Abstract
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy worldwide and is responsible as one of the main causes of mortality in both men and women. Despite massive efforts to raise public awareness on early screening and significant advancements in the treatment for CRC, the majority of cases are still being diagnosed at the advanced stage. This contributes to low survivability due to this cancer. CRC patients present various genetic changes and epigenetic modifications. The most common genetic alterations associated with CRC are p53 and KRAS mutations. Gene therapy targeting defect genes such as TP53 (tumor suppressor gene encodes for p53) and KRAS (oncogene) in CRC potentially serves as an alternative treatment avenue for the disease in addition to the standard therapy. For the last decade, significant developments have been seen in gene therapy for translational purposes in treating various cancers. This includes the development of vectors as delivery vehicles. Despite the optimism revolving around targeted gene therapy for cancer treatment, it also has various limitations, such as a lack of availability of related technology, high cost of the involved procedures, and ethical issues. This article will provide a review on the potentials and challenges of gene therapy targeting p53 and KRAS for the treatment of CRC.
1. Introduction
Cancer is one of the major health issues affecting men and women globally. It is estimated that 19.3 million new cancer cases and nearly 10.0 million cancer deaths occurred in 2020. Colorectal cancer (CRC) was recorded as the third most commonly diagnosed malignancy worldwide, following breast and lung cancers. CRC qualified as the second leading cause of cancer death after lung cancer. Developed countries possess the highest risk of colon and rectal cancer [1].
Surgical resection and conventional therapy such as chemotherapy have long since been considered as the ‘gold standard’ management for CRC [2]. Despite many initiatives to increase public awareness for early screening of CRC and advancements in cancer therapy, the majority of cases are diagnosed at the advanced stage, which contribute to the poor prognosis of this cancer [3,4].
Cancer arises from the accumulation of somatic mutations and other genetic changes which affect cell division checkpoints and result in both abnormal cell growth and eventually tumorigenesis. The process of carcinogenesis occurs, in which abnormal cells acquire advantageous biological capabilities known as the hallmarks of cancer [5]. Similar to other types of cancer, genetic and epigenetic changes are implicated in the progression of CRC. Mutations of TP53 (tumor suppressor) and KRAS (oncogene) are among the most commonly reported genetic alterations in CRC [6].
Conventional treatments such as chemotherapy impose serious adverse side effects on patients [7]. It is imperative that safer and more effective options be included as part of cancer management. One of the proposed therapies for CRC is targeted gene therapy. This approach aims to improve the life expectancy of the patients and reduce the risk of disease recurrence. Advancement in the field of gene therapy offers promise to various innovative treatments that are likely to become prominent in improving the survivability of cancer patients. Gene transfer is one of the treatment modalities that introduces new genes into a malignant cell or into the surrounding tissue to induce cell death or slow cancer proliferation. Flexibility of this technique and the ability to utilize a wide range of genes and vectors have solidified its successful outcomes in many clinical trials. As these gene therapy technologies in oncology mature, they may be used independently or in combination with current or conventional treatments for cancer management [8,9].
2. Colorectal Carcinogenesis
CRC is a cancer that originated from the colon and rectum. It starts with transformation from a normal colonic crypt to benign intestinal polyps. A small number of these (adenomatous polyps) will progress into advanced adenoma and become cancerous growths (malignant neoplasia) and metastases. The majority of polyps such as inflammatory and hyperplastic polyps are benign and not categorized as pre-malignant lesions [10]. Most CRCs are adenocarcinomas. The gradual transition occurs in CRC development, also referred to as ‘multistep tumorigenesis’, and specific genetic changes in tumor suppressors or oncogenes are associated with every stage [11,12,13].
CRC can be classified into four groups based on their consensus molecular subtypes (CMSs), namely [14];
- (a)
- CMS1: microsatellite instability (MSI) and strong activation of immune components
- (b)
- CMS2: exhibits epithelial differentiation and upregulation of the wingless-related integration site (WNT) and MYC signaling;
- (c)
- CMS3: prominent metabolic dysregulation; and
- (d)
- CMS4: evident expression of genes compatible with stromal invasion, epithelial–mesenchymal transition (EMT), transforming growth factor-beta (TGF-β), and angiogenesis.
A small fraction of mutations, referred to as ‘drivers’, are responsible for the cellular transformations that result in cancer formation. Driver mutations characterize molecular phenotypes of tumors and act as predictors in clinical outcomes for patients [15]. Cells harboring driver mutations acquire selective advantage, leading to their proliferation, and these mutations have been positively selected during cancer evolution. They are considered as a part of the ‘cancer genes’. Other than driver mutations, our cells also accumulate other mutations referred to as ‘passenger’ mutations, which may not seemingly affect the cancer progression. Despite this, passenger mutations attract more interest as it can impact epigenetics, immunogenicity, or the response to therapy for cancer patients [16]. Three key players in promoting CRC progression are tumor suppressor genes, oncogenes, and mismatch-repair genes. Mutations in those genes may occur during the gestational period or may be acquired over the lifetime (inherited versus somatic mutation). Although many scientists have focused more on the roles of somatic mutation rather than germline mutation in the predisposition to cancer, it is undeniable that the hereditary aspect of mutations (germline mutation) possesses an essential role in cancer development and metastasis, which presents clinical applications in cancer studies. CRC represents one of the largest percentages of familial cases [17,18]. It is also worthy to note that the majority of CRC cases (70%) arise from sporadic mutation and are significantly influenced by external risk factors such as lifestyle, lack of physical activity, and unhealthy diet [4,17].
It is an established notion that CRC arises from accumulations of genetic and epigenetic changes. These modifications promote the formation of carcinoma from normal mucosa by altering the signaling pathways involved in the regulation of cancer behaviors [19,20]. Six key driver genes in CRC, namely adenomatous polyposis coli (APC), KRAS, BRAF, PIK3CA, SMAD4, and TP53, were identified by using bioinformatics tools [21]. Figure 1 illustrates the colorectal progression and respective mutations that are involved [10,22,23,24].
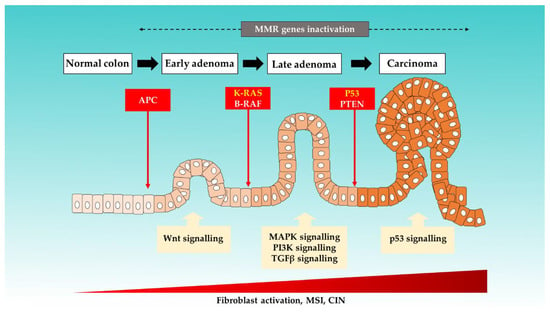
Figure 1.
The proposed mechanisms of sporadic events for CRC progression from the normal colon.
Three major molecular pathways have been identified in CRC carcinogenesis: chromosome instability (CIN), as well as the MSI and CpG island methylation pathways (CIMP) [25]. In up to 80% of sporadic CRC cases, the tumor arises from the CIN pathway and progresses through the classic adenoma–carcinoma sequence. This pathway is triggered by multistep mutational events involving multiple genes, such as KRAS oncogene, and tumor suppressor genes, such as APC and TP53. APC is found to be mutated in the majority (80–90%) of inherited and sporadic CRC. APC mutations disrupt the APC complex formation, thus resulting in the b-vatenin translocation to the nucleus. This subsequently induces overactivation of Wnt-signalling and drives both cell proliferation and activation [12,26,27].
CRC formation also involves a dynamic interaction between malignant cells, components of the microenvironment (stromal and vascular endothelial cells), and the immune system [28,29,30]. Increased activation of fibroblasts occurs alongside CRC progression. Accumulation of cancer-associated fibroblasts (CAFs), which is a major component of the tumor microenvironment, contributes to poor prognosis and recurrence of CRC [31]. These stromal cells support cancer cell growth and differentiation via secretion of cytokine, and vice-versa [32]. It is worth noting that variation in the genetic makeup also (i.e., polymorphism) explains the differences in responsiveness to infection and regarding the risk of developing CRC between different populations or races [33,34].
3. The p53 and KRAS Mutations in CRC
Cancer profiling can be performed by analyzing the genetic alterations or changes that contribute to its carcinogenesis. Table 1 summarizes main genetic mutations detected in several of the most commonly diagnosed solid tumors [35,36,37,38,39]. The listed genes serve as possible targets for designing targeted therapy against different cancer types. We note that p53 and KRAS mutations are not only reported to be prevalent in CRC but also in many other cancers, such as malignancy of the breast, lung, and pancreas.

Table 1.
Common genetic mutations in solid tumors.
Although Table 1 lists the common mutations involved in solid tumors, the frequency and mutational profiling of cancer patients may differ greatly depending on their genetic makeup and tumor heterogeneity. Taking TP53 as an example, mutation sites of this gene are diverse between cancer subjects. Thus, it is important to analyze different tumors thoroughly in order to cater to more personalized therapy based on patients’ mutational profiles.
The Cancer Genome Atlas Network reported the most frequently mutated genes in the CIN pathway, which include APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, TCF7L2 and NRAS [40]. One of these genes, namely TP53, is the second most frequent gene to be mutated in colorectal cancer, affecting approximately 60% of CRC patients. The TP53 gene, located at the short arm of chromosome 17, encodes the important tumor suppressor protein p53. p53 acts as a transcription factor that regulates cell cycles and apoptosis by binding to specific sequences at various genes including p21, Bax, and Bcl-2 in response to cellular stressors such as DNA damage or hypoxia [41]. Activated p53 via different target molecules can (a) induce temporary cell cycle arrest to allow for repair of DNA damage; (b) induce apoptosis; and (c) abolish further cell division [42]. Loss of p53 transcriptional activity therefore results in unregulated cell growth and promoted tumorigenesis in various organs including the colon. In the majority of cases, the TP53 mutations in CRC cancer cells are missense-type mutations in the DNA-binding domain [41]. This mutation produces mutant p53 with a reduced capacity to bind to the specific sequence that regulates the p53 transcriptional pathway. In approximately 70% of CRC tumors that carry the missense mutations, the wild type TP53 is lost via loss of heterozygosity (LOH) due to chromosomal instability [25]. It is proposed that the missense-type mutations of p53 drive the progression of adenoma to adenocarcinoma and the additional loss of wild type TP53 results in a more advanced progression of CRC [41].
Among the genes of interest stated by the Cancer Genome Atlas Network (2012) [40], KRAS gene mutation testing is the most useful assessment, routinely used currently to predict response to anti-EGFR therapy. KRAS (Kirsten ras sarcoma viral oncogene) is a guanosine triphosphate (GTP)-binding protein located downstream from EGFR in the KRAS-BRAF-MEK-ERK pathway, which is one of the most important mitogen-activated protein kinase (MAPK)-signaling pathways. The ERK-MAPK pathway is important in cell proliferation and differentiation. Mutations in KRAS result in EGFR-independent activation of the MAPK pathway by reducing the GTPase activity that leads to an increase of cell proliferation potentially occurring in the early stage of colorectal carcinogenesis [43]. It is also proposed that the ERK-MAPK pathway is involved in the progression and oncogenic behavior of human CRC.
Various studies have demonstrated that mutations in the KRAS are implicated in approximately 30–40% of CRC patients [44,45,46]. The proportion of KRAS mutations differed between regions and races. For example, in Malaysia, the prevalence is lower (approximately 20%), as previously reported [47,48,49]. More than 90% of the activating mutations occurred at codon 12 and codon 13 in exon 1, with single-base substitution of glycine to aspartate (G12D) and glycine to valine (G12V) on codon 12 being the most common mutations observed [45]. Mutations at codons 61 and 146 have also been recorded, although these account for only about 1–4% of KRAS mutations and its clinical significance remain unclear [50].
Associations between the KRAS mutational status and clinicopathological features of CRC have been studied but there is no consensus obtained regarding its relation. Hayama et al. (2019) and Scott et al. (2020) [42,46], for instance, demonstrated that KRAS-mutated CRC were more frequently observed at the right side of the colon, while Zulhabri et al. (2012) [48] reported that these mutations are more common on left-sided CRC. Other studies including the collaborative RASCAL study [51] and study by Phipps et al. (2013) [52] showed no association between KRAS mutations and tumor site. Zulhabri et al. (2012) also explored the relationship between different ethnicities and KRAS mutational profiles but found no significant association between the variables [48]. The missense-type p53 mutation expression or suppression of TGF-β induces submucosal invasion. Additional KRAS gene mutation promotes more malignant transformation (epithelial–mesenchymal transition and metastasis) [53].
Molecular characterization of cancer-associated mutations over the more recent years has been proven to provide essential information on disease prognosis and response to treatment such as chemotherapy. Two previously unreported prognostic associations in CRC, namely the TP53 mutation and the total mutation burden, confirmed correlations with the KRAS homolog and B-Raf proto-oncogenes (BRAF) were identified through multigene panels. This finding has provided insight into the importance of information acquired from modest-sized genetic and molecular testing in clinical practice [54]. This is supported by a more recent study, in which it was reported that multiple variables, such as molecular markers (e.g., KRAS mutations), tumor location with the other clinical-pathological variables, and microsatellite status, could be used in combination to determine the prognosis and act as predictive factors in stage I-III CRC [55]. Another study by Tortola et al. (2016) also reported the correlation between TP53 mutations alone or in combination with KRAS mutations, with poor outcomes in CRC patients, although they did not recommend the routine use of these mutations as prognostic markers in clinics [6].
KRAS mutations are associated with more aggressive tumor phenotypes and poor prognosis in CRC patients in comparison to those with wild type-KRAS [56,57]. Mutations in codon 12 and 13 of KRAS, BRAF, and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) also act as a predictor for resistance to anti-EGFR therapies. Thus, it has been proposed that these patients with KRAS or NRAS mutations should not be treated with therapies targeting EGFR. Instead, a BRAF inhibitor should be included in the anti-EGFR therapy for patients with BRAF mutations to overcome treatment resistance [58,59].
4. Gene Therapy for Cancer
Management of CRC involves (a) surgery; (b) conventional chemotherapy; (c) immunotherapy; and d) various gene therapy technologies such as gene correction and virus-directed enzyme–prodrug therapy [10]. Gene therapy represents a promising approach as it allows for a more personalized treatment. This would ensure the success of treatment clinically. In brief, gene therapy can be referred to as the delivery of specific genetic material (genes, gene segments, or oligonucleotides) to change the encoding of a gene product or to alter the biological properties of the host cells for the treatment of various pathological conditions or diseases [60]. Gene transfer therapies are performed either through in vivo or ex vivo methods.
Viral and non-viral delivery vehicles are used to administer drugs or genes of interest depending on the desired specificity of the gene therapy. Targeted gene therapy is preferred over the recombinant therapeutic use of peptides, where the latter is usually characterized by low availability, lack of stability, a significant toxicity effect, and expensive cost [61].
Targeted gene therapy represents a novel treatment avenue to manage a disease with no definitive cure. There has been good progress in gene therapy for cancer in the last three decades, wherein a number of drugs have been approved and many are still at the stage of clinical trials. In the future, tumor genomic profiling coupled with the analysis of host humoral and cellular immunity will ensure the selection of the most appropriate patient and treatment modalities for gene therapy [62].
Gene therapy serves as a treatment avenue that can be used in combination with existing and conventional therapies. Strategies in gene therapy usually involve the use of interventional genetic techniques aiming to a) boost the immunological response to the tumor and b) deliver cytotoxic or lethal agents, and/or genes to neoplastic cells, performed via various clinical trials [63]. These are performed through different mechanisms such as including or replacing defect genes, knocking down genes, deactivating malfunction genes, or inserting new genes, which are administered using delivery vehicles or vectors [64]. Figure 2 summarizes the vectors and strategies in gene therapy for cancer [60,65,66,67,68,69,70]. Several treatment modalities which can potentially be incorporated into gene therapy have also been proposed for cancer management, including [71]:
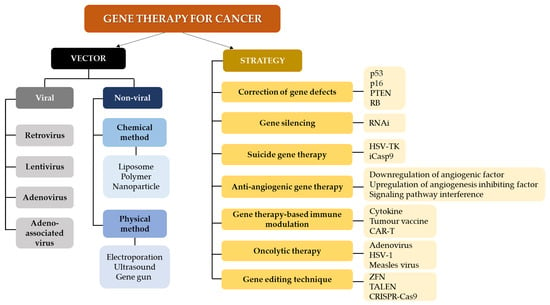
Figure 2.
Gene therapy approaches and vectors for cancer treatment. Methods for gene therapy in cancer include RNA interference (RNAi), herpes simplex virus thymidine kinase (HSV-TK), inducible caspase 9; CAR-T (iCasp9): chimeric antigen receptor T-cell therapy (CAR-T), herpes simplex virus 1 (HSV-1); zinc finger nucleases (ZFN), transcription activator -like effector nucleases (TALEN) and clustered regularly interspaced short palindromic repeats-Cas9 (CRISPR-Cas9).
- (a)
- Gene-directed enzyme prodrug therapy (GDEPT), which aims to reduce the toxicity effect after chemotherapy;
- (b)
- Cancer drug-resistance gene transfer to elevate the chemotherapy and radiation effects onto the tumor cells without compromising the normal tissues; and
- (c)
- Combination of diagnostic and therapeutic systems (theranostic).
As stated in the previous section, CRC progression is also heavily influenced by the tumor microenvironment. Besides targeting cancer cells alone, blocking the angiogenesis process is one the prime methods for CRC treatment. Targeted therapy using AAV to deliver anti-angiogenic genes, namely angiostatin and endostatin, has resulted in tumor shrinkage with little side effects [72].
According to ClinicalTrials.gov, in August 2021, there were 140 clinical trials involving CRC ((accessed on 30 August 2021) https://www.clinicaltrials.gov). This data signifies a massive interest and effort from the medical community to establish gene therapy as a key treatment modality for CRC. Different strategies have been explored in targeted cancer gene therapy for CRC using various vectors and delivery approaches in vitro and in vivo. Table 2 summarizes examples of the viral and non-viral delivery systems used for CRC gene therapy in pre-clinical and clinical stages [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88]. It is worth noting that encouraging data on gene therapy efficacy was gathered from these studies and the majority of the stated treatment strategies produced minimal or no side effects.
Table 2.
Viral and non-viral delivery approaches for CRC gene therapy.
5. Gene Therapy Targeting p53 and KRAS in CRC
The principle of targeting driver mutations for CRC treatment has intrigued the scientific community for ages. These mutations, which play a vital role in carcinogenesis, represent specific targets of a tumor and generally are expressed by the majority, if not all, neoplastic cells [89]. Gene therapy targeting p53 has been explored for various solid tumors. The p53-based gene therapy in oncology commonly aims to administer wild type-p53 or to suppress mutant p53 expression in p53-defective cancer cells [90,91]. Suppression of mutant p53 function is proposed to be a robust approach to block the malignant development of CRC [41,92].
The first-ever gene therapy product to treat head and neck squamous cell carcinoma (HNSCC), namely Gendicine (recombinant human p53 adenovirus), was developed by Shenzhen SiBiono GeneTech Co. Ltd. Gendicine entered the commercial market in 2004 following approval by the China Food and Drug Administration (CFDA) a year earlier. Gendicine functions through adenoviruses carrying the p53 gene. The wild type p53 protein expressed by Gendicine-transduced cells acts as a tumor suppressor, activated by cellular stress, and mediates cell-cycle arrest and DNA repair, or, depending on the cellular stress state, will promote programmed cell death (apoptosis, senescence, and/or autophagy). In short, by restoring p53 activity and thus putting a ‘brake’ to the cell division process, cancer development is subsequently blocked. The commercial success of Gendicine was evidently shown by its safety record, reporting that it has been used in more than 30,000 patients, involving no less than 30 published clinical trials. In combination with conventional treatment, namely chemotherapy and radiotherapy, Gendicine has exhibited significantly better response rates compared to the standard therapies alone, with no significant adverse effects reported [69]. In China, Gendicine has been clinically used to treat malignancies other than HNSCC for more than a decade now mainly for patients with advanced or unresectable cancerous tumors. However, standard treatment regimens utilizing p53 gene therapy still need to be established for advanced use in clinical practice [93]. This successful track record of Gendicine has cemented the idea of targeted treatment in oncology and has provided valuable insight into the potentials of gene therapy for treating malignancies.
Besides Gendicine, targeted gene therapies for cancer include Rexin-G® and VB-111, which are used for the treatment of pancreatic cancer and glioblastoma, respectively. Rexin-G® is the first tumor-targeted gene therapy vector-bearing cytocidal cyclin G1 construct that has been tested in the clinical setting [94,95]. VB-111 (ofranergene obadenovec), in contrast, is a non-replicating adenovirus carrying a Fas-chimera transgene (Fas and human tumor necrosis factor receptor 1) that induces targeted apoptosis of tumor angiogenic blood vessels and promotes a tumor-specific immune response [96,97].
The curative effect of the combination of gene therapy using the p53 gene and standard treatment in treating esophageal cancer has also been explored by Cui et al. (2015) [9]. In this study, middle to advanced stage esophageal cancer patients were treated with a combination of gene therapy, with a recombinant human adenovirus-p53 (rAd-p53) vector, and chemotherapy. Shrinkage of the tumors was reported post-injection and besides self-limiting fever, no side effects were recorded.
A number of cancer and/or tumor-specific promoter systems have been designed to target malignant cells. This potentially highlights the potential of including the promoter for tumor-suppressor genes delivered to cancer cells using a suitable genetic vector [98]. A more recent development in CRC gene therapy is the establishment of nanoparticles (NPs) from mesoporous silica nanoparticles (MSNs) as novel genetic vehicles, which are reported to effectively bind and target CRC cells [99,100].
Compared to gene therapy methods involving p53, the approach of targeting KRAS is not fully developed in oncology practice. However, several studies have reported on the potential of targeted gene therapy against KRAS. In pancreatic cancer, where high prevalence of KRAS mutation was found (95% of the cases), overexpression of the pro-apoptotic protein, which is the p53-upregulated modulator of apoptosis (PUMA) under a Ras responsive promoter, was proved to be effective in selectively killing Ras-transformed cells in vitro. This method was suggested to be a useful, efficient, and safe strategy to eradicate the Ras-mutated tumor cells [101,102].
6. Challenges
6.1. Inaccessible Genetic Testing
CRC screening is usually performed using stool-based tests such as the fecal immunochemical test (FIT) and endoscopic methods (i.e., colonoscopy). Both approaches evidently lower the risk for mortality due to CRC and their benefits outweigh the limitations presented by each screening [103]. It is worthy to note that to cater to more effective targeted therapy in CRC subjects, genetic testing to identify mutational profiles, including MSI status and both p53 and KRAS mutations, is required. In many countries, the cost for mutational screening is considered expensive and may not be accessible to the majority of the population due to a lack of technologies and facilities [104]. This hinders the progress of targeted gene therapy in CRC.
6.2. Variability in Response and Efficacy
A major issue with gene therapy in oncology concerns resistance to treatment with cancer recurrence and low survival rates. Tumor cell resistance following the therapy potentially is a result of an intrinsic mechanism or is acquired through the dysregulation and release of Bcl-2, which inhibits the apoptosis process [72]. This also may relate to the rate of success for gene therapy and selection of cancer patients. Many candidates enrolled in clinical trials are those with advanced stages of cancer. The efficacy of gene therapy most likely will be influenced by the severity of the disease as the cancer progresses and the complexity of both its genetic profile (mutational signatures) and immune system also increases, which can contribute to unsuccessful therapy in those subjects in comparison to having patients with earlier stages of malignancy as candidates.
Despite the success of gene therapy using adenoviral vectors expressing wild-type p53 in many clinical trials, there are questions regarding the variability and insufficient gene delivery to every neoplastic cell, as well as regarding the possible effects of antibodies to adenovirus in humans, which may have limited its clinical application [105]. Although reintroducing wild-type p53 through gene therapy is a seemingly straightforward and principally sound approach, technical difficulties, which affect the efficacy of the gene delivery and present possible safety issues with the vector, may present as major drawbacks to this method [106]. Moreover, most of the molecules, such as inhibitors or compounds (synthetic or natural agents) used to reactivate and restore p53 function in CRC cells, were only tested in vitro and in animal models but not yet in clinical trials [107].
In terms of specificity, although KRAS mutation is an early driver for colorectal carcinogenesis, which makes it an attractive target for treatment, these events may not be the main initiators compared to APC loss or β-catenin mutations in MMR-deficient tumors. The extent of the dependency of these tumors on KRAS is still yet to be elucidated [107]. The lack of this fundamental knowledge on the KRAS pathway in CRC may have influenced the success rate of targeted gene therapy against this mutation. Several groups are studying to directly target KRAS mutations and thus its biological activity by either using molecules and oligonucleotides to bind the KRAS-mutated sites or KRAS synthesis inhibition at the DNA level [108,109,110]. Other proposed strategies targetting KRAS in CRC include using compounds to target G4 motifs present in the human KRAS promoter [111,112,113] and microRNAs to regulate the expression of KRAS or associated genes in the KRAS-driven pathway [114,115,116]. Although promising results were reported in inactivating mutations of KRAS or in the downregulation of mRNA and proteins of KRAS, and in the downstream genes in the KRAS pathway, these works are perfomed at the pre-clinical stage. Large scale randomized clinical trials are needed to verify the efficacy and specificity of the KRAS-targetting strategies and drugs [112].
6.3. Promising and Alternative Treatment Options Available for CRC
Some have proposed that the combination of gene therapy with standard treatments (chemotherapy and/or radiotherapy), immunotherapy, and stem cell therapy in the future are projected to warrant a cancer cure [70]. Despite the enthusiasm surrounding the targeted gene therapy, standard treatments such as chemotherapy, radiotherapy, and surgical resection still represent the gold standard methods for CRC management in many countries. 5-fluorouracil (5-FU), irinotecan, and oxaliplatin are among cytotoxic drugs that are commonly included in chemotherapy regiments, although adverse effects were recorded post-treatment in many CRC patients. Conventional methods are preferred due to cost efficiency, availability of drugs, and availability of experienced personnel, including oncologists, and technologies in hospitals and treatment facilities. Despite the wide application of chemotherapy, studies are currently being performed to improve this approach and reduce the side effects that may be experienced by the patients. For example, a treatment regimen with oxaliplatin in addition to fluoropyrimidine has been established as standard adjuvant chemotherapy since 2004 for patients with stage III CRC [117,118,119].
Genetic testing and mutational analysis helped to pave a way to more targeted treatment against many types of cancer including CRC. Targeted therapies in CRC also showed concrete positive results. For instance, better survival rates and responsiveness were recorded in metastatic CRC patients with the BRAF V600E mutation after a treatment consisting of the combination of encorafenib, cetuximab, and binimetinib [120]. This is a major outbreak in CRC therapy, as conventional chemotherapy for subjects with BRAF V600E mutations were not successful [121].
Moreover, recent breakthroughs in immunotherapy for the past decade have hailed it as the ‘Holy Grail’ for cancer treatment, including CRC. This was significantly verified by tumor profiling. The number and type of genomic changes have been proven to be essential to dictate the efficacy of immune checkpoint inhibitors, such as those involved in the PD-1/PDL-1 pathway [122,123]. Le et al. (2015) reported the clinical success of immune checkpoint blockage (ICB) by using pembrolizumab based on the mismatch-repair status in CRC patients [124]. This is supported by André et al. (2020), wherein a longer survivability was found in metastatic CRC subjects with MSI-H (microsatellite high) treated with Pembrolizumab compared to chemotherapy. Fewer adverse effects from the treatment also were found [119]. These findings solidified immunotherapy as the prime treatment for CRC.
Some have argued that KRAS mutation is ‘undruggable’, as there is no clinically proven anti-KRAS therapy despite decades of research on this oncogene. However, the previous case report by Tran et al. (2016) exhibited an encouraging outcome whereby high effectiveness of antitumor therapy post-adoptive transfer of tumor-infiltrating CD8+ (expanded ex vivo) in a metastatic CRC patient with mutant KRAS G12D expression was demonstrated [125]. This personalized cell therapy approach in targeting mutant KRAS in colorectal tumors, utilizing the T-cell response, potentially will block the progression of various solid tumors. These reports suggested that CRC treatment using different modalities such as immunotherapy and targeted therapy may be the preferred options rather than gene therapy targeting p53 and KRAS mutations directly.
6.4. Safety
As stated in the previous section, the more common method for gene therapy targeting p53 in cancer involves administrating wild-type p53 or suppressing p53 activity in defect cells. An obvious drawback of this approach concerns the overexpression of wild-type p53, which could induce adverse effects on normal cells [126,127].
A successful gene therapy also warrants the use of a safe vector. The safety issue of the delivery vehicle, such as an adenovirus, has been raised due to several reported incidences during clinical trials and due to a lack of understanding on its mechanism of action [128,129]. The United States Food and Drug Administration (USFDA) has disapproved the use of Introgen′s Advexin, which utilizes p53, due to safety concerns. Currently, there is no record of the Gendicine™ clinical data submission or approval status from the USFDA [130].
6.5. Ethical Concerns
Until 2017, nearly 2600 clinical trials involving gene therapy were completed, ongoing, or approved globally [131]. Ethically, gene therapy in germ cells has been controversial and, at the moment, is banned from human trials [132]. Additional ethical issues revolving around gene therapy include subject selection and consent, inadequate protection to subjects involved, and the question of bias or conflict of interest from the physician’s perspectives [133].
6.6. Cost-Effectiveness
Gene therapies are costly and this factor poses a great challenge in its implementation. For example, Zolgensma, a prescription gene therapy for spinal muscular atrophy (SMA), costs USD 2.1 million per patient, which ranks it as the most expensive drug in the world [134]. For cancer, Yescarta, which is a cell-based gene therapy used to treat Non-Hodgkin lymphoma patients, comes with a price of USD 0.373 million for a one-time dose [135]. The staggering prices of these approved therapies question the affordability of the treatment for patients, especially those from poor countries.
The expensive expenditure in gene therapy originates from the personalized nature of this technology. Often the vectors and compounds are produced, tested, and manufactured on a smaller scale unlike those drugs in standard chemotherapy. It is worth noting that the development of a targeted gene therapy involves an extensive research and development (R&D) process, which adds to its overall cost and high per-patient price [136]. One also may also argue that a successful gene therapy administered at a single time may seem expensive but is cost-effective when compared to the costs of years of expensive and ongoing treatment for cancer patients. This highlights the definition of ‘values’ acquired from gene therapy, which lead to a better quality-of-life [137]. Gene therapy has been proven to improve body function, reduce suffering, and improve psychological stress due to the illness [137,138]. Efforts to establish a consensus is needed among the stakeholders, including policy makers and analysts, pharmaceutical companies, payers (both public and private), and key personnel from providers and organizations to further refine solutions and opportunities in regard to gene therapy access to patients [134].
7. Potential Outlook of Cancer Gene Therapy
A number of driver gene mutations, including of TP53 and KRAS as stated in Table 1, resulted in immunogenic neoantigens. Among “hotspot” missense mutations of R175, G245, R248, R249, R273, and R282 in the TP53 are found mutated in various types of cancers. These p53 missense mutations uniquely influence tumor behavior and promote carcinogenesis, thus providing insight into more targeted cancer treatment [139]. For instance, targeting p53 gene mutations of R175H and R273H has been reported to restore p53 mutants’ DNA binding activity and to abolish tumor development [140]. Additionally, a recent breakthrough discovery on significant anticancer properties of recently approved FDA drugs, namely Sotorasib (AMG 510), in subjects with solid tumors (non-small cell lung carcinoma, colon, etc.) harboring the KRAS p.G12C mutation has shed light on cancer gene therapy. Sotorasib acts as a covalent inhibitor that specifically and irreversibly extinguishes KRASG12C activity [141]. These encouraging discoveries indicate the prospect of cancer gene therapy as a cancer gene vaccine and immunotherapy targeting mutation sites of TP53 and KRAS.
Currently, the major immunotherapy types for cancer treatment include (a) monoclonal antibodies targeting specific antigens [142]; (b) ICB to reinvigorate T-cell immunity [143]; (c) CAR-T cell therapy [144]; (d) oncolytic viruses to eliminate specific cancer cells; and (e) cancer vaccines [145,146]. One of these strategies, namely therapeutic DNA cancer vaccines, may represent a very promising strategy to activate patients’ immune system to combat cancer [147,148]. It is worth noting that until now, pre-clinical and clinical trials indicated that therapeutic cancer vaccines on their own may not significantly improve cancer outcome. A DNA vaccine is proposed to be coupled with other personalized modes of treatment to be provided as a combined therapy after the analysis of a patient’s genetic profile and biomarkers that predict treatment response to a specific drug or agent. This will ensure better efficacy of the therapy and reduce the risk of adverse effects of the treatment [147].
8. Conclusions
There are currently significant advancements in gene therapy for CRC. Despite many promising results from pre-clinical studies, the efficacy of targeted gene therapy in oncology must be tested and further verified in large randomized clinical trials. Two of the major players in colorectal carcinogenesis, specifically TP53 and KRAS genes, serve as robust targets for gene therapy. However, more fundamental studies are required to dissect the roles and regulations of p53 and KRAS pathways in tumorigenesis. Targeted gene therapy also needs to be scrutinized more carefully to ensure the best strategies for combating CRC. Currently, this approach is still far from the gold standard of CRC therapy, mainly due to its high cost and the availability of other treatment modalities such as immunotherapy that warrant promising results in CRC patients.
Author Contributions
Conceptualization, M.M.; writing—original draft preparation H.H.H. and M.M.; writing—review and editing, H.H.H. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the PPSP Incentive Postgraduate Studies Development Fund, School of Medical Sciences, USM (grant account number: 1001/PPSP/8070011).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality morldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Brown, K.G.M.; Solomon, M.J.; Mahon, K.; O’Shannassy, S. Management of colorectal cancer. BMJ 2019, 366, l4561. [Google Scholar] [CrossRef]
- Andrew, A.S.; Parker, S.; Anderson, J.C.; Rees, J.R.; Robinson, C.; Riddle, B.; Butterly, L.F. Risk Factors for Diagnosis of Colorectal Cancer at a Late Stage: A Population-Based Study. J. Gen. Intern. Med. 2018, 33, 2100–2105. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Tortola, S.; Marcuello, E.; González, I.; Reyes, G.; Arribas, R.; Aiza, G.; Sancho, F.J.; Peinado, M.A.; Capella, G. p53 and K-ras Gene Mutations Correlate with Tumor Aggressiveness but Are Not of Routine Prognostic Value in Colorectal Cancer. J. Clin. Oncol. 1999, 17, 1375. [Google Scholar] [CrossRef]
- Pearce, A.; Haas, M.; Viney, R.; Pearson, S.-A.; Haywood, P.; Brown, C.; Ward, R. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. PLoS ONE 2017, 12, e0184360. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.; Burmester, J.K. Gene Therapy for Cancer Treatment: Past, Present and Future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Shen, X.; Chen, D. Combined chemotherapy and gene therapy of esophageal cancer with human adenoviral p53 administered by endoscopic injection combined with chemotherapy. J. Clin. Oncol. 2015, 33 (Suppl. S15), e15097. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Brown, A.-L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef]
- Wodarz, D.; Newell, A.C.; Komarova, N.L. Passenger mutations can accelerate tumour suppressor gene inactivation in cancer evolution. J. R. Soc. Interface 2018, 15, 20170967. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and Familial Colon Cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef]
- Milanese, J.-S.; Wang, E. Germline mutations and their clinical applications in cancer. Breast Cancer Manag. 2019, 8, BMT23. [Google Scholar] [CrossRef]
- Goel, A.; Boland, C.R. Epigenetics of Colorectal Cancer. Gastroenterology 2012, 143, 1442–1460.e1441. [Google Scholar] [CrossRef]
- Hong, S.N. Genetic and epigenetic alterations of colorectal cancer. Intest. Res. 2018, 16, 327–337. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Wickham, R.; Lassere, Y. The ABCs of Colorectal Cancer. Semin. Oncol. Nurs. 2007, 23, 1–8. [Google Scholar] [CrossRef]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.; Wolf, C.R. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433–9438. [Google Scholar] [CrossRef]
- Grazioso, T.P.; Brandt, M.; Djouder, N. Diet, Microbiota, and Colorectal Cancer. iScience 2019, 21, 168–187. [Google Scholar] [CrossRef]
- Hardiman, K. Update on Sporadic Colorectal Cancer Genetics. Clin. Colon Rectal Surg. 2018, 31, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The Intestinal Crypt, A Prototype Stem Cell Compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Perochon, J.; Carroll, L.R.; Cordero, J.B. Wnt Signalling in Intestinal Stem Cells: Lessons from Mice and Flies. Genes 2018, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Peddareddigari, V.G.; Wang, D.; DuBois, R.N. The Tumor Microenvironment in Colorectal Carcinogenesis. Cancer Microenviron. 2010, 3, 149–166. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Driving immune evasion. Nat. Rev. Cancer 2018, 18, 67. [Google Scholar] [CrossRef]
- Mascaux, C.; Angelova, M.; Vasaturo, A.; Beane, J.; Hijazi, K.; Anthoine, G.; Buttard, B.; Rothe, F.; Willard-Gallo, K.; Haller, A.; et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nat. Cell Biol. 2019, 571, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Berral-González, A.; López-Cade, I.; Galindo-Pumariño, C.; Bueno-Fortes, S.; Martín-Merino, M.; Carrato, A.; Ocaña, A.; De La Pinta, C.; López-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 1–6. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Marks, L.; May, G.H.W.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723. [Google Scholar] [CrossRef]
- Law, P.J.; The PRACTICAL Consortium; Timofeeva, M.; Fernandez-Rozadilla, C.; Broderick, P.; Studd, J.; Fernandez-Tajes, J.; Farrington, S.; Svinti, V.; Palles, C.; et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nat. Cell Biol. 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef]
- Munro, A.J.; Lain, S.; Lane, D. P53 abnormalities and outcomes in colorectal cancer: A systematic review. Br. J. Cancer 2005, 92, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Andreyev, H.N.; Norman, A.; Cunningham, D.; Oates, J.; Dix, B.; Iacopetta, B.; Young, J.; Walsh, T.P.; Ward, R.; Hawkins, N.; et al. Kirsten ras mutations in patients with colorectal cancer: The ‘RASCAL II’ study. Br. J. Cancer 2001, 85, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Hayama, T.; Hashiguchi, Y.; Okamoto, K.; Okada, Y.; Ono, K.; Shimada, R.; Ozawa, T.; Toyoda, T.; Tsuchiya, T.; Iinuma, H.; et al. G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int. J. Colorectal Dis. 2019, 34, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Goffredo, P.; Ginader, T.; Hrabe, J.; Gribovskaja-Rupp, I.; Kapadia, M.R.; Weigel, R.J.; Hassan, I. The Impact of KRAS Mutation on the Presentation and Prognosis of Non-Metastatic Colon Cancer: An Analysis from the National Cancer Database. J. Gastrointest. Surg. 2020, 24, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Murad, N.A.A.; Othman, Z.; Khalid, M.; Razak, Z.A.; Hussain, R.; Nadesan, S.; Sagap, I.; Rose, I.M.; Ngah, W.Z.W.; Jamal, R. Missense Mutations in MLH1, MSH2, KRAS, and APC Genes in Colorectal Cancer Patients in Malaysia. Dig. Dis. Sci. 2012, 57, 2863–2872. [Google Scholar] [CrossRef]
- Zulhabri, O.; Rahman, J.; Ismail, S.; Isa, M.R.; Zurinah, W.N.W. Predominance of G to A codon 12 mutation K-ras gene in Dukes’ B colorectal cancer. Singap. Med. J. 2012, 53, 26–31. [Google Scholar]
- Yip, W.K.; Choo, C.W.; Leong, V.C.-S.; Leong, P.P.; Jabar, M.F.; Seow, H.F. Molecular alterations of Ras-Raf-mitogen-activated protein kinase and phosphatidylinositol 3-kinase-Akt signaling pathways in colorectal cancers from a tertiary hospital at Kuala Lumpur, Malaysia. APMIS 2013, 121, 954–966. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol. Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Andreyev, H.J.N.; Norman, A.R.; Clarke, P.; Cunningham, D.; Oates, J.R. Kirsten ras Mutations in Patients With Colorectal Cancer: The Multicenter “RASCAL” Study. J. Natl. Cancer Inst. 1998, 90, 675–684. [Google Scholar] [CrossRef]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Win, A.K.; Baron, J.A.; Lindor, N.M.; Potter, J.D.; A Newcomb, P. KRAS-mutation status in relation to colorectal cancer survival: The joint impact of correlated tumour markers. Br. J. Cancer 2013, 108, 1757–1764. [Google Scholar] [CrossRef]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2017, 78, 1334–1346. [Google Scholar] [CrossRef]
- Domingo, E.; Camps, C.; Kaisaki, P.J.; Parsons, M.J.; Mouradov, D.; Pentony, M.M.; Makino, S.; Palmieri, M.; Ward, R.L.; Hawkins, N.J.; et al. Mutation burden and other molecular markers of prognosis in colorectal cancer treated with curative intent: Results from the QUASAR 2 clinical trial and an Australian community-based series. Lancet Gastroenterol. Hepatol. 2018, 3, 635–643. [Google Scholar] [CrossRef]
- Gallo, G.; Sena, G.; Vescio, G.; Papandrea, M.; Sacco, R.; Trompetto, M.; Sammarco, G. The prognostic value of KRAS and BRAF in stage I-III colorectal cancer. A systematic review. Ann. Ital. Chir. 2019, 8, 127–137. [Google Scholar]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: Results from the MRC FOCUS trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Foltran, L.; De Maglio, G.; Pella, N.; Ermacora, P.; Aprile, G.; Masiero, E.; Giovannoni, M.; Iaiza, E.; Cardellino, G.G.; Lutrino, S.E.; et al. Prognostic role of KRAS, NRAS, BRAF and PIK3CA mutations in advanced colorectal cancer. Future Oncol. 2015, 11, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology Provisional Clinical Opinion: Testing for KRAS Gene Mutations in Patients With Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Chen, D.; Gu, K.; Wang, H. Optimizing sequential treatment with anti-EGFR and VEGF mAb in metastatic colorectal cancer: Current results and controversies. Cancer Manag. Res. 2019, 11, 1705–1716. [Google Scholar] [CrossRef]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.R.; Kundu, S.K.; Nam, J.-S.; Sharma, G.; Doss, C.G.P.; Lee, S.-S.; Chakraborty, C. Next Generation Delivery System for Proteins and Genes of Therapeutic Purpose: Why and How? BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Belete, T.M. The Current Status of Gene Therapy for the Treatment of Cancer. Biol. Targets Ther. 2021, 15, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Chung-Faye, G.A.; Kerr, D.J.; Young, L.; Searle, P.F. Gene therapy strategies for colon cancer. Mol. Med. Today 2000, 6, 82–87. [Google Scholar] [CrossRef]
- Steele, F.R. A Moving Target Gene Therapy: Therapeutic Mechanisms and Strategies. Mol. Ther. 2001, 3, 429–430. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, Y.; Jiang, W.; Dai, W.; Jiang, Y. Anti-Tumor Effect of a Novel Soluble Recombinant Human Endostatin: Administered as a Single Agent or in Combination with Chemotherapy Agents in Mouse Tumor Models. PLoS ONE 2014, 9, e107823. [Google Scholar] [CrossRef]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef]
- Porteus, M. Genome Editing: A New Approach to Human Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 163–190. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53(Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar]
- Amer, M.H. Gene therapy for cancer: Present status and future perspective. Mol. Cell. Ther. 2014, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Pastoriza, J.; Quinn, T.; Libutti, S.K. Chapter 2—Targeting Tumor Vasculature Using Adeno-Associated Virus Phage Vectors Coding Tumor Necrosis Factor-α. In Gene Therapy of Cancer, 3rd ed.; Lattime, E.C., Gerson, S.L., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 19–33. [Google Scholar]
- Vidic, S.; Markelc, B.; Sersa, G.; Coer, A.; Kamensek, U.; Tevz, G.; Kranjc, S.; Cemazar, M. MicroRNAs targeting mutant K-ras by electrotransfer inhibit human colorectal adenocarcinoma cell growth in vitro and in vivo. Cancer Gene Ther. 2010, 17, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hendricks, W.; Liu, G.; McCaffery, J.M.; Kinzler, K.W.; Huso, D.L.; Vogelstein, B.; Zhou, S. A nanoparticle formulation that selectively transfects metastatic tumors in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 14717–14722. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.-Q.; Lu, Y.-X.; Wu, Q.-N.; Liu, J.; Zeng, Z.-L.; Mo, H.-Y.; Chen, Y.; Tian, T.; Wang, Y.; Kang, T.-B.; et al. Disrupting G6PD-mediated Redox homeostasis enhances chemosensitivity in colorectal cancer. Oncogene 2017, 36, 6282–6292. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, X.; Zheng, S.; Wang, B.; Li, Y.; Zhao, C.; Muftuoglu, Y.; Chen, S.; Li, Y.; Yao, H.; et al. Modified nanoparticle mediated IL-12 immunogene therapy for colon cancer. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1993–2004. [Google Scholar] [CrossRef]
- Pishavar, E.; Ramezani, M.; Hashemi, M. Co-delivery of doxorubicin and TRAIL plasmid by modified PAMAM dendrimer in colon cancer cells, in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2019, 45, 1931–1939. [Google Scholar] [CrossRef]
- Gao, Y.; Men, K.; Pan, C.; Li, J.; Wu, J.; Chen, X.; Lei, S.; Gao, X.; Duan, X. Functionalized DMP-039 Hybrid Nanoparticle as a Novel mRNA Vector for Efficient Cancer Suicide Gene Therapy. Int. J. Nanomed. 2021, 16, 5211–5232. [Google Scholar] [CrossRef]
- Chung-Faye, G.; Chen, M.; Green, N.; Burton, A.; Anderson, D.; Mautner, V.; Searle, P.; Kerr, D. In vivo gene therapy for colon cancer using adenovirus-mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther. 2001, 8, 1547–1554. [Google Scholar] [CrossRef]
- Reid, T.R.; Freeman, S.; Post, L.; McCormick, F.; Sze, D.Y. Effects of Onyx-015 among metastatic colorectal cancer patients that have failed prior treatment with 5-FU/leucovorin. Cancer Gene Ther. 2005, 12, 673–681. [Google Scholar] [CrossRef]
- Tu, S.P.; Cui, J.T.; Liston, P.; Jiang, X.H.; Xu, R.; Lin, M.C.; Zhu, Y.B.; Zou, B.; Ng, S.S.; Jiang, S.H.; et al. Gene therapy for colon cancer by adeno-associated viral vector-mediated transfer of survivin Cys84Ala mutant. Gastroenterology 2005, 128, 361–375. [Google Scholar] [CrossRef]
- He, L.F.; Wang, Y.G.; Xiao, T.; Zhang, K.J.; Li, G.C.; Gu, J.F.; Chu, L.; Tang, W.H.; Tan, W.-S.; Liu, X.Y. Suppression of cancer growth in mice by adeno-associated virus vector-mediated IFN-beta expression driven by hTERT promoter. Cancer Lett. 2009, 286, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.P.; Xue, Z.; Sun, P.H.; Zhu, L.M.; Jiang, S.H.; Qiao, M.M.; Chi, A.L. Adeno-associated virus-mediated survivin mutant Thr34Ala cooperates with oxaliplatin to inhibit tumor growth and angiogenesis in colon cancer. Oncol. Rep. 2011, 25, 1039–1046. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, Y.; Guo, Z.; Zhang, D.; Zhang, W.; Yan, Q.; Shi, X.; Zhang, M.; Zhao, Y.; Zhang, Y.; Jiang, B.; et al. A novel colon cancer gene therapy using rAAV-mediated expression of human shRNA-FHL2. Int. J. Oncol. 2013, 43, 1618–1626. [Google Scholar] [CrossRef]
- Koon, H.W.; Cheng, M.; Ho, S.; Yoo, J.H.; Bakirtzi, K.; Tran, D.; Su, B.; Tran, D.; Ichikawa, R.; Kubota, Y. Cathelicidin suppresses colon cancer development by inhibition of cancer associated fibroblasts. Clin. Exp. Gastroenterol. 2014, 8, 13–29. [Google Scholar] [CrossRef][Green Version]
- Xing, M.; Wang, X.; Chi, Y.; Zhou, D. Gene therapy for colorectal cancer using adenovirus-mediated full-length antibody, cetuximab. Oncotarget 2016, 7, 28262–28272. [Google Scholar] [CrossRef]
- U.S National Library of Medicine. Chemoradiation with Enadenotucirev as a Radiosensitiser in Locally Advanced Rectal Cancer (CEDAR). Available online: https://clinicaltrials.gov/ct2/show/NCT03916510 (accessed on 24 October 2021).
- U.S National Library of Medicine. Mechanism of Action Trail of ColoAd1 (MOA). Available online: https://www.clinicaltrials.gov/ct2/show/NCT02053220 (accessed on 24 October 2021).
- McGranahan, N.; Favero, F.; De Bruin, E.C.; Birkbak, N.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef]
- Roth, J.; Nguyen, D.; Lawrence, D.; Kemp, B.; Carrasco, C.; Ferson, D.; Hong, W.; Komaki, R.; Lee, J.; Nesbitt, J.; et al. Retrovirus–mediated wild–type P53 gene transfer to tumors of patients with lung cancer. Nat. Med. 1996, 2, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Matsubara, H.; Shiratori, T.; Shimizu, T.; Miyazaki, S.; Okazumi, S.; Nabeya, Y.; Shuto, K.; Hayashi, H.; Tanizawa, T.; et al. Phase I/II adenoviral p53 gene therapy for chemoradiation resistant advanced esophageal squamous cell carcinoma. Cancer Sci. 2006, 97, 554–561. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. [Google Scholar] [CrossRef]
- Xia, Y.; Li, X.; Sun, W. Applications of recombinant adenovirus-p53 gene therapy for cancers in the clinic in China. Curr. Gene Ther. 2020, 20, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Cornelio, G.; Lorenzo, C.; Levy, J.; Reed, R.; Liu, L.; Hall, F. First clinical experience using a ‘pathotropic’ injectable retroviral vector (Rexin-G) as intervention for stage IV pancreatic cancer. Int. J. Oncol. 2004, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Hall, F.L. Rexin-G, a targeted genetic medicine for cancer. Expert Opin. Biol. Ther. 2010, 10, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, S.; Shaish, A.; Varda-Bloom, N.; Levanon, K.; Breitbart, E.; Goldberg, I.; Barshack, I.; Hodish, I.; Yaacov, N.; Bangio, L.; et al. Transcription-controlled gene therapy against tumor angiogenesis. J. Clin. Investig. 2004, 113, 1017–1024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brenner, A.J.; Peters, K.B.; Vredenburgh, J.; Bokstein, F.; Blumenthal, D.T.; Yust-Katz, S.; Peretz, I.; Oberman, B.; Freedman, L.S.; Ellingson, B.M.; et al. Safety and efficacy of VB-111, an anticancer gene therapy, in patients with recurrent glioblastoma: Results of a phase I/II study. Neuro-Oncology 2020, 22, 694–704. [Google Scholar] [CrossRef]
- Montaño-Samaniego, M.; Bravo-Estupiñan, D.M.; Méndez-Guerrero, O.; Alarcón-Hernández, E.; Ibáñez-Hernández, M. Strategies for Targeting Gene Therapy in Cancer Cells With Tumor-Specific Promoters. Front. Oncol. 2020, 10, 2671. [Google Scholar] [CrossRef]
- Yue, J.; Luo, S.; Lu, M.; Shao, D.; Wang, Z.; Dong, W. A comparison of mesoporous silica nanoparticles and mesoporous organosilica nanoparticles as drug vehicles for cancer therapy. Chem. Biol. Drug Des. 2018, 92, 1435–1444. [Google Scholar] [CrossRef]
- Yan, L.-J.; Guo, X.-H.; Wang, W.-P.; Hu, Y.-R.; Duan, S.-F.; Liu, Y.; Sun, Z.; Huang, S.-N.; Li, H.-L. Gene therapy and photothermal therapy of layer-by-layer assembled AuNCs/PEI/miRNA/HA nanocomplexes. Curr. Cancer Drug Targets 2019, 19, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Lisiansky, V.; Naumov, I.; Shapira, S.; Kazanov, D.; Starr, A.; Arber, N.; Kraus, S. Gene therapy of pancreatic cancer targeting the K-Ras oncogene. Cancer Gene Ther. 2012, 19, 862–869. [Google Scholar] [CrossRef]
- Murugan, A.K.; Grieco, M.; Tsuchida, N. RAS mutations in human cancers: Roles in precision medicine. Semin. Cancer Biol. 2019, 59, 23–35. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Vilahur, N.; Bianchini, F.; Guha, N.; Straif, K. The IARC Perspective on Colorectal Cancer Screening. N. Engl. J. Med. 2018, 378, 1734–1740. [Google Scholar] [CrossRef]
- Unim, B.; Pitini, E.; De Vito, C.; D’Andrea, E.; Marzuillo, C.; Villari, P. Cost-Effectiveness of RAS Genetic Testing Strategies in Patients With Metastatic Colorectal Cancer: A Systematic Review. Value Health 2020, 23, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Lu, X. A new way to target p53-defective colorectal cancer. Future Oncol. 2015, 11, 3101–3104. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based Cancer Therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. p53mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- McCormick, F. KRAS as a Therapeutic Target. Clin. Cancer Res. 2015, 21, 1797–1801. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Inoue, T.; Taylor, R.D.; Watanabe, T.; Koshikawa, N.; Yoda, H.; Shinohara, K.-I.; Takatori, A.; Sugimoto, H.; Maru, Y.; et al. Inhibition of KRAS codon 12 mutants using a novel DNA-alkylating pyrrole–imidazole polyamide conjugate. Nat. Commun. 2015, 6, 6706. [Google Scholar] [CrossRef]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef]
- Lavrado, J.; Borralho, P.M.; Ohnmacht, S.A.; Castro, R.E.; Rodrigues, C.M.P.; Moreira, R.; dos Santos, D.J.V.A.; Neidle, S.; Paulo, A. Synthesis, G-Quadruplex Stabilisation, Docking Studies, and Effect on Cancer Cells of Indolo[3,2-b]quinolines with One, Two, or Three Basic Side Chains. ChemMedChem 2013, 8, 1648–1661. [Google Scholar] [CrossRef]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef]
- Kaiser, C.E.; Van Ert, N.A.; Agrawal, P.; Chawla, R.; Yang, D.; Hurley, L.H. Insight into the Complexity of the i-Motif and G-Quadruplex DNA Structures Formed in the KRAS Promoter and Subsequent Drug-Induced Gene Repression. J. Am. Chem. Soc. 2017, 139, 8522–8536. [Google Scholar] [CrossRef]
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar] [CrossRef]
- Liao, W.-T.; Ye, Y.-P.; Zhang, N.-J.; Li, T.-T.; Wang, S.-Y.; Cui, Y.-M.; Qi, L.; Wu, P.; Jiao, H.-L.; Xie, Y.-J.; et al. MicroRNA-30b functions as a tumour suppressor in human colorectal cancer by targeting KRAS, PIK3CD and BCL2. J. Pathol. 2014, 232, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Rachagani, S.; Macha, M.; Menning, M.S.; Dey, P.; Pai, P.; Smith, L.M.; Mo, Y.-Y.; Batra, S.K. Changes in microRNA (miRNA) expression during pancreatic cancer development and progression in a genetically engineered KRASG12D;Pdx1-Cre mouse (KC) model. Oncotarget 2015, 6, 40295–40309. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef]
- André, T.; de Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-Raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial Therapy with FOLFOXIRI and Bevacizumab for Metastatic Colorectal Cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.J.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D. p53—Master and commander. N. Engl. J. Med. 2007, 357, 2539–2541. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar] [CrossRef]
- Wilson, J.M. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2009, 96, 151–157. [Google Scholar] [CrossRef]
- Arnold, C. Record number of gene-therapy trials, despite setbacks. Nat. Med. 2021, 27, 1312–1315. [Google Scholar] [CrossRef]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef] [PubMed]
- de Wert, G.; Pennings, G.; Clarke, A.; Eichenlaub-Ritter, U.; van El, C.G.; Forzano, F.; Goddijn, M.; Heindryckx, B.; Howard, H.C.; Radojkovic, D.; et al. Human germline gene editing. Recommendations of ESHG and ESHRE. Eur. J. Hum. Genet. 2018, 26, 445–449. [Google Scholar] [CrossRef]
- Riva, L.; Petrini, C. A few ethical issues in translational research for gene and cell therapy. J. Transl. Med. 2019, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Humer, C. Novartis $2 Million Gene Therapy for Rare Disorder Is World’s Most Expensive Drug. 2019. Available online: https://www.reuters.com/article/us-novartis-genetherapy/novartis-2-million-gene-therapy-for-rare-disorder-is-worlds-most-expensive-drug-idUSKCN1SU1ZP (accessed on 15 August 2021).
- Andrews, M. Staggering Prices Slow Insurers’ Coverage of CAR-T Cancer Therapy. 2018. Available online: https://khn.org/news/staggering-prices-slow-insurers-coverage-of-car-t-cancer-therapy/ (accessed on 1 September 2021).
- Hampson, G.; Towse, A.; Pearson, S.D.; Dreitlein, W.B.; Henshall, C. Gene therapy: Evidence, value and affordability in the US health care system. J. Comp. Eff. Res. 2018, 7, 15–28. [Google Scholar] [CrossRef]
- Salzman, R.; Cook, F.; Hunt, T.; Malech, H.L.; Reilly, P.; Foss-Campbell, B.; Barrett, D. Addressing the Value of Gene Therapy and Enhancing Patient Access to Transformative Treatments. Mol. Ther. 2018, 26, 2717–2726. [Google Scholar] [CrossRef]
- Hemp, P. Presenteeism: At work—But out of it. Harv. Bus. Rev. 2004, 82, 49–58. [Google Scholar]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Gasser, M.; Waaga-Gasser, A.M. Therapeutic antibodies in cancer therapy. Adv. Exp. Med. Biol. 2016, 917, 95–120. [Google Scholar]
- Clarke, J.M.; George, D.J.; Lisi, S.; Salama, A. Immune Checkpoint Blockade: The New Frontier in Cancer Treatment. Target. Oncol. 2018, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Mackall, C.L. T-cell adoptive immunotherapy for acute lymphoblastic leukemia. Hematology 2013, 2013, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J. Therapeutic Cancer Vaccines: Current Status and Moving Forward. J. Natl. Cancer Inst. 2012, 104, 599–613. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Redman, J.M.; Collins, J.M.; Bilusic, M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum. Vaccines Immunother. 2017, 13, 2561–2574. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).