
Expression of Human Mutant Preproinsulins Induced Unfolded Protein Response, Gadd45 Expression, JAK-STAT Activation, and Growth Inhibition in Drosophila



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
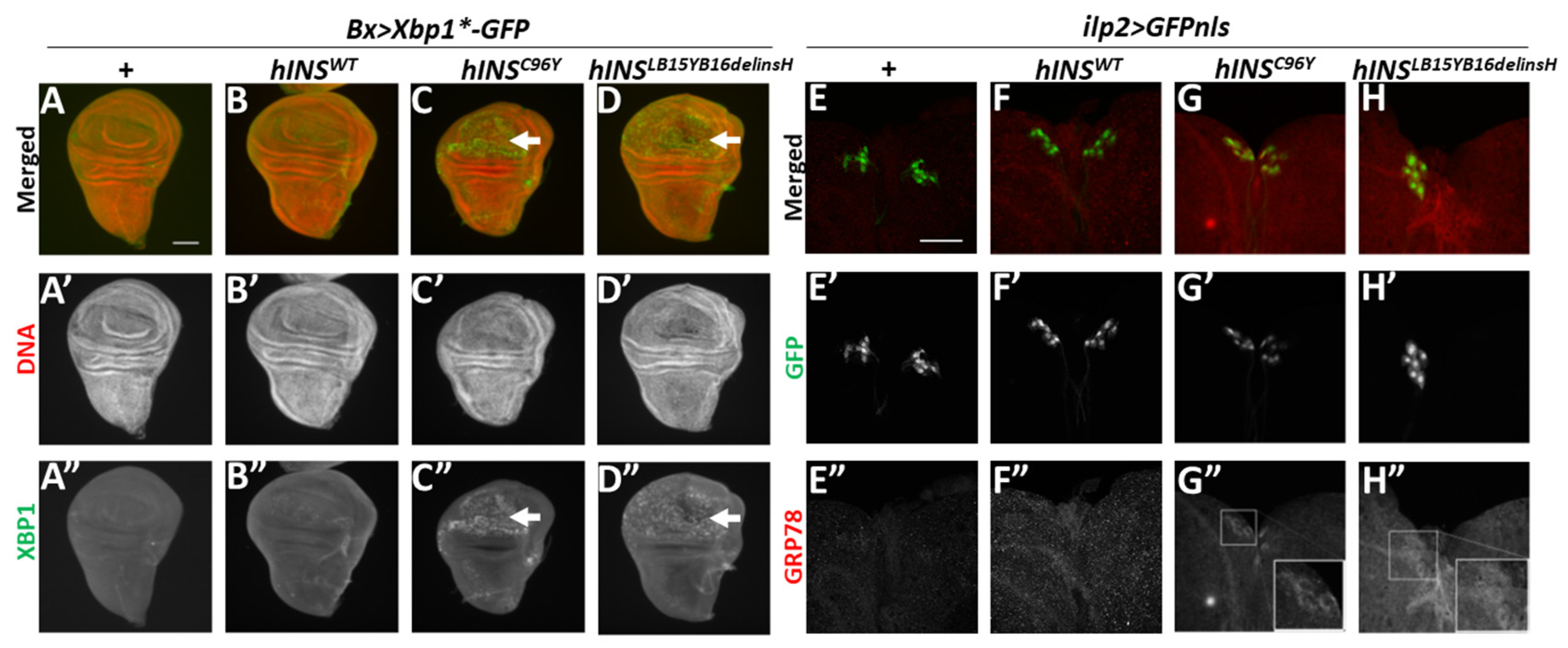
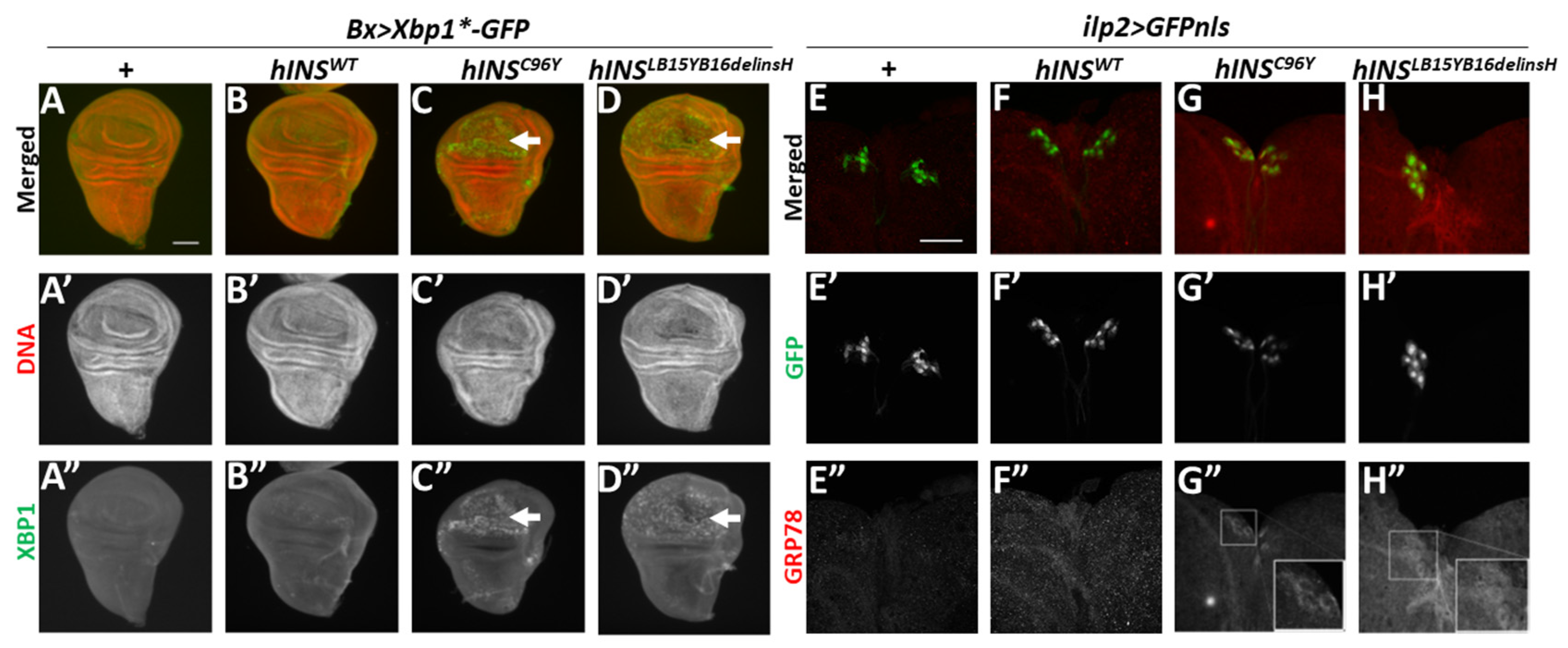
2.1. Ectopic Expression of Human Mutant Preproinsulins Induced the Expression of Two Types of ER Stress Marker in Drosophila Tissues
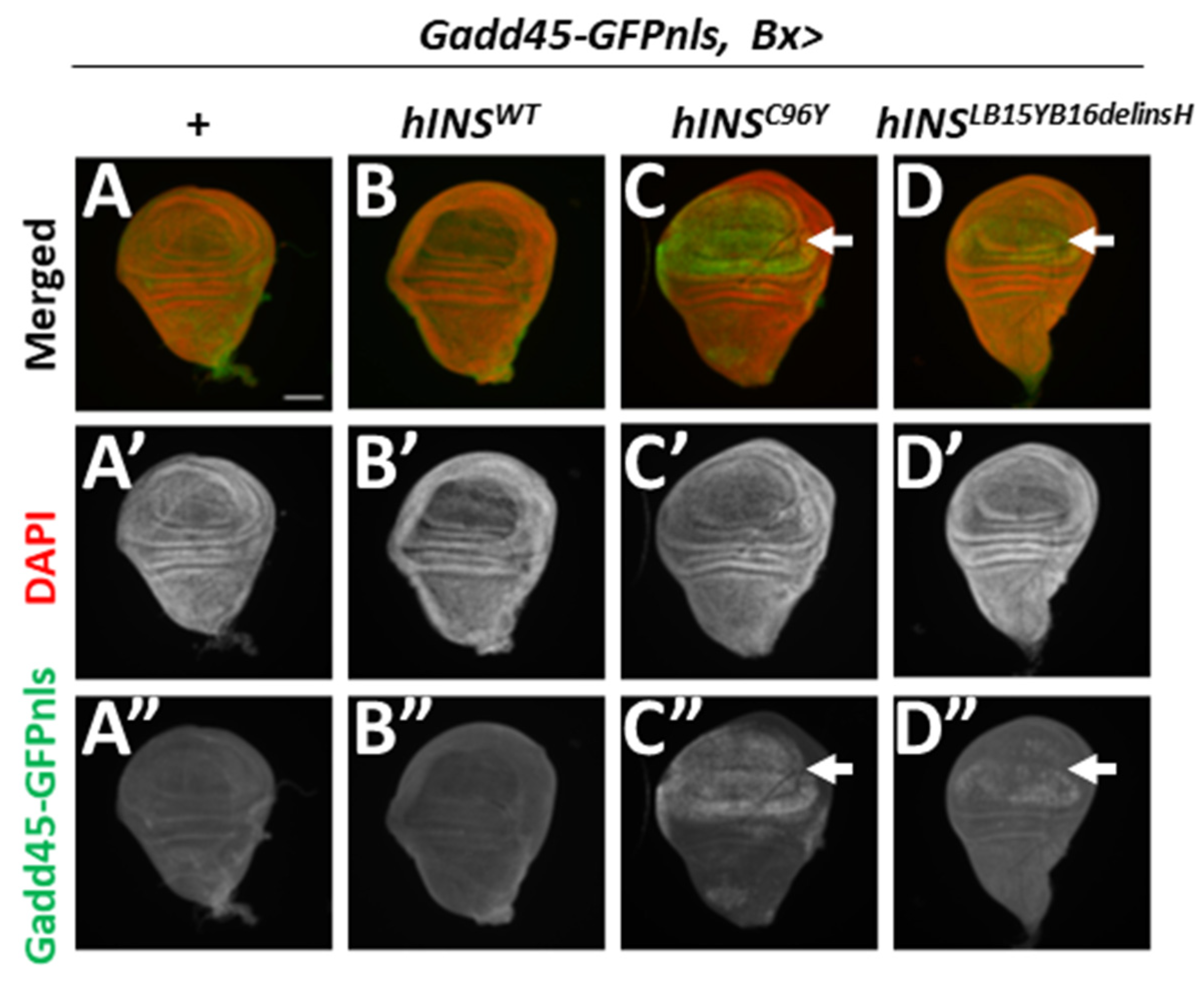
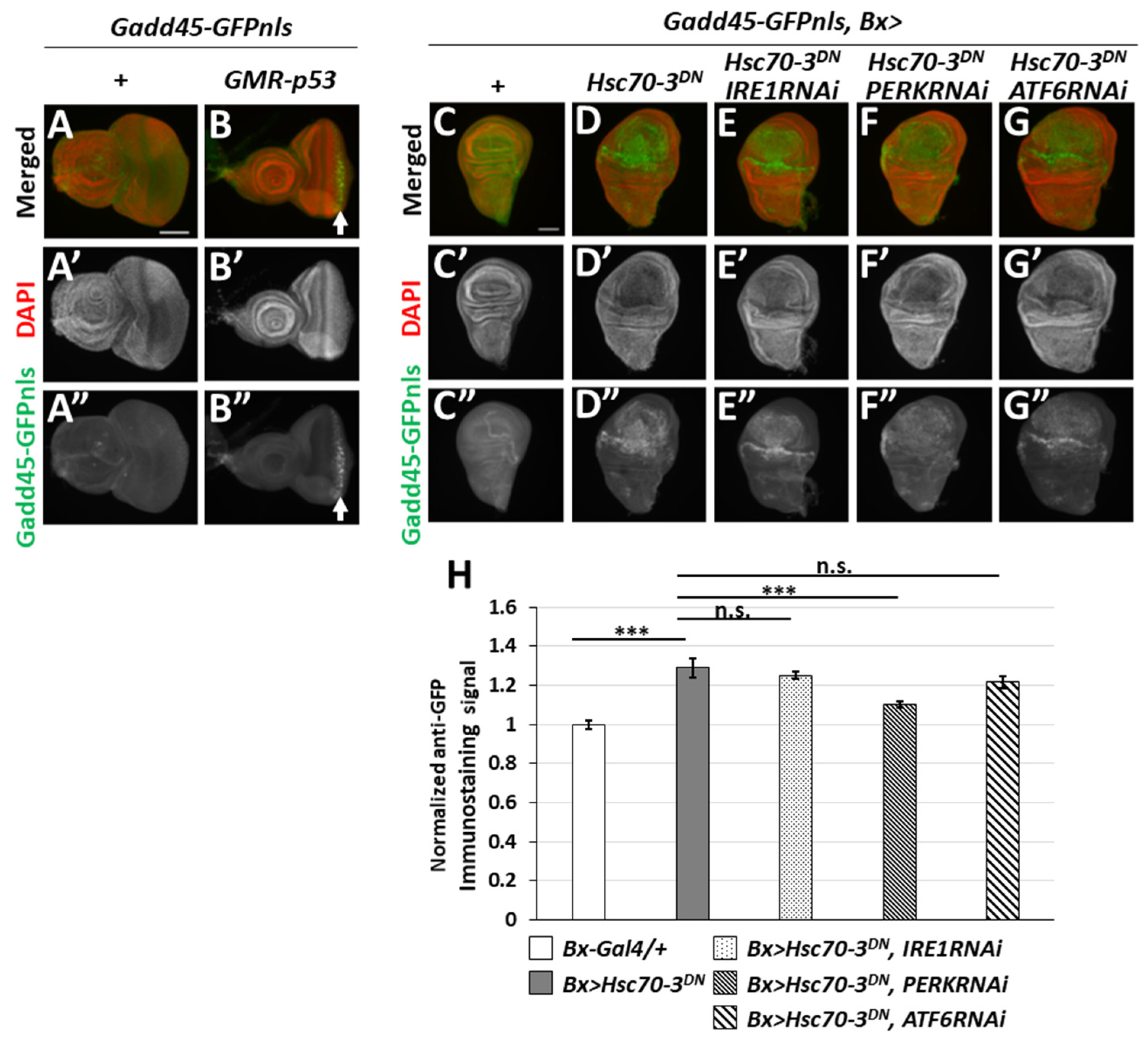
2.2. ER-Stress Induced the Expression of Gadd45 via UPR Pathway Mediated by PERK
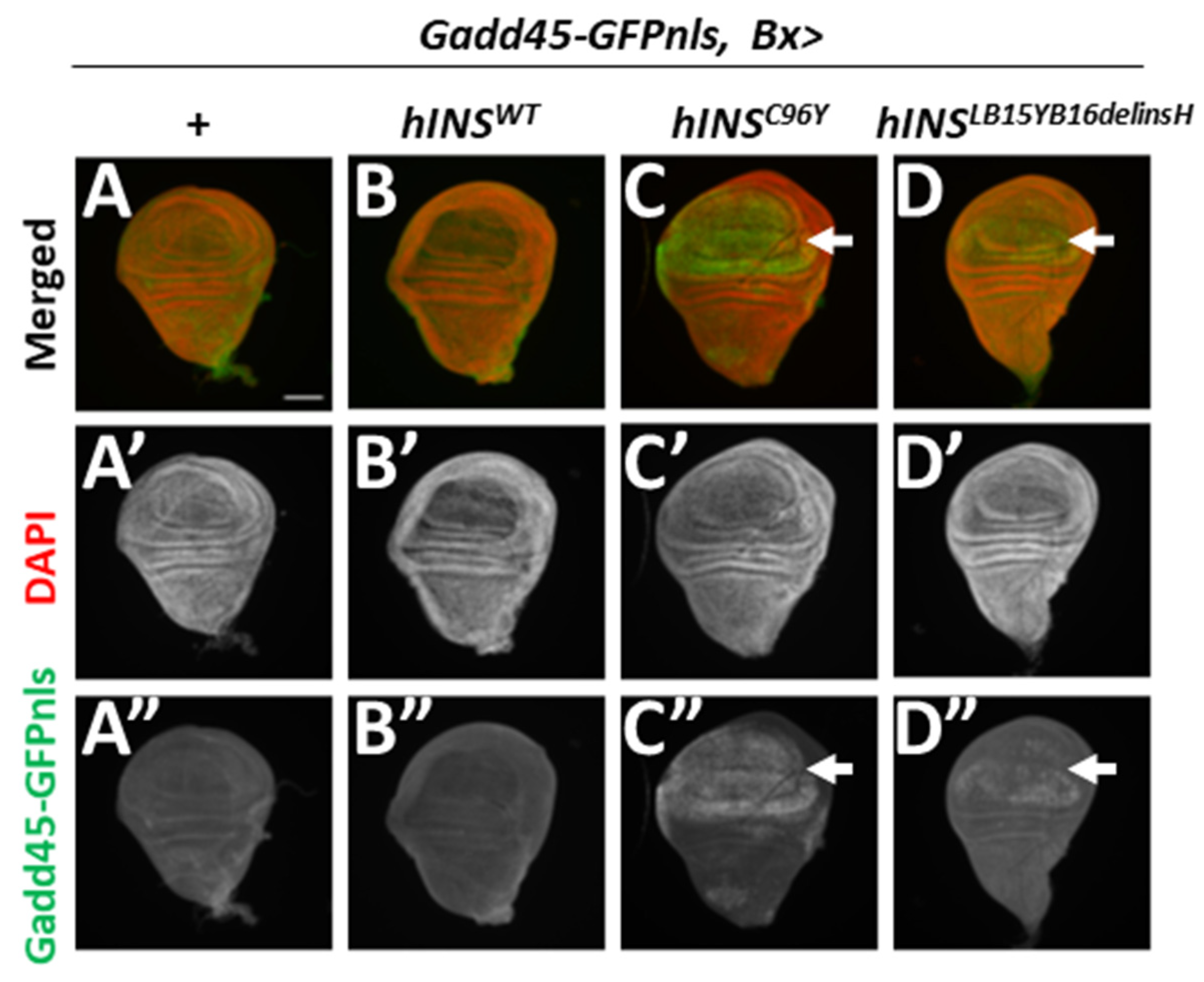
2.3. Human Mutant Preproinsulin-Induced ER Stress Also Induced Gadd45 Expression
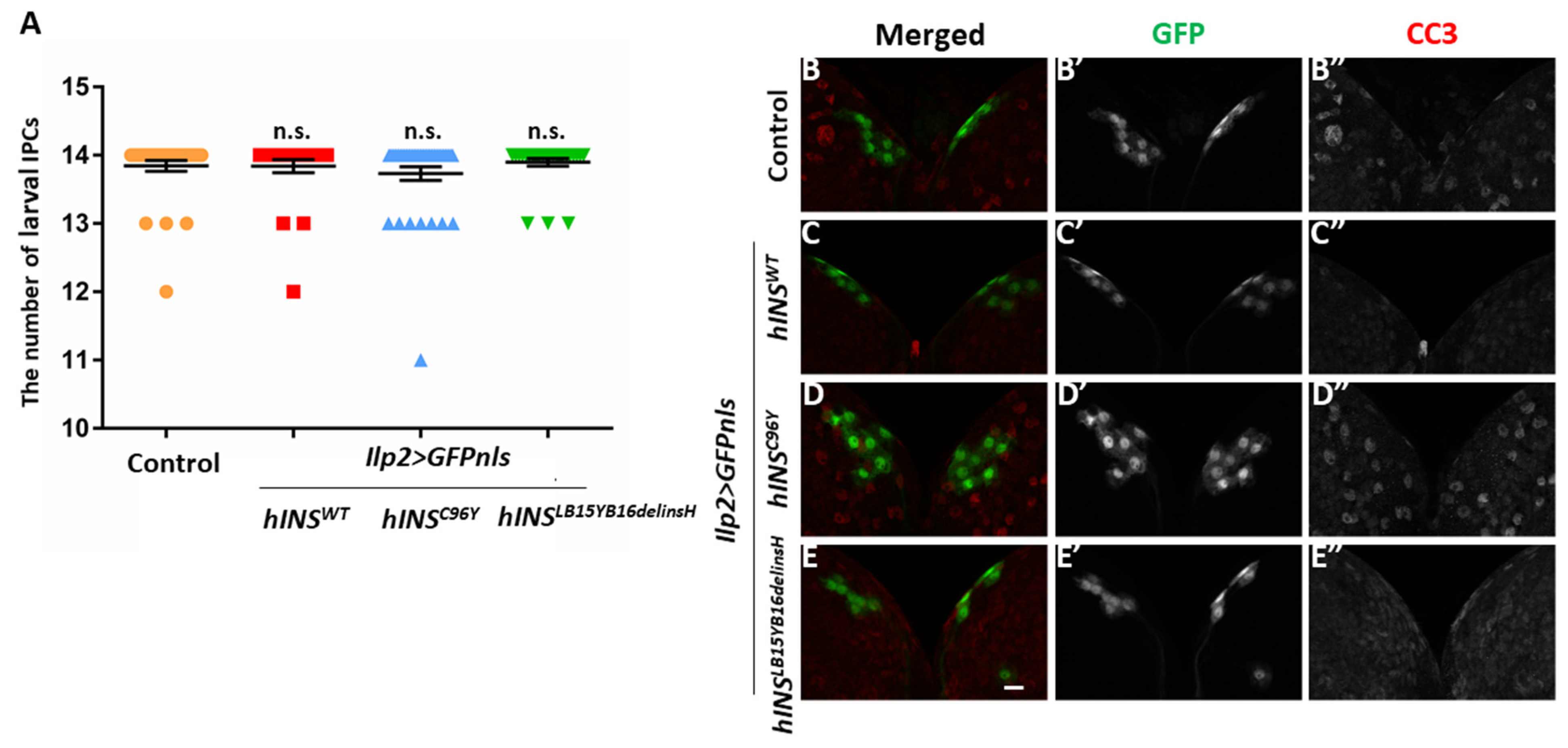
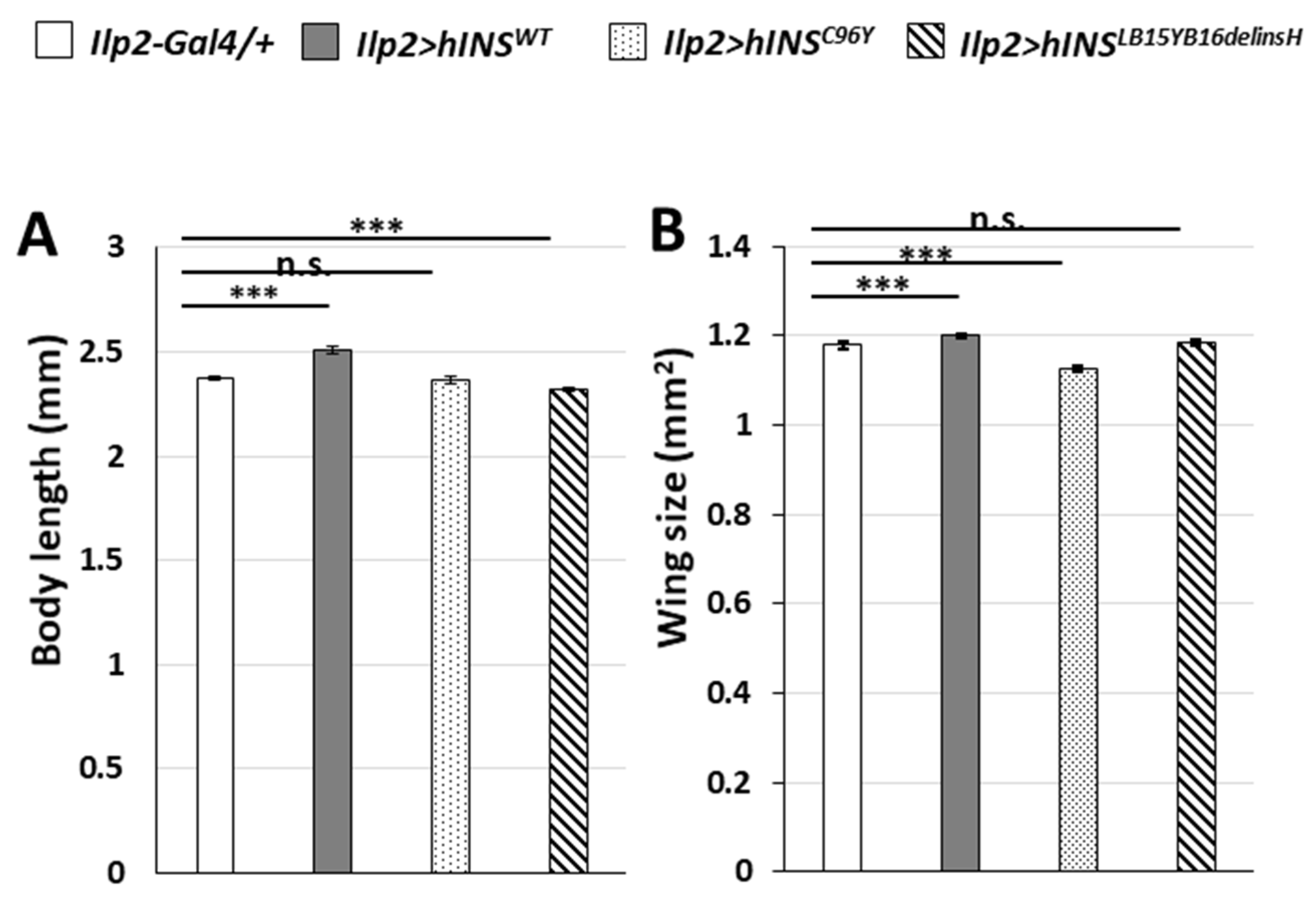
2.4. Induction of ER Stress by a Constitutive Expression of Human Mutant Preproinsulins in IPCs Resulted in Growth Inhibition of Drosophila
2.5. The Ectopic Expression of Human Mutant Preproinsulins Failed to Induce Apoptosis in IPCs
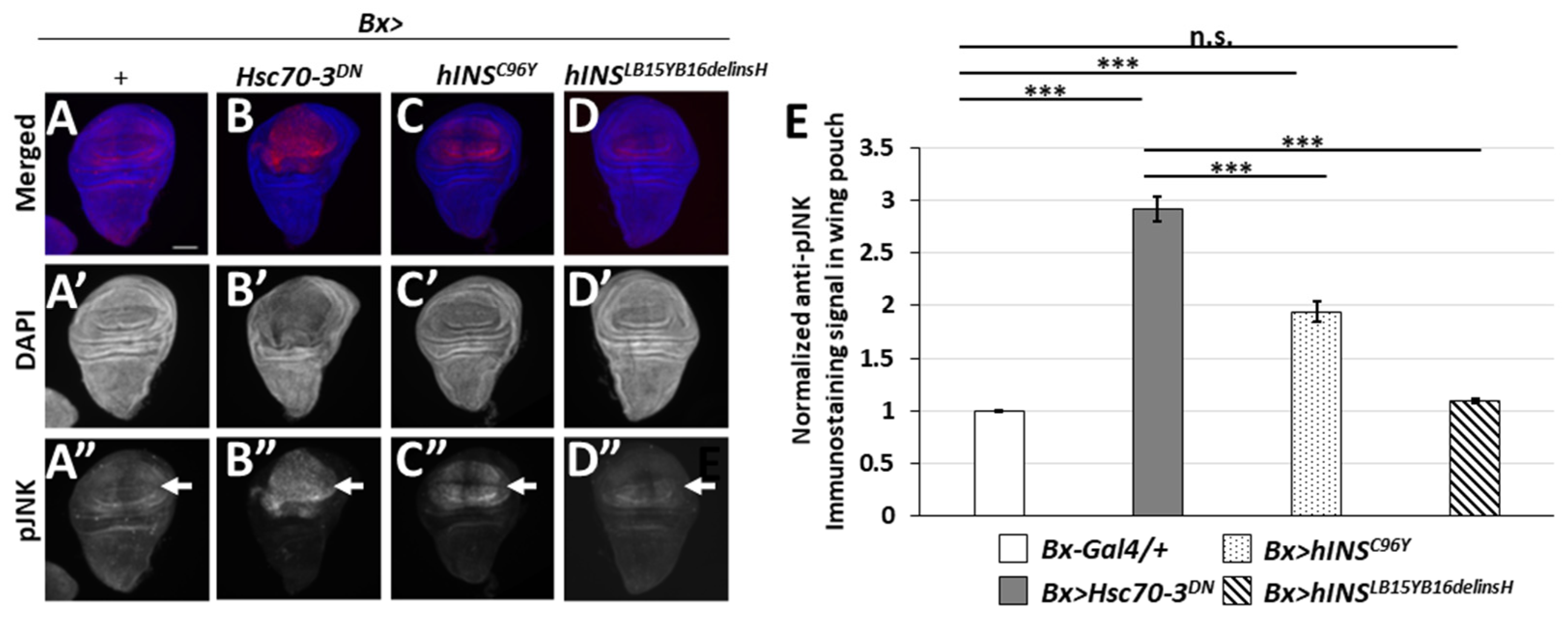
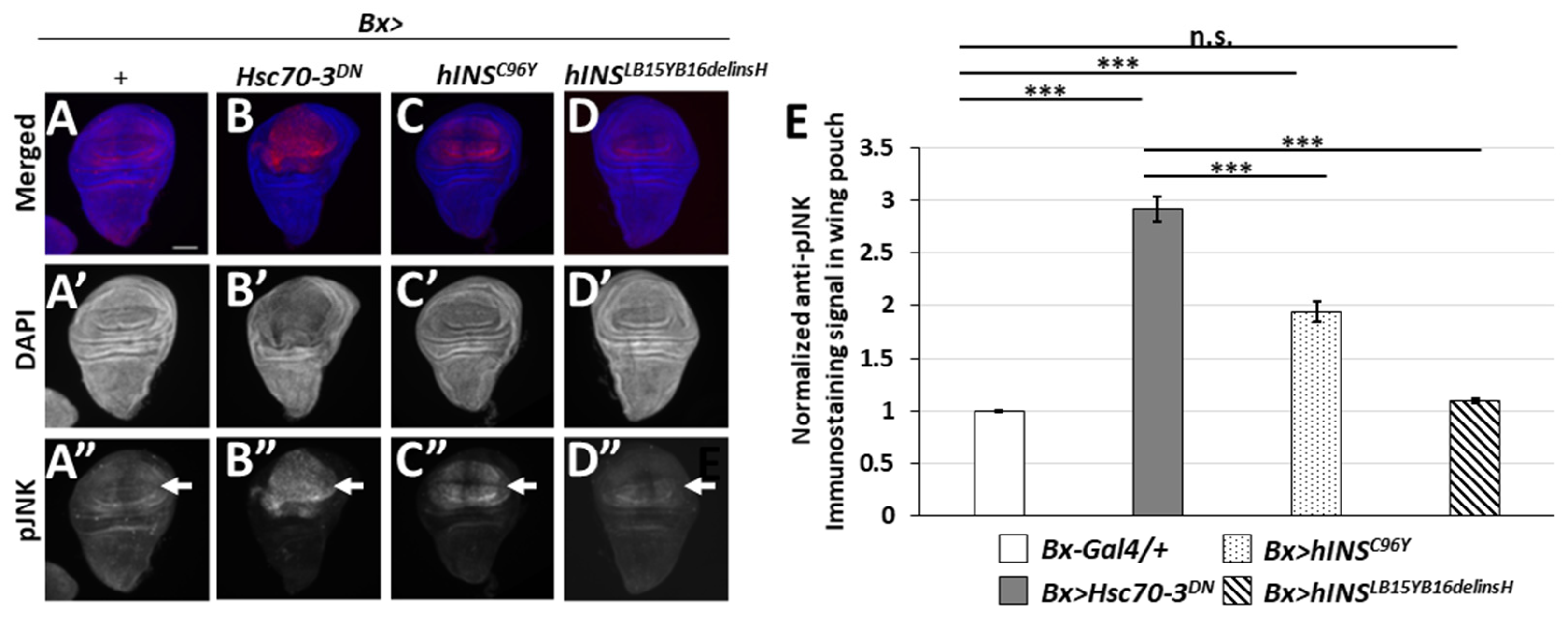
2.6. The Ectopic Expression of the Human Mutant Preproinsulins Activated the JNK Pathway Less Efficiently Than That of the Stronger ER Stress Inducer, Hsc70-3DN
2.7. ER Stress Accumulated in the Wing Disc Cells Resulted in the Activation of the JAK/STAT Pathway
3. Discussion
3.1. Expression of Human Mutant Preproinsulin in IPCs Gives Rise to the Drosophila Growth Inhibition Reminiscent of Undernutrition in Diabetes
3.2. Gadd45 Transcription Is Induced in Response to ER Stress in Drosophila Tissues
3.3. Gadd45-GFP Is a Useful Reporter for the Detection of ER Stress Accumulation and UPR Induction
3.4. Accumulation of ER Stress by hINSC96Y Expression Triggers Activation of JNK and JAK-STAT Signaling Pathways
4. Materials and Methods
4.1. Drosophila Stocks and Husbandry
4.2. GFP Reporter Assay
4.3. Immunostaining Procedures
4.4. Counting of IPCs
4.5. Measurement of Adult Body Length and Wing Area
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collares, C.V.; Evangelista, A.F.; Xavier, D.J.; Rassi, D.M.; Arns, T.; Foss-Freitas, M.C.; Foss, M.C.; Puthier, D.; Sakamoto-Hojo, E.T.; A Passos, G.; et al. Identifying common and specific microRNAs expressed in peripheral blood mononuclear cell of type 1, type 2, and gestational diabetes mellitus patients. BMC Res. Notes 2013, 6, 491. [Google Scholar] [CrossRef] [Green Version]
- Cerf, M.E. Beta Cell Dysfunction and Insulin Resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, C.; Porzio, O.; Liu, M.; Massa, O.; Vasta, M.; Salardi, S.; Beccaria, L.; Monciotti, C.; Toni, S.; Pedersen, O.; et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Investig. 2008, 118, 2148–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Støy, J.; Edghill, E.L.; Flanagan, S.E.; Ye, H.; Paz, V.P.; Pluzhnikov, A.; Below, J.E.; Hayes, M.G.; Cox, N.J.; Lipkind, G.M.; et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 15040–15044. [Google Scholar] [CrossRef] [Green Version]
- Støy, J.; Steiner, D.F.; Park, S.-Y.; Ye, H.; Philipson, L.H.; Bell, G.I. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev. Endocr. Metab. Disord. 2010, 11, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Mari, A.; Nofrate, V.; Sosenko, J.M.; Skyler, J.S.; Group, for the D.-1 S. Progression to diabetes in relatives of type 1 diabetic patients: Mechanisms and Mode of Onset. Diabetes 2010, 59, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Ize-Ludlow, D.; Lightfoot, Y.L.; Parker, M.; Xue, S.; Wasserfall, C.; Haller, M.J.; Schatz, D.; Becker, D.J.; Atkinson, M.A.; Mathews, C.E. Progressive erosion of β-cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes 2011, 60, 2086–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskinen, P.; Korhonen, S.; Kupila, A.; Veijola, R.; Erkkilä, S.; Savolainen, H.; Arvilommi, P.; Simell, T.; Ilonen, J.; Knip, M.; et al. First-phase insulin response in young healthy children at genetic and immunological risk for Type I diabetes. Diabetologia 2002, 45, 1639–1648. [Google Scholar] [PubMed] [Green Version]
- Sreenan, S.; Pick, A.J.; Levisetti, M.; Baldwin, A.C.; Pugh, W.; Polonsky, K.S. Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes 1999, 48, 989–996. [Google Scholar] [CrossRef]
- Lombardi, A.; Tomer, Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J. Autoimmun. 2017, 80, 48–55. [Google Scholar] [CrossRef]
- O’Sullivan-Murphy, B.; Urano, F. ER stress as a trigger for β-cell dysfunction and autoimmunity in type 1 diabetes. Diabetes 2012, 61, 780–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Moskalev, A.A.; Smit-McBride, Z.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Tacutu, R.; Fraifeld, V.E. Gadd45 proteins: Relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 2012, 11, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.-M.; Yun, M.; Lee, S.-G.; Han, J. ER stress induces cell cycle arrest at the G2/M phase through eIF2α phosphorylation and GADD45α. Int. J. Mol. Sci. 2019, 20, 6309. [Google Scholar] [CrossRef] [Green Version]
- Boot-Handford, R.P.; Briggs, M.D. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010, 339, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.H.; Katsube, H.; Hinami, Y. Drosophila models to investigate Insulin action and mechanisms underlying human diabetes mellitus. In Drosophila Models for Human Diseases; Yamaguchi, M., Ed.; Springer: Singapore, 2018; pp. 235–256. [Google Scholar]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Teleman, A.A. Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 2009, 425, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Broughton, S.J.; Piper, M.D.W.; Ikeya, T.; Bass, T.M.; Jacobson, J.; Driege, Y.; Martinez, P.; Hafen, E.; Withers, D.J.; Leevers, S.J.; et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA 2005, 102, 3105–3110. [Google Scholar] [CrossRef] [Green Version]
- Colombani, J.; Andersen, D.S.; Léopold, P. Secreted Peptide Dilp8 Coordinates Drosophila Tissue Growth with Developmental Timing. Science 2012, 336, 582–585. [Google Scholar] [CrossRef]
- Garelli, A.; Gontijo, A.M.; Miguela, V.; Caparros, E.; Dominguez, M. Imaginal Discs Secrete Insulin-Like Peptide 8 to Mediate Plasticity of Growth and Maturation. Science 2012, 336, 579–582. [Google Scholar] [CrossRef]
- Ikeya, T.; Galic, M.; Belawat, P.; Nairz, K.; Hafen, E. Nutrient-Dependent Expression of Insulin-like Peptides from Neuroendocrine Cells in the CNS Contributes to Growth Regulation in Drosophila. Curr. Biol. 2002, 12, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Rulifson, E.J.; Kim, S.K.; Nusse, R. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 2002, 296, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Katsube, H.; Hinami, Y.; Yamazoe, T.; Inoue, Y.H. Endoplasmic reticulum stress-induced cellular dysfunction and cell death in insulin-producing cells results in diabetes-like phenotypes in Drosophila. Biol. Open 2019, 8, bio046524. [Google Scholar] [CrossRef] [Green Version]
- Ham, H.; Woolery, A.R.; Tracy, C.; Stenesen, D.; Krämer, H.; Orth, K. Unfolded protein response-regulated Drosophila fic (dFic) protein reversibly AMPylates BiP chaperone during endoplasmic reticulum homeostasis. J. Biol. Chem. 2014, 289, 36059–36069. [Google Scholar] [CrossRef] [Green Version]
- Carrier, F.; Georgel, P.T.; Pourquier, P.; Blake, M.; Kontny, H.U.; Antinore, M.J.; Gariboldi, M.; Myers, T.G.; Weinstein, J.N.; Pommier, Y.; et al. Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol. Cell Biol. 1999, 19, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Papa, F.R. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, A007666. [Google Scholar] [CrossRef] [Green Version]
- Willsey, H.R.; Zheng, X.; Carlos Pastor-Pareja, J.; Willsey, A.J.; Beachy, P.A.; Xu, T. Localized JNK signaling regulates organ size during development. eLife 2016, 5, e11491. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Luo, K.L.; Shi, L. Endoplasmic Reticulum Stress Interacts with Inflammation in Human Diseases. J. Cell. Physiol. 2016, 231, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.; Ekas, L.A.; Ayala-Camargo, A.; Flaherty, M.S.; Lee, H.; Perrimon, N.; Baeg, G.-H. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 2007, 7, 323–331. [Google Scholar] [CrossRef]
- Johnstone, K.; Wells, R.E.; Strutt, D.; Zeidler, M.P. Localised JAK/STAT pathway activation is required for Drosophila wing hinge development. PLoS ONE 2013, 8, e65076. [Google Scholar] [CrossRef]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Domingos, P.M.; Kang, M.-J.; Steller, H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007, 26, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.-J.; Chung, J.; Ryoo, H.D. CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 2012, 14, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, D.Y.; Wang, Y.Y.; Zheng, Y.T. Activation of c-Jun N-terminal kinases by ribotoxic stresses. Cell. Mol. Immunol. 2005, 2, 419–425. [Google Scholar] [PubMed]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med Biol. 2013, 793, 1–19. [Google Scholar]
- Schäfer, A. Gadd45 proteins: Key players of repair-mediated DNA demethylation. Adv. Exp. Med. Biolol. 2013, 793, 35–50. [Google Scholar]
- Saha, A.; Kuzuhara, T.; Echigo, N.; Fujii, A.; Suganuma, M.; Fujiki, H. Apoptosis of human lung cancer cells by curcumin mediated through up-regulation of Growth arrest and DNA damage inducible genes 45 and 153. Biol. Pharm. Bull. 2010, 33, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takekawa, M.; Saito, H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 1998, 95, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Gupta, S.K.; Hoffman, B.; Liebermann, D.A. Gadd45a and Gadd45b Protect Hematopoietic Cells from UV-induced Apoptosis via Distinct Signaling Pathways, including p38 Activation and JNK Inhibition. J. Biol. Chem. 2006, 281, 17552–17558. [Google Scholar] [CrossRef] [Green Version]
- Ammendrup, A.; Maillard, A.; Nielsen, K.; Andersen, N.A.; Serup, P.; Madsen, O.D.; Mandrup-Poulsen, T.; Bonny, C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000, 49, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Døssing, M.G.; Papa, S.; Franzoso, G.; Billestrup, N.; Mandrup-Poulsen, T. Growth arrest- and DNA-damage-inducible 45β gene inhibits c-Jun N-terminal kinase and extracellular signal-regulated kinase and decreases IL-1β-induced apoptosis in insulin-producing INS-1E cells. Diabetologia 2006, 49, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bgatova, N.; Dubatolova, T.; Omelyanchuk, L.; Plyusnina, E.; Shaposhnikov, M.; Moskalev, A. Gadd45 expression correlates with age dependent neurodegeneration in Drosophila melanogaster. Biogerontology 2014, 16, 53–61. [Google Scholar] [CrossRef]
- Plyusnina, E.N.; Shaposhnikov, M.V.; Moskalev, A.A. Increase of Drosophila melanogaster lifespan due to D-GADD45 overexpression in the nervous system. Biogerontology 2011, 12, 211–226. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Xie, J.; Zhang, D.; Han, Y.; Wang, C. Polypeptide from Chlamys farreri suppresses ultraviolet-B irradiation-induced apoptosis through restoring ER redox homeostasis, scavenging ROS generation, and suppressing the PERK-eIF2a-CHOP pathway in HaCaT cells. J. Photochem. Photobiol. B 2015, 151, 10–16. [Google Scholar] [CrossRef]
- Katsube, H.; Yamazoe, T.; Inoue, Y.H. Association of oxidative stress with ER stress are involved in a loss of IPCs and their dysfunction in Drosophila diabetes model. Dis. Model Mech. 2021. submitted for publication. [Google Scholar]
- Lin, W.-C.; Chuang, Y.-C.; Chang, Y.-S.; Lai, M.-D.; Teng, Y.-N.; Su, I.-J.; Wang, C.C.C.; Lee, K.-H.; Hung, J.-H. Endoplasmic reticulum stress stimulates p53 expression through NF-κB activation. PLoS ONE 2012, 7, e39120. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D. JAK–STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [Green Version]
- Ahmed-De-Prado, S.; Diaz-Garcia, S.; Baonza, A. JNK and JAK/STAT signalling are required for inducing loss of cell fate specification during imaginal wing discs regeneration in Drosophila melanogaster. Dev. Biol. 2018, 441, 31–41. [Google Scholar] [CrossRef]
- Bosch, M.; Serras, F.; Martín-Blanco, E.; Baguñà, J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev. Biol. 2005, 280, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Katsuyama, T.; Comoglio, F.; Seimiya, M.; Cabuy, E.; Paro, R. During Drosophila disc regeneration, JAK/STAT coordinates cell proliferation with Dilp8-mediated developmental delay. Proc. Natl. Acad. Sci. USA 2015, 112, E2327–E2336. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Hirai, J.; Yasukawa, T.; Nakahara, Y.; Inoue, Y.H. A correlation of reactive oxygen species accumulation by depletion of superoxide dismutases with age-dependent impairment in the nervous system and muscles of Drosophila adults. Biogerontology 2015, 16, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, A.J.; Brook, W.J.; Cohen, S.M.; Bell, J.B. Distinguishable functions for engrailed and invected in anterior–posterior patterning in the Drosophila wing. Nature 1995, 376, 424–427. [Google Scholar] [CrossRef]
- Nagy, P.; Varga, A.; Pircs, K.; Hegedűs, K.; Juhász, G. Myc-driven overgrowth requires unfolded protein response-mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003664. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Barolo, S.; Carver, L.A.; Posakony, J.W. GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. BioTechniques 2000, 29, 726–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamazoe, T.; Nakahara, Y.; Katsube, H.; Inoue, Y.H. Expression of Human Mutant Preproinsulins Induced Unfolded Protein Response, Gadd45 Expression, JAK-STAT Activation, and Growth Inhibition in Drosophila. Int. J. Mol. Sci. 2021, 22, 12038. https://doi.org/10.3390/ijms222112038
Yamazoe T, Nakahara Y, Katsube H, Inoue YH. Expression of Human Mutant Preproinsulins Induced Unfolded Protein Response, Gadd45 Expression, JAK-STAT Activation, and Growth Inhibition in Drosophila. International Journal of Molecular Sciences. 2021; 22(21):12038. https://doi.org/10.3390/ijms222112038
Chicago/Turabian StyleYamazoe, Tatsuki, Yasuyuki Nakahara, Hiroka Katsube, and Yoshihiro H. Inoue. 2021. "Expression of Human Mutant Preproinsulins Induced Unfolded Protein Response, Gadd45 Expression, JAK-STAT Activation, and Growth Inhibition in Drosophila" International Journal of Molecular Sciences 22, no. 21: 12038. https://doi.org/10.3390/ijms222112038
APA StyleYamazoe, T., Nakahara, Y., Katsube, H., & Inoue, Y. H. (2021). Expression of Human Mutant Preproinsulins Induced Unfolded Protein Response, Gadd45 Expression, JAK-STAT Activation, and Growth Inhibition in Drosophila. International Journal of Molecular Sciences, 22(21), 12038. https://doi.org/10.3390/ijms222112038