
Enterococcus faecium Regulates Honey Bee Developmental Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
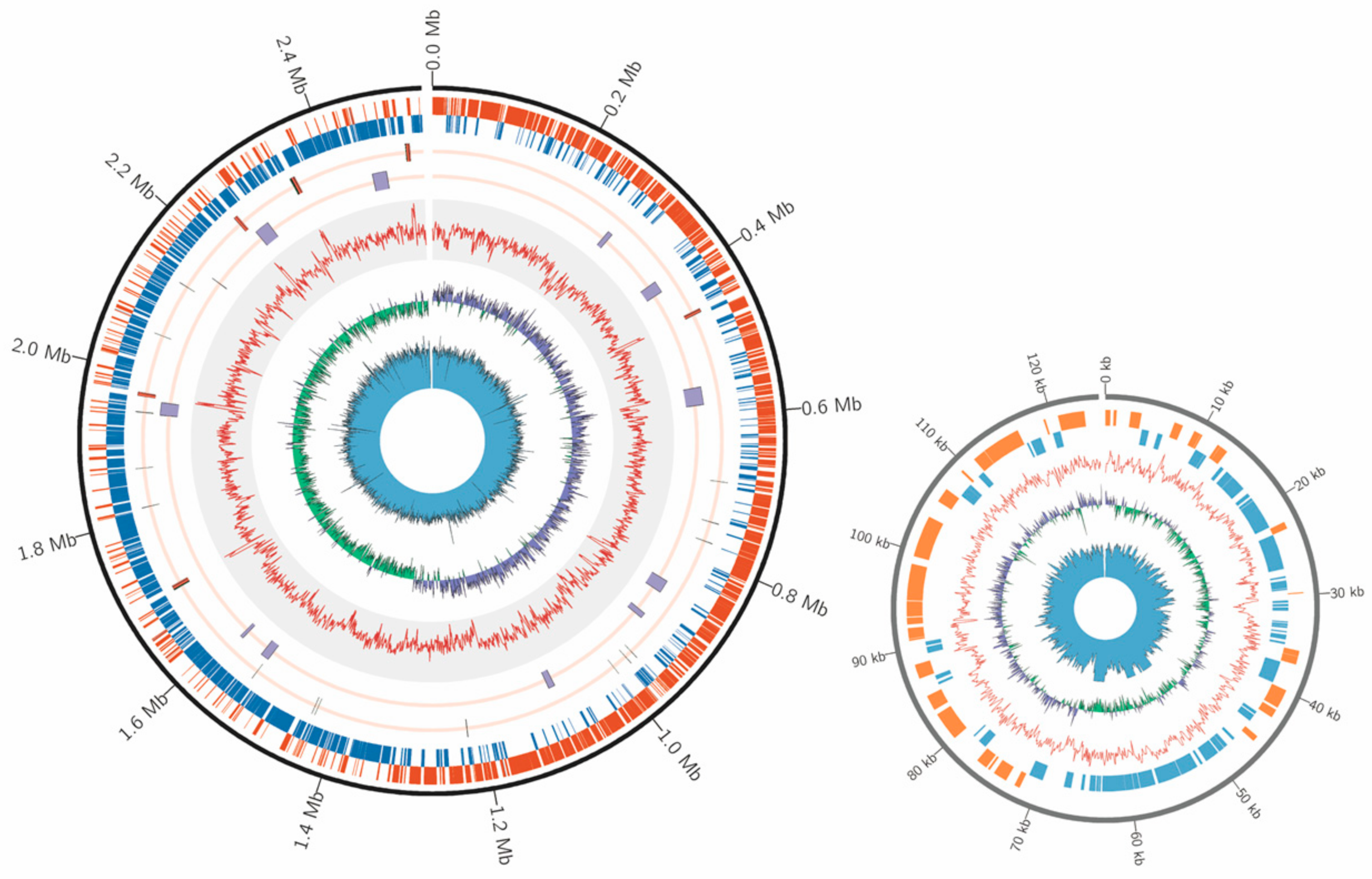
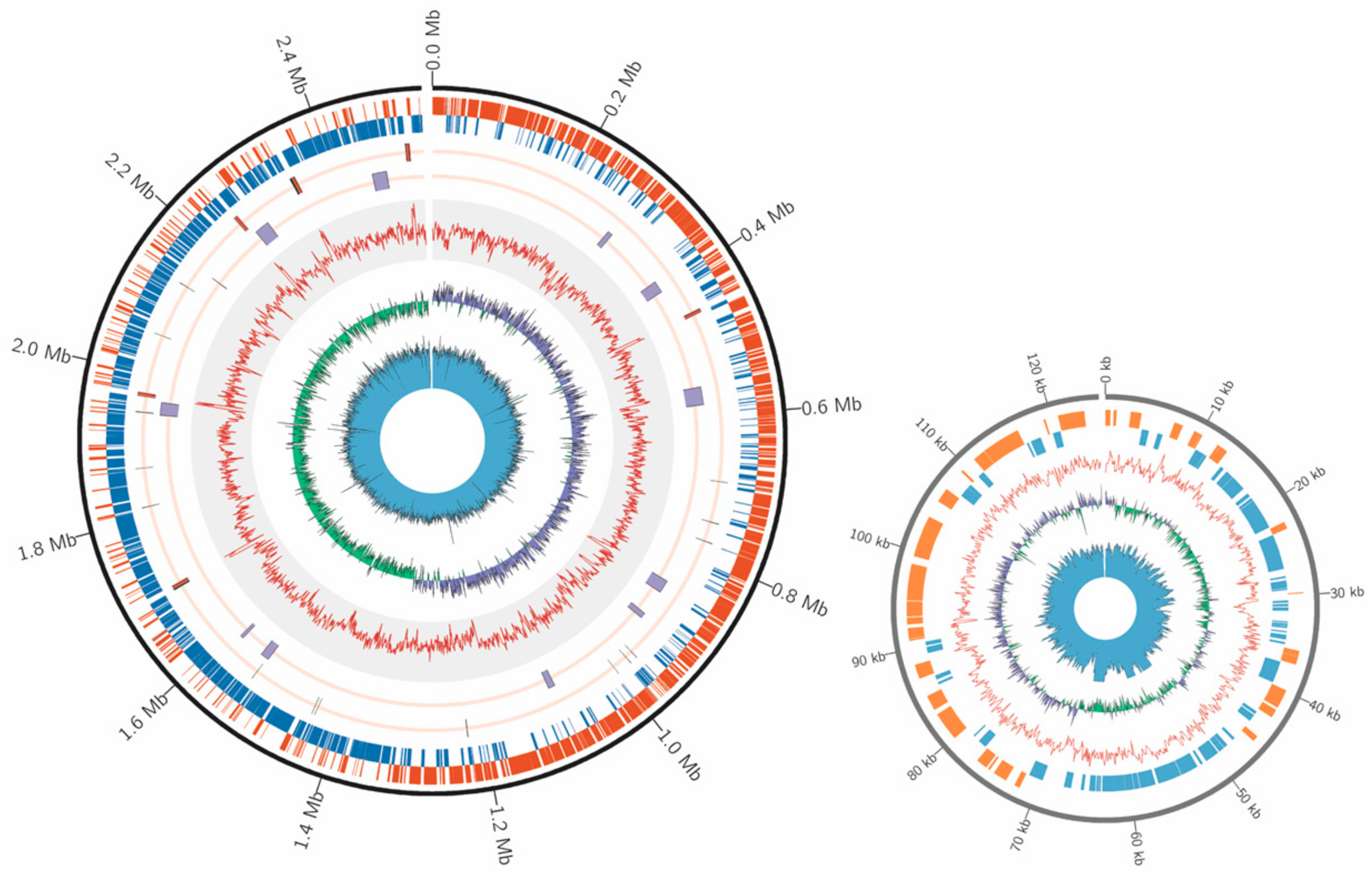
2.1. Draft Genome Sequence of E. faecium Isolated from Honey Bee Gut
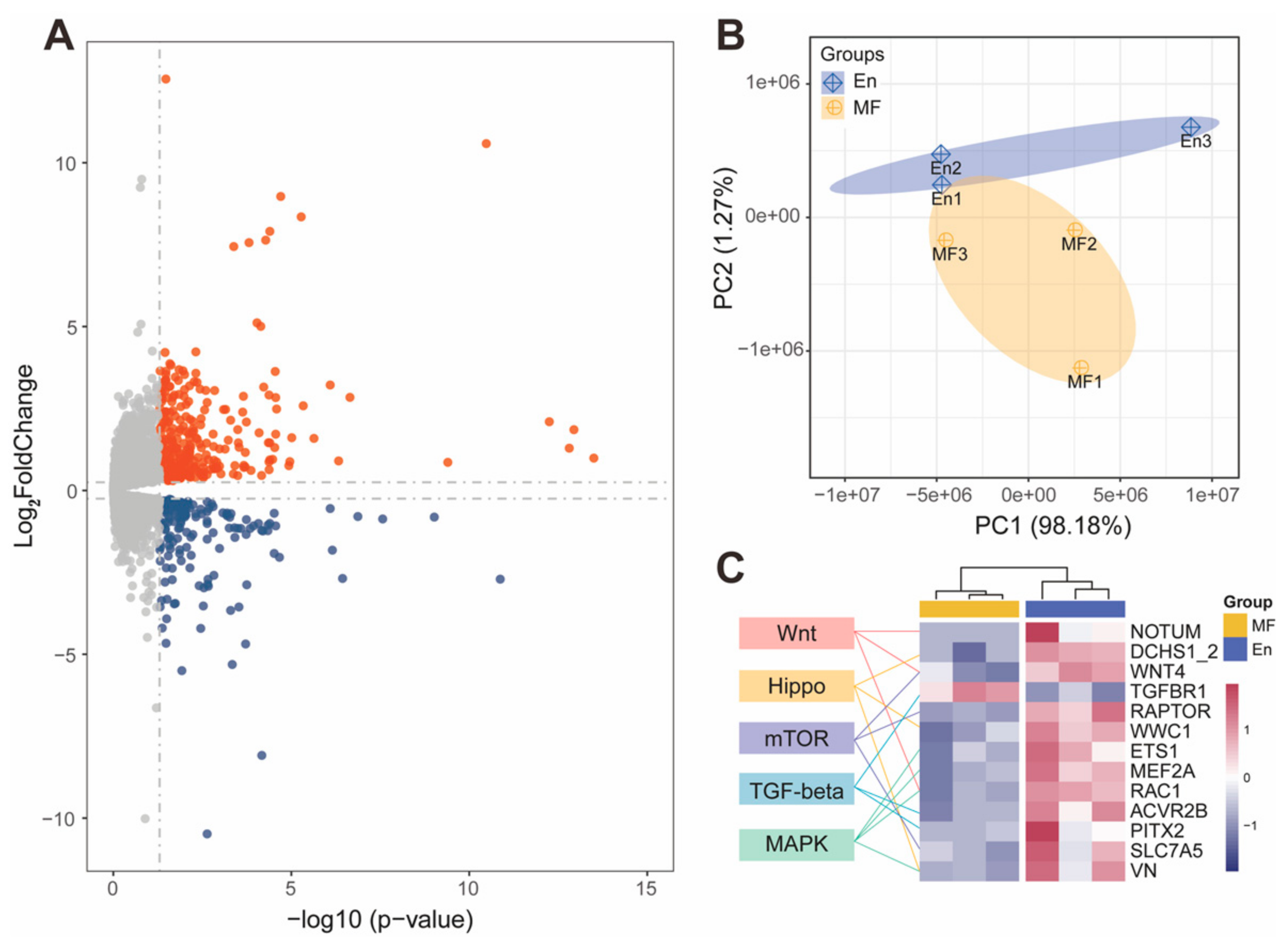
2.2. E. faecium Increased Gut Weight of Honey Bee and Activated Host Developmental Gene Expression
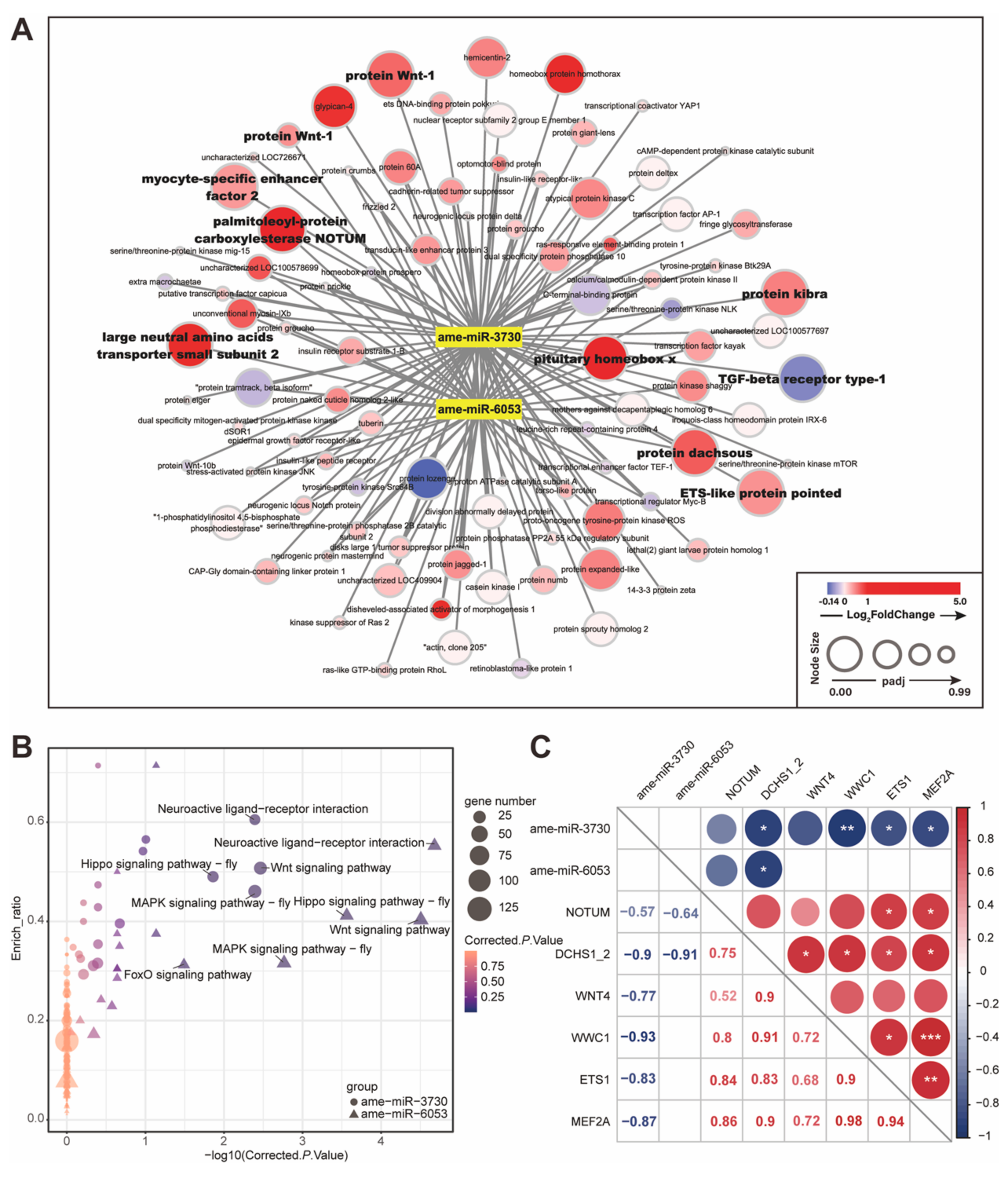
2.3. MicroRNAs Participated in Host Developmental Pathway Regulation during E. faecium Colonization
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain Isolation and Culture
4.2. Bacterial DNA Extraction and Genome Sequencing
4.3. Bacterial Genome Assembly
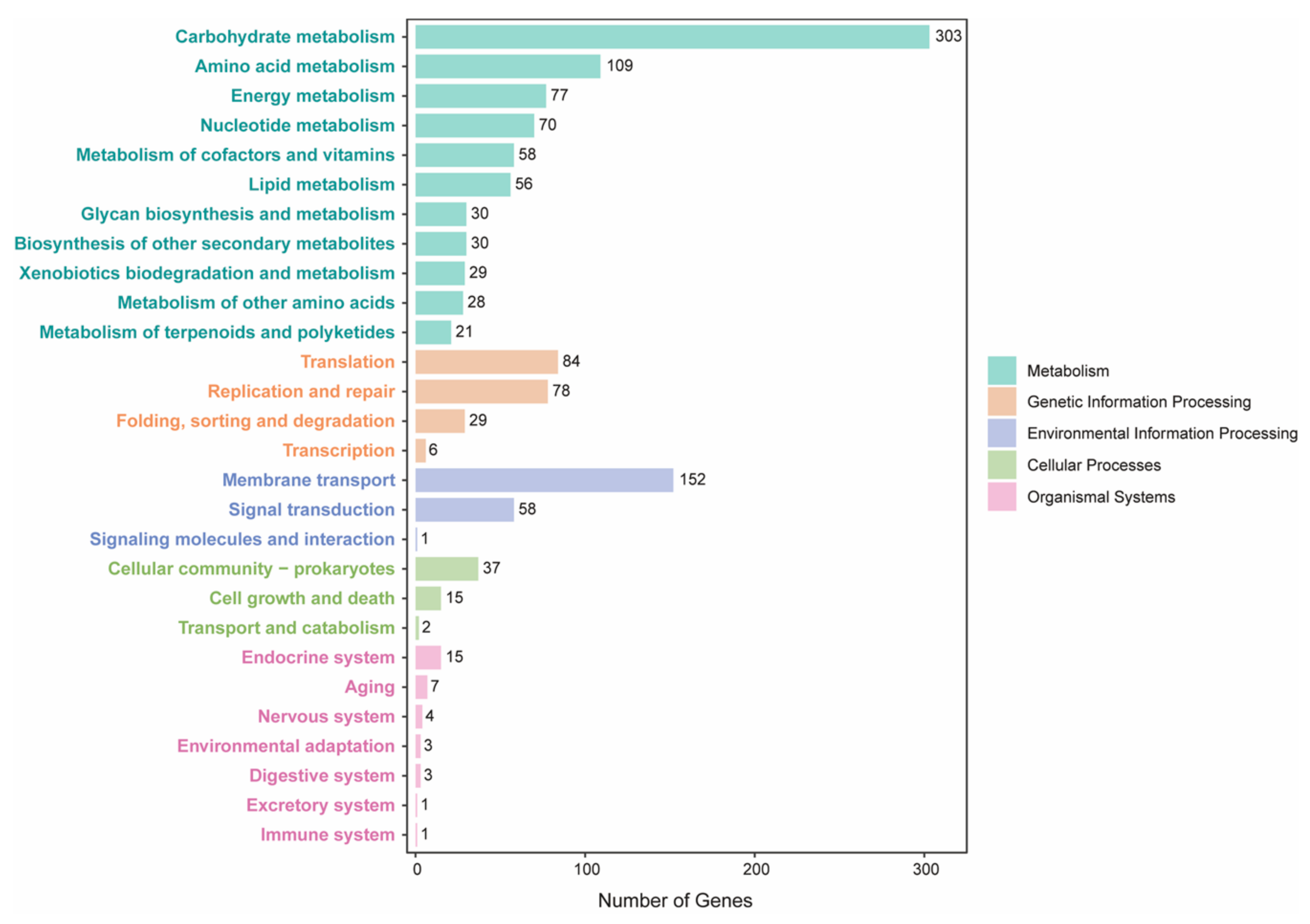
4.4. Structural and Functional Annotations for Bacterial Genomes
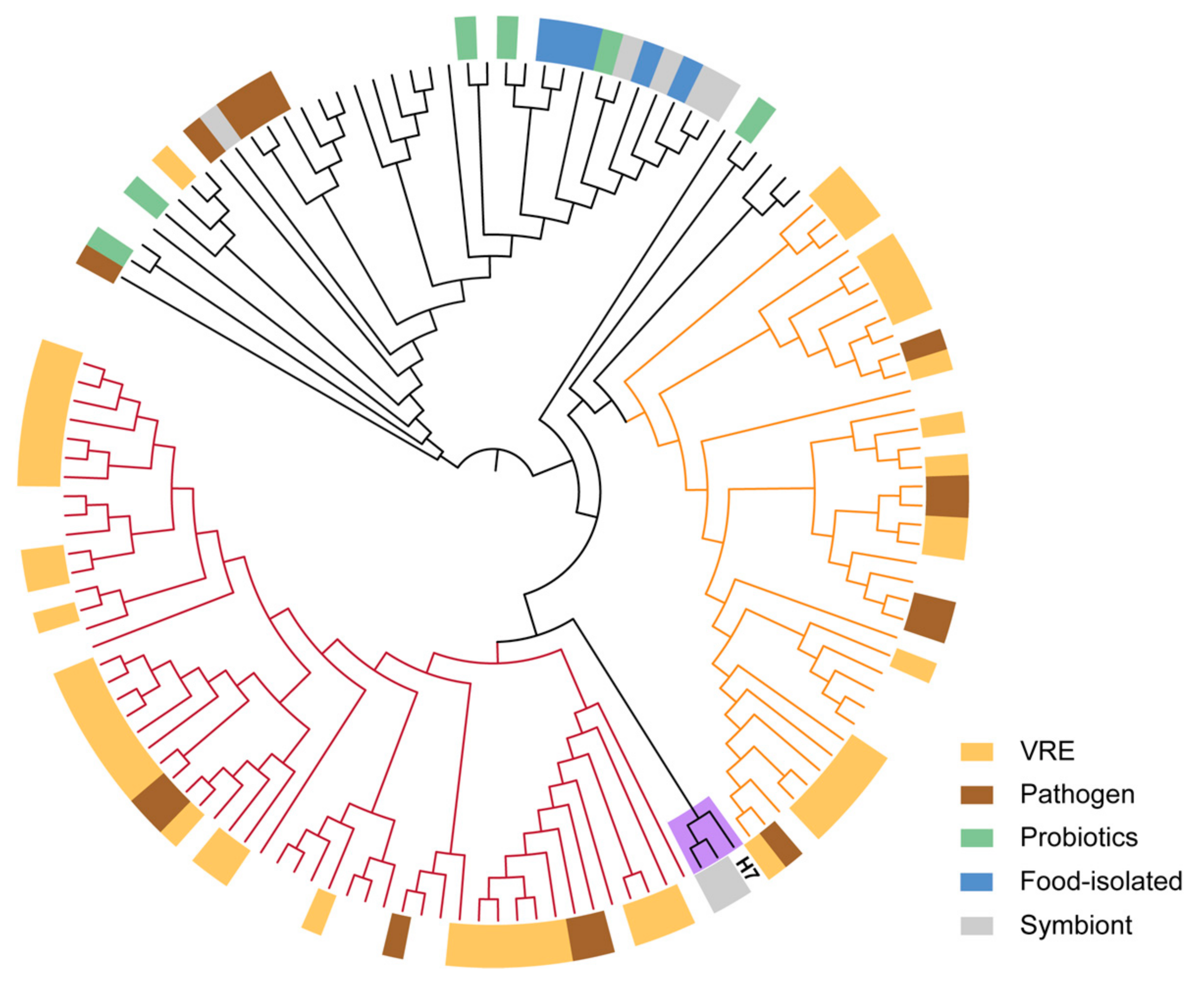
4.5. Phylogenetic Analysis
4.6. Generation of Microbiota-Free (MF) and E. faecium-Colonized (En) Bees
4.7. Extraction of DNA and RNA from Honey Bee Hindgut
4.8. Quantitative PCR (qPCR) for Determination of Gut Bacterial Loads
4.9. 16S Amplicon Sequence Analysis
4.10. RNA-Seq Analysis
4.11. Small RNA-Seq Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanengelsdorp, D.; Meixner, M.D. A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Invertebr. Pathol. 2010, 103, S80–S95. [Google Scholar] [CrossRef] [PubMed]
- Kaznowski, A.; Szymas, B.; Jazdzinska, E.; Kazimierczak, M.; Paetz, H.; Mokracka, J. The effects of probiotic supplementation on the content of intestinal microflora and chemical composition of worker honey bees (Apis mellifera). J. Apic. Res. 2005, 44, 10–14. [Google Scholar] [CrossRef]
- Renata Rogala, B.S. Nutritional value for bees of pollen substitute enriched with synthetic amino acids. Part II. Biological methods. J. Apic. Sci. 2004, 48, 29–36. [Google Scholar]
- Alberoni, D.; Gaggìa, F.; Baffoni, L.; Di Gioia, D. Beneficial microorganisms for honey bees: Problems and progresses. Appl. Microbiol. Biotechnol. 2016, 100, 9469–9482. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, D.; Castelli, L.; Porrini, M.P.; Garrido, P.M.; Eguaras, M.J.; Zunino, P.; Antúnez, K. Lactobacillus kunkeei strains decreased the infection by honey bee pathogens Paenibacillus larvae and Nosema ceranae. Benef. Microbes 2018, 9, 279–290. [Google Scholar] [CrossRef]
- Daisley, B.A.; Pitek, A.P.; Chmiel, J.A.; Al, K.F.; Chernyshova, A.M.; Faragalla, K.M.; Burton, J.P.; Thompson, G.J.; Reid, G. Novel probiotic approach to counter Paenibacillus larvae infection in honey bees. ISME J. 2020, 14, 476–491. [Google Scholar] [CrossRef] [Green Version]
- Dimov, S.G.; Guyrova, A.; Vladimirova, A.; Dimitrov, M.; Peykov, S.; Strateva, T. WGS-based characterization of the potentially beneficial Enterococcus faecium EFD from a beehive. Mol. Biol. Rep. 2020, 47, 6445–6449. [Google Scholar] [CrossRef] [PubMed]
- Pătruică, S.; Hutu, I. Economic benefits of using prebiotic and probiotic products as supplements in stimulation feeds administered to bee colonies. Turk. J. Vet. Anim. Sci. 2013, 37, 259–263. [Google Scholar]
- Pătruică, S.; Mot, D. The effect of using prebiotic and probiotic products on intestinal micro-flora of the honeybee (Apis mellifera carpatica). Bull. Entomol. Res. 2012, 102, 619–623. [Google Scholar] [CrossRef]
- Baffoni, L.; Gaggia, F.; Alberoni, D.; Cabbri, R.; Nanetti, A.; Biavati, B.; Di Gioia, D. Effect of dietary supplementation of Bifidobacterium and Lactobacillus strains in Apis mellifera L. against Nosema ceranae. Benef. Microbes 2016, 7, 45–51. [Google Scholar] [CrossRef]
- Yoshiyama, M.; Wu, M.; Sugimura, Y.; Takaya, N.; Kimoto-Nira, H.; Suzuki, C. Inhibition of Paenibacillus larvae by lactic acid bacteria isolated from fermented materials. J. Invertebr. Pathol. 2013, 112, 62–67. [Google Scholar] [CrossRef]
- Sabaté, D.C.; Cruz, M.S.; Benítez-Ahrendts, M.R.; Audisio, M.C. Beneficial effects of Bacillus subtilis subsp. subtilis Mori2, a honey-associated strain, on honeybee colony performance. Probiotics Antimicrob. Proteins 2012, 4, 39–46. [Google Scholar] [CrossRef]
- García-Solache, M.; Rice, L.B. The Enterococcus: A model of adaptability to its environment. Clin. Microbiol. Rev. 2019, 32, e00058-18. [Google Scholar] [CrossRef] [Green Version]
- Feizabadi, F.; Sharifan, A.; Tajabadi, N. Isolation and identification of lactic acid bacteria from stored Apis mellifera honey. J. Apic. Res. 2021, 60, 421–426. [Google Scholar] [CrossRef]
- Elzeini, H.M.; Ali, A.A.; Nasr, N.F.; Elenany, Y.E.; Hassan, A.A.M. Isolation and identification of lactic acid bacteria from the intestinal tracts of honey bees, Apis mellifera L. in Egypt. J. Apic. Res. 2021, 60, 349–357. [Google Scholar] [CrossRef]
- Audisio, M.C.; Terzolo, H.R.; Apella, M.C. Bacteriocin from honeybee beebread Enterococcus avium, active against Listeria monocytogenes. Appl. Environ. Microbiol. 2005, 71, 3373–3375. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Mundt, J.O. Enterococci in insects. Appl. Microbiol. 1972, 24, 575–580. [Google Scholar] [CrossRef]
- Tajabadi, N.; Mardan, M.; Shuhaimi, M.; Abdul Manap, M.Y. Isolation and identification of Enterococcus sp. from honey stomach of honeybee based on biochemical and 16S rrna sequencing analysis. Int. J. Probiotics Prebiotics 2011, 6, 95–100. [Google Scholar]
- Carina Audisio, M.; Torres, M.J.; Sabaté, D.C.; Ibarguren, C.; Apella, M.C. Properties of different lactic acid bacteria isolated from Apis mellifera L. bee-gut. Microbiol. Res. 2011, 166, 1–13. [Google Scholar] [CrossRef]
- Zommiti, M.; Cambronel, M.; Maillot, O.; Barreau, M.; Sebei, K.; Feuilloley, M.; Ferchichi, M.; Connil, N. Evaluation of probiotic properties and safety of Enterococcus faecium isolated from artisanal tunisian meat “Dried Ossban”. Front. Microbiol. 2018, 9, 1685. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, E.; Marchioni, E.; Aoude-Werner, D.; Hasselmann, C.; Ennahar, S. Smearing of soft cheese with Enterococcus faecium WHE 81, a multi-bacteriocin producer, against Listeria monocytogenes. Food Microbiol. 2009, 26, 16–20. [Google Scholar] [CrossRef]
- Hegarty, J.W.; Guinane, C.M.; Ross, R.P.; Hill, C.; Cotter, P.D. Bacteriocin production: A relatively unharnessed probiotic trait? F1000Research 2016, 5, 2587. [Google Scholar] [CrossRef]
- Martín, M.; Gutiérrez, J.; Criado, R.; Herranz, C.; Cintas, L.M.; Hernández, P.E. Cloning, production and expression of the bacteriocin enterocin A produced by Enterococcus faecium PLBC21 in Lactococcus lactis. Appl. Microbiol. Biotechnol. 2007, 76, 667–675. [Google Scholar] [CrossRef]
- Torres, C.; Alonso, C.A.; Ruiz-Ripa, L.; León-Sampedro, R.; Del Campo, R.; Coque, T.M. Antimicrobial resistance in Enterococcus spp. of animal origin. Microbiol. Spectr. 2018, 6, 6.4.24. [Google Scholar] [CrossRef]
- Zheng, A.; Luo, J.; Meng, K.; Li, J.; Bryden, W.L.; Chang, W.; Zhang, S.; Wang, L.X.N.; Liu, G.; Yao, B. Probiotic (Enterococcus faecium) induced responses of the hepatic proteome improves metabolic efficiency of broiler chickens (Gallus gallus). BMC Genom. 2016, 17, 89. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhen, W.; Geng, Y.; Wang, Z.; Guo, Y. Pretreatment with probiotic Enterococcus faecium NCIMB 11181 ameliorates necrotic enteritis-induced intestinal barrier injury in broiler chickens. Sci. Rep. 2019, 9, 10256. [Google Scholar] [CrossRef]
- Pollmann, M.; Nordhoff, M.; Pospischil, A.; Tedin, K.; Wieler, L.H. Effects of a probiotic strain of Enterococcus faecium on the rate of natural chlamydia infection in swine. Infect. Immun. 2005, 73, 4346–4353. [Google Scholar] [CrossRef] [Green Version]
- Costa Sousa, N.; Couto, M.V.S.; Abe, H.A.; Paixão, P.E.G.; Cordeiro, C.A.M.; Monteiro Lopes, E.; Ready, J.S.; Jesus, G.F.A.; Martins, M.L.; Mouriño, J.L.P.; et al. Effects of an Enterococcus faecium-based probiotic on growth performance and health of Pirarucu, Arapaima gigas. Aquac. Res. 2019, 50, 3720–3728. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Guan, L.L. Noncoding RNAs: Regulatory molecules of host–microbiome crosstalk. Trends Microbiol. 2021, 29, 713–724. [Google Scholar] [CrossRef]
- Li, M.; Chen, W.D.; Wang, Y.D. The roles of the gut microbiota–miRNA interaction in the host pathophysiology. Mol. Med. 2020, 26, 101. [Google Scholar] [CrossRef]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casaus, P.; Nilsen, T.; Cintas, L.M.; Nes, I.F.; Hernández, P.E.; Holo, H. Enterocin B, a new bacteriocin from Enterococcus faecium T136 which can act synergistically with enterocin A. Microbiology 1997, 143, 2287–2294. [Google Scholar] [CrossRef] [Green Version]
- van Hal, S.J.; Willems, R.J.L.; Gouliouris, T.; Ballard, S.A.; Coque, T.M.; Hammerum, A.M.; Hegstad, K.; Westh, H.T.; Howden, B.P.; Malhotra-Kumar, S.; et al. The global dissemination of hospital clones of Enterococcus faecium. Genome Med. 2021, 13, 52. [Google Scholar] [CrossRef]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Werner, G.; Coque, T.M.; Hammerum, A.M.; Hope, R.; Hryniewicz, W.; Johnson, A.; Klare, I.; Kristinsson, K.G.; Leclercq, R.; Lester, C.H.; et al. Emergence and spread of vancomycin resistance among enterococci in Europe. Eurosurveillance 2008, 13, 19046. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Powell, J.E.; Steele, M.I.; Dietrich, C.; Moran, N.A. Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 4775–4780. [Google Scholar] [CrossRef] [Green Version]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Lucchetta, E.; Rafel, N.; Ohlstein, B. Maintenance of the adult Drosophila intestine: All roads lead to homeostasis. Curr. Opin. Genet. Dev. 2016, 40, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Shivdasani, A.A.; Ingham, P.W. Regulation of stem cell maintenance and transit amplifying cell proliferation by tgf-beta signaling in Drosophila spermatogenesis. Curr. Biol. 2003, 13, 2065–2072. [Google Scholar] [CrossRef] [Green Version]
- Hanchi, H.; Mottawea, W.; Sebei, K.; Hammami, R. The genus Enterococcus: Between probiotic potential and safety concerns-an update. Front. Microbiol. 2018, 9, 1791. [Google Scholar] [CrossRef]
- Mason, K.L.; Stepien, T.A.; Blum, J.E.; Holt, J.F.; Labbe, N.H.; Rush, J.S.; Raffa, K.F.; Handelsman, J. From commensal to pathogen: Translocation of Enterococcus faecalis from the midgut to the hemocoel of Manduca sexta. mBio 2011, 2, e00065-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doron, S.; Snydman, D.R. Risk and safety of probiotics. Clin. Infect. Dis. 2015, 60, S129–S134. [Google Scholar] [CrossRef] [Green Version]
- Engel, P.; Martinson, V.G.; Moran, N.A. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. USA 2012, 109, 11002–11007. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Perreau, J.; Powell, J.E.; Han, B.; Zhang, Z.; Kwong, W.K.; Tringe, S.G.; Moran, N.A. Division of labor in honey bee gut microbiota for plant polysaccharide digestion. Proc. Natl. Acad. Sci. USA 2019, 116, 25909–25916. [Google Scholar] [CrossRef]
- Kwong, W.K.; Mancenido, A.L.; Moran, N.A. Immune system stimulation by the native gut microbiota of honey bees. R. Soc. Open Sci. 2017, 4, 170003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zheng, Y.; Chen, Y.; Chen, G.; Zheng, H.; Hu, F. Apis cerana gut microbiota contribute to host health though stimulating host immune system and strengthening host resistance to Nosema ceranae. R. Soc. Open Sci. 2020, 7, 192100. [Google Scholar] [CrossRef] [PubMed]
- Kešnerová, L.; Mars, R.A.T.; Ellegaard, K.M.; Troilo, M.; Sauer, U.; Engel, P. Disentangling metabolic functions of bacteria in the honey bee gut. PLoS Biol. 2017, 15, e2003467. [Google Scholar] [CrossRef] [Green Version]
- Broderick, N.A.; Buchon, N.; Lemaitre, B.; McFall-Ngai, M.J. Microbiota-induced changes in Drosophila melanogaster host gene expression and gut morphology. mBio 2014, 5, e01117-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing complexity of Ras signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar] [CrossRef] [Green Version]
- McKenna, L.B.; Schug, J.; Vourekas, A.; McKenna, J.B.; Bramswig, N.C.; Friedman, J.R.; Kaestner, K.H. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 2010, 139, 1654–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foronda, D.; Weng, R.; Verma, P.; Chen, Y.W.; Cohen, S.M. Coordination of insulin and Notch pathway activities by microRNA miR-305 mediates adaptive homeostasis in the intestinal stem cells of the Drosophila gut. Genes Dev. 2014, 28, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Vieira, J.; Freitas, F.C.P.; Cristino, A.S.; Moda, L.M.R.; Martins, J.R.; Bitondi, M.M.G.; Simões, Z.L.P.; Barchuk, A.R. miRNA-34 and miRNA-210 target hexamerin genes enhancing their differential expression during early brain development of honeybee (Apis mellifera) castes. Insect Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Lourenço, A.P.; Guidugli-Lazzarini, K.R.; Freitas, F.C.P.; Bitondi, M.M.G.; Simões, Z.L.P. Bacterial infection activates the immune system response and dysregulates microRNA expression in honey bees. Insect Biochem. Mol. Biol. 2013, 43, 474–482. [Google Scholar] [CrossRef]
- Li, L.; Liu, F.; Li, W.; Li, Z.; Pan, J.; Yan, L.; Zhang, S.; Huang, Z.Y.; Su, S. Differences in microRNAs and their expressions between foraging and dancing honey bees, Apis mellifera L. J. Insect Physiol. 2012, 58, 1438–1443. [Google Scholar] [CrossRef]
- Shi, Y.Y.; Zheng, H.J.; Pan, Q.Z.; Wang, Z.L.; Zeng, Z.J. Differentially expressed microRNAs between queen and worker larvae of the honey bee (Apis mellifera). Apidologie 2015, 46, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Kwong, W.K.; Moran, N.A. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order ‘Enterobacteriales’ of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 2013, 63, 2008–2018. [Google Scholar]
- Winnepenninckx, B.; Backeljau, T.; De Wachter, R. Extraction of high molecular weight DNA from molluscs. Trends Genet. 1993, 9, 407. [Google Scholar] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.A.O. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 5, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Hunt, M.; Silva, N.D.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar]
- van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Powell, J.E.; Martinson, V.G.; Urban-Mead, K.; Moran, N.A. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera. Appl. Environ. Microbiol. 2014, 80, 7378–7387. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.R.; Alaux, C.; Costa, C.; Csáki, T.; Doublet, V.; Eisenhardt, D.; Fries, I.; Kuhn, R.; McMahon, D.P.; Medrzycki, P.; et al. Standard methods for maintaining adult Apis mellifera in cages under in vitro laboratory conditions. J. Apic. Res. 2013, 52, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Luo, S.; Zhou, X. Enterococcus faecium Regulates Honey Bee Developmental Genes. Int. J. Mol. Sci. 2021, 22, 12105. https://doi.org/10.3390/ijms222212105
Du Y, Luo S, Zhou X. Enterococcus faecium Regulates Honey Bee Developmental Genes. International Journal of Molecular Sciences. 2021; 22(22):12105. https://doi.org/10.3390/ijms222212105
Chicago/Turabian StyleDu, Yating, Shiqi Luo, and Xin Zhou. 2021. "Enterococcus faecium Regulates Honey Bee Developmental Genes" International Journal of Molecular Sciences 22, no. 22: 12105. https://doi.org/10.3390/ijms222212105
APA StyleDu, Y., Luo, S., & Zhou, X. (2021). Enterococcus faecium Regulates Honey Bee Developmental Genes. International Journal of Molecular Sciences, 22(22), 12105. https://doi.org/10.3390/ijms222212105