarcinoma model | - | [132] | ||
| GPC3 | 41BB | IL15, IL21, iCasp9 | Liver cancer Rhabdomyosarcoma | - | - | NCT04715191 | [136] |
| N/A | N/A | Knockout of PD1 | Esophageal cancer Renal cell carcinoma Castration resistant prostate cancer Metastatic NSCLC Advanced stage EBV | - - - - - | - - - - - | NCT03081715 NCT02867332 NCT02867345 NCT02793856 NCT03044743 | |
| CD19 | CD28 | Knockout of NR4A | Lymphoma model, Melanoma model | - | [120] | ||
| CD19 | 41BB | Knockout of TCR/β2 microglobulin/PD1 | ALL model | - | [116] | ||
| HER2 | CD28 | Knockout of A2AR | Ovarian carcinoma model | - | [112] | ||
| HER2 | CD28 | Knockout of PTPN2 | Breast cancer model | - | [121] | ||
| EGFR | 41BB | Knockout of DGK | Glioblastoma model | - | [122] | ||
| HER2 | 41BB | Overexpression ofc-Jun | Osteosarcoma model | - | [126] | ||
| GPC3 | 41BB | Overexpression ofCXCR2 | Hepatocellular carcinoma model | - | [128] | ||
| CD70 | 41BB | Overexpression of CXCR1 or CXCR2 | Glioblastoma model Pancreatic tumor model Ovarian tumor model | - | [129] | ||
| PSMA | 41BB | Overexpression of TGF-β DNRII | Prostate cancer | - | - | NCT03089203 | [125] |
| CD19 | CD28 (Yescarta®) | Lenzilumab | DLBCL | - | - | ZUMA-19 | [50] |
| CD19 | 41BB | Dasatinib | Lymphoma | - | [141] | ||
| CD133/HER2 | CD28, 41BB | Galunisertib | Glioblastoma, Breast cancer cell | * | [144] | ||
| HER2 | CD28 | Cell-carrier-delivered oncolytic adenovirus | NSCLC | - | - | NCT03740256 | [147] |
| EGFR | 41BB | Anti-PD1 scFv secretion | Lung gastric liver cancer | - | - | NCT02862028 | [152,154] |
| MSLN | 41BB | Anti-PD1 Ab expression | Refractory prostate cancer | - | - | NCT03030001 | [153] |
| MSLN | 41BB | Anti-CTLA4/PD1 Ab expression | Non-hematologic malignancies | - | - | NCT03182803 | [159] |
| EGFR | 41BB | Anti-CTLA4/PD1 Ab expression | NSCLC | - | - | NCT03182816 | [160] |
| CD19 | 41BB | Pembrolizumab | DLBCL | - | - | NCT02650999 | [161] |
| CD19 | 41BB | Pembrolizumab | NHL | - | - | NCT02030834 | [162] |
| GD2 | CD28,OX40 | Pembrolizumab | Neuroblastoma | - | - | NCT01822652 | [163] |
| CD19 | 41BB | alemtuzumab | B-ALL | - | - | NCT02808442 NCT02746952 | [96] |
| N/A | TIL | Ipilimumab | Metastatic melanoma | - | - | NCT02027935 NCT03296137 | [158] |
| N/A | TIL | Nivolumab | Metastatic melanoma | - | - | NCT03638375 | |
| N/A | TIL | Nivolumab, Pembrolizumab | Metastatic melanoma | - | - | NCT03645928 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, H.; Jung, H.; Noh, J.-Y. Emerging Approaches for Solid Tumor Treatment Using CAR-T Cell Therapy. Int. J. Mol. Sci. 2021, 22, 12126. https://doi.org/10.3390/ijms222212126
Chung H, Jung H, Noh J-Y. Emerging Approaches for Solid Tumor Treatment Using CAR-T Cell Therapy. International Journal of Molecular Sciences. 2021; 22(22):12126. https://doi.org/10.3390/ijms222212126
Chicago/Turabian StyleChung, Hyunmin, Haiyoung Jung, and Ji-Yoon Noh. 2021. "Emerging Approaches for Solid Tumor Treatment Using CAR-T Cell Therapy" International Journal of Molecular Sciences 22, no. 22: 12126. https://doi.org/10.3390/ijms222212126
APA StyleChung, H., Jung, H., & Noh, J.-Y. (2021). Emerging Approaches for Solid Tumor Treatment Using CAR-T Cell Therapy. International Journal of Molecular Sciences, 22(22), 12126. https://doi.org/10.3390/ijms222212126