
Alzheimer’s Disease and Diabetes Mellitus in Comparison: The Therapeutic Efficacy of the Vanadium Compound



Abstract
1. Introduction
2. The Pharmacological Research of Vanadium Compounds in Treating Diabetes
3. The Correlations between AD and Diabetes
3.1. Insulin Degrading Enzyme (IDE)
3.2. Glycogen Synthase Kinase 3β
3.3. Ferroptosis
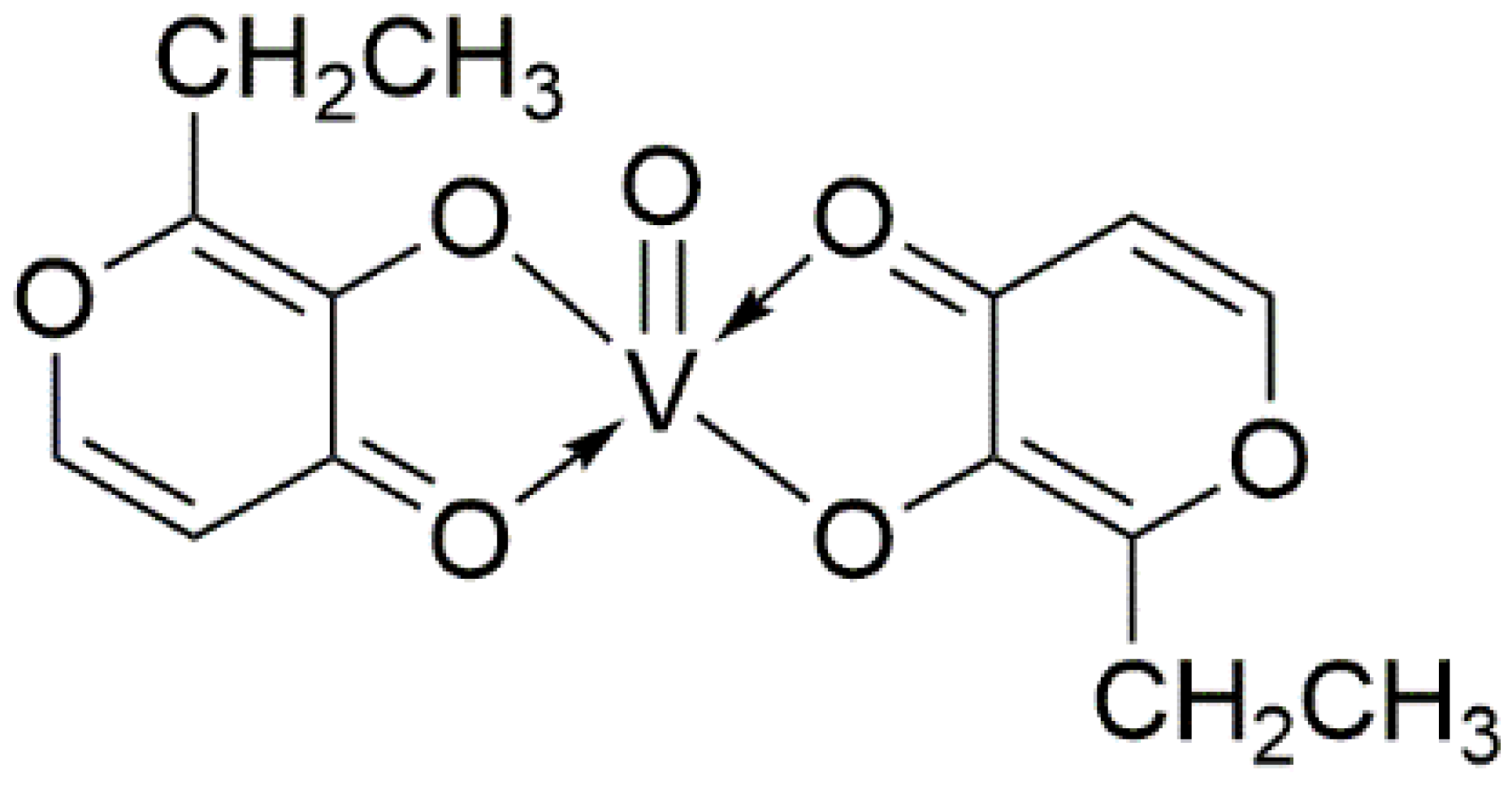
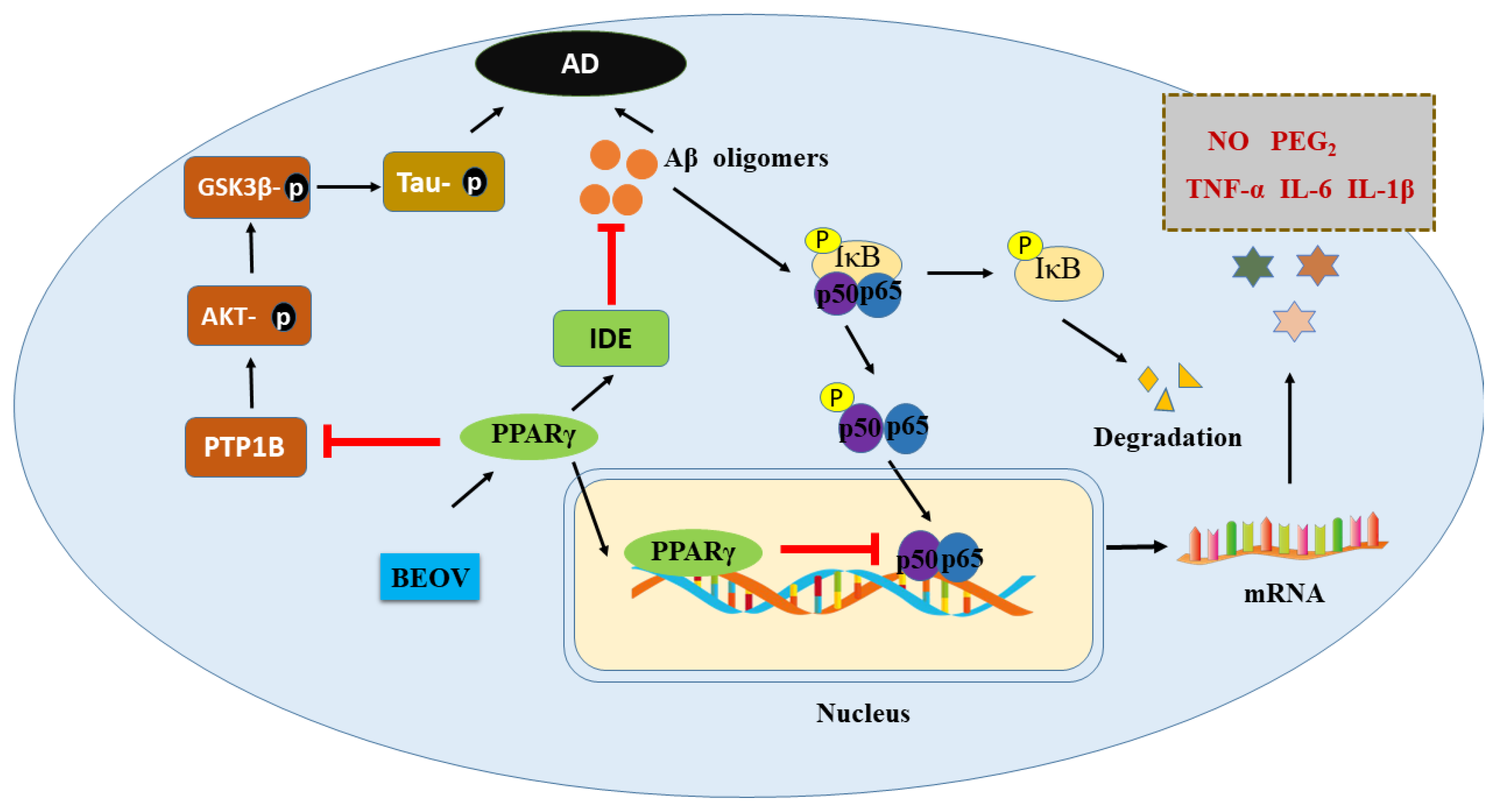
4. The Protective Efficacy of Vanadium Compound on AD Mouse Models
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arvanitakis, Z.; Shah, R.C.; Bennett, D.A. Diagnosis and Management of Dementia: Review. JAMA 2019, 322, 1589–1599. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Ribe, E.M.; Lovestone, S. Insulin signalling in Alzheimers disease and diabetes: From epidemiology to molecular links. J. Intern. Med. 2016, 280, 430–442. [Google Scholar] [CrossRef]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef]
- Rosales-Corral, S.; Tan, D.X.; Manchester, L.; Reiter, R.J. Diabetes and Alzheimer Disease, Two Overlapping Pathologies with the Same Background: Oxidative Stress. Oxidative Med. Cell. Longev. 2015, 2015, 985845. [Google Scholar] [CrossRef]
- Ng, R.C.; Cheng, O.Y.; Jian, M.; Kwan, J.S.C.; Ho, P.W.L.; Cheng, K.K.Y.; Yeung, P.K.K.; Zhou, L.L.; Hoo, R.L.C.; Chung, S.K.; et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 2016, 11, 71. [Google Scholar] [CrossRef]
- Hosoi, M.; Hori, K.; Konishi, K.; Tani, M.; Tomioka, H.; Kitajima, Y.; Akashi, N.; Inamoto, A.; Minami, S.; Izuno, T.; et al. Plasma Cholinesterase Activity in Alzheimer’s Disease. Neurodegener. Dis. 2015, 15, 188–190. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Cook, D.G.; Leverenz, J.B.; Mcmillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon 4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- He, L.; Wang, X.S.; Zhu, D.S.; Zhao, C.; Du, W.H. Methionine oxidation of amyloid peptides by peroxovanadium complexes: Inhibition of fibril formation through a distinct mechanism. Metallomics 2015, 7, 1562–1572. [Google Scholar] [CrossRef]
- Dong, Y.Q.; Stewart, T.; Zhang, Y.; Shi, M.; Tan, C.; Li, X.; Yuan, L.; Mehrotra, A.; Zhang, J.; Yang, X.D. Anti-diabetic vanadyl complexes reduced Alzheimer’s disease pathology independent of amyloid plaque deposition. Sci. China-Life Sci. 2019, 62, 126–139. [Google Scholar] [CrossRef]
- He, Z.; Li, X.; Han, S.; Ren, B.; Hu, X.; Li, N.; Du, X.; Ni, J.; Yang, X.; Liu, Q. Bis(ethylmaltolato)oxidovanadium (IV) attenuates amyloid-beta-mediated neuroinflammation by inhibiting NF-kappaB signaling pathway via a PPARgamma-dependent mechanism. Metallomics 2021, 13, mfab036. [Google Scholar] [CrossRef]
- He, Z.J.; Wang, M.H.; Zhao, Q.H.; Li, X.Q.; Liu, P.G.; Ren, B.Y.; Wu, C.; Du, X.B.; Li, N.; Liu, Q. Bis(ethylmaltolato)oxidovanadium (IV) mitigates neuronal apoptosis resulted from amyloid-beta induced endoplasmic reticulum stress through activating peroxisome proliferator-activated receptor gamma. J. Inorg. Biochem. 2020, 208, 111073. [Google Scholar] [CrossRef]
- He, Z.J.; Han, S.X.; Zhu, H.Z.; Hu, X.; Li, X.Q.; Hou, C.F.; Wu, C.; Xie, Q.G.; Li, N.; Du, X.B.; et al. The Protective Effect of Vanadium on Cognitive Impairment and the Neuropathology of Alzheimer’s Disease in APPSwe/PS1dE9 Mice. Front. Mol. Neurosci. 2020, 13, 21. [Google Scholar] [CrossRef]
- Huang, M.; Wu, Y.; Wang, N.; Wang, Z.; Zhao, P.; Yang, X. Is the hypoglycemic action of vanadium compounds related to the suppression of feeding? Biol. Trace Elem. Res. 2014, 157, 242–248. [Google Scholar] [CrossRef]
- Zaporowska, H.; Wasilewski, W. Hematological Effects of Vanadium on Living Organisms. Comp. Biochem. Physiol. C-Pharmacol. Toxicol. Endocrinol. 1992, 102, 223–231. [Google Scholar] [CrossRef]
- Beauge, L.A.; Glynn, I.M. A modifier of (Na+ + k+) atpase in commercial ATP. Nature 1977, 268, 355–356. [Google Scholar] [CrossRef]
- Beauge, L.A.; Glynn, I.M. Commercial ATP containing traces of vanadate alters the response of (Na+ + K+) ATPase to external potassium. Nature 1978, 272, 551–552. [Google Scholar] [CrossRef]
- Shechter, Y.; Karlish, S.J. Insulin-like stimulation of glucose oxidation in rat adipocytes by vanadyl (IV) ions. Nature 1980, 284, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Huyer, G.; Liu, S.; Kelly, J.; Moffat, J.; Payette, P.; Kennedy, B.; Tsaprailis, G.; Gresser, M.J.; Ramachandran, C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.L.; Zhang, C.Y.; Yu, S.W.; Yang, X.D.; Wang, K. Vanadyl bisacetylacetonate protects beta cells from palmitate-induced cell death through the unfolded protein response pathway. J. Biol. Inorg. Chem. 2011, 16, 789–798. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, X.D. Vanadium compounds modulate PPAR gamma activity primarily by increasing PPAR gamma protein levels in mouse insulinoma NIT-1 cells. Metallomics 2013, 5, 836–843. [Google Scholar] [CrossRef]
- Wu, Y.L.; Huang, M.L.; Zhao, P.; Yang, X.D. Vanadyl acetylacetonate upregulates PPAR gamma and adiponectin expression in differentiated rat adipocytes. J. Biol. Inorg. Chem. 2013, 18, 623–631. [Google Scholar] [CrossRef]
- Liu, J.C.; Yu, Y.; Wang, G.; Wang, K.; Yang, X.G. Bis(acetylacetonato)-oxovanadium(IV), bis(maltolato)-oxovanadium(IV) and sodium metavanadate induce antilipolytic effects by regulating hormone-sensitive lipase and perilipin via activation of Akt. Metallomics 2013, 5, 813–820. [Google Scholar] [CrossRef]
- Wu, J.X.; Hong, Y.H.; Yang, X.G. Bis(acetylacetonato)-oxidovanadium(IV) and sodium metavanadate inhibit cell proliferation via ROS-induced sustained MAPK/ERK activation but with elevated AKT activity in human pancreatic cancer AsPC-1 cells. J. Biol. Inorg. Chem. 2016, 21, 919–929. [Google Scholar] [CrossRef]
- Harland, B.F.; Harden-Williams, B.A. Is vanadium of human nutritional importance yet? J. Am. Diet Assoc. 1994, 94, 891–894. [Google Scholar] [CrossRef]
- Ladagu, A.D.; Olopade, F.E.; Folarin, O.R.; Elufioye, T.O.; Wallach, J.V.; Dybek, M.B.; Olopade, J.O.; Adejare, A. Novel NMDA-receptor antagonists ameliorate vanadium neurotoxicity. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1729–1738. [Google Scholar] [CrossRef]
- Colin-Barenque, L.; Bizarro-Nevares, P.; Gonzalez Villalva, A.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Jimenez-Martinez, R.; Rodriguez-Rangel, D.S.; Resendiz, S.; Fortoul, T.I. Neuroprotective effect of carnosine in the olfactory bulb after vanadium inhalation in a mouse model. Int. J. Exp. Pathol. 2018, 99, 180–188. [Google Scholar] [CrossRef]
- Yang, X.G.; Wang, K. Chemical, biochemical, and biological behaviors of vanadate and its oligomers. Prog. Mol. Subcell Biol. 2013, 54, 1–18. [Google Scholar]
- Llobet, J.M.; Domingo, J.L. Acute toxicity of vanadium compounds in rats and mice. Toxicol. Lett. 1984, 23, 227–231. [Google Scholar] [CrossRef]
- Bishayee, A.; Waghray, A.; Patel, M.A.; Chatterjee, M. Vanadium in the detection, prevention and treatment of cancer: The in vivo evidence. Cancer Lett. 2010, 294, 1–12. [Google Scholar] [CrossRef]
- Scibior, A.; Pietrzyk, L.; Plewa, Z.; Skiba, A. Vanadium: Risks and possible benefits in the light of a comprehensive overview of its pharmacotoxicological mechanisms and multi-applications with a summary of further research trends. J. Trace Elem. Med. Biol. 2020, 61, 126508. [Google Scholar] [CrossRef]
- Scibior, A.; Kurus, J. Vanadium and Oxidative Stress Markers—In Vivo Model: A Review. Curr. Med. Chem. 2019, 26, 5456–5500. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.B.; Ye, L.H.; Liu, H.X.; Xia, Q.; Zhang, Y.; Yang, X.D.; Wang, K. Vanadium compounds induced mitochondria permeability transition pore (PTP) opening related to oxidative stress. J. Inorg. Biochem. 2010, 104, 371–378. [Google Scholar] [CrossRef]
- Gerhardsson, L.; Lundh, T.; Minthon, L.; Londos, E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 25, 508–515. [Google Scholar] [CrossRef]
- Szabo, S.T.; Harry, G.J.; Hayden, K.M.; Szabo, D.T.; Birnbaum, L. Comparison of Metal Levels between Postmortem Brain and Ventricular Fluid in Alzheimer’s Disease and Nondemented Elderly Controls. Toxicol. Sci. 2016, 150, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Mcneill, J.H.; Yuen, V.G.; Hoveyda, H.R.; Orvig, C. Bis(Maltolato)Oxovanadium(Iv) Is a Potent Insulin Mimic. J. Med. Chem. 1992, 35, 1489–1491. [Google Scholar] [CrossRef]
- Thompson, K.H.; Orvig, C. Vanadium compounds in the treatment of diabetes. Met. Ions Complexes Medicat. 2004, 41, 221–252. [Google Scholar]
- Wang, N.; Wang, Z.; Niu, X.; Yang, X. Synthesis, characterization and anti-diabetic therapeutic potential of novel aminophenol-derivatized nitrilotriacetic acid vanadyl complexes. J. Inorg. Biochem. 2015, 152, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Yang, J.; Yang, X. Synthesis and anti-diabetic activity of new N,N-dimethylphenylenediamine-derivatized nitrilotriacetic acid vanadyl complexes. J. Inorg. Biochem. 2017, 177, 291–299. [Google Scholar] [CrossRef]
- Du, J.; Feng, B.; Dong, Y.; Zhao, M.; Yang, X. Vanadium coordination compounds loaded on graphene quantum dots (GQDs) exhibit improved pharmaceutical properties and enhanced anti-diabetic effects. Nanoscale 2020, 12, 9219–9230. [Google Scholar] [CrossRef]
- Setyawati, I.A.; Thompson, K.H.; Yuen, V.G.; Sun, Y.; Battell, M.; Lyster, D.M.; Vo, C.; Ruth, T.J.; Zeisler, S.; Mcneill, J.H.; et al. Kinetic analysis and comparison of uptake, distribution, and excretion of 48V-labeled compounds in rats. J. Appl. Physiol. 1998, 84, 569–575. [Google Scholar] [CrossRef]
- Rennhard, H.H. The metabolism of ethyl maltol and maltol in the dog. J. Agric. Food Chem. 1971, 19, 152–154. [Google Scholar] [CrossRef]
- Thompson, K.H.; Liboiron, B.D.; Sun, Y.; Bellman, K.D.; Setyawati, I.A.; Patrick, B.O.; Karunaratne, V.; Rawji, G.; Wheeler, J.; Sutton, K.; et al. Preparation and characterization of vanadyl complexes with bidentate maltol-type ligands; in vivo comparisons of anti-diabetic therapeutic potential. J. Biol. Inorg. Chem. 2003, 8, 66–74. [Google Scholar] [CrossRef]
- Liboiron, B.D.; Thompson, K.H.; Hanson, G.R.; Lam, E.; Aebischer, N.; Orvig, C. New insights into the interactions of serum proteins with bis(maltolato)oxovanadium(IV): Transport and biotransformation of insulin-enhancing vanadium pharmaceuticals. J. Am. Chem. Soc. 2005, 127, 5104–5115. [Google Scholar] [CrossRef]
- Thompson, K.H.; Orvig, C. Vanadium in diabetes: 100 years from Phase 0 to Phase, I. J. Inorg. Biochem. 2006, 100, 1925–1935. [Google Scholar] [CrossRef]
- Thompson, K.H.; Lichter, J.; Lebel, C.; Scaife, M.C.; Mcneill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef]
- De La Torre, A.; Granero, S.; Mayayo, E.; Corbella, J.; Domingo, J.L. Effect of age on vanadium nephrotoxicity in rats. Toxicol. Lett. 1999, 105, 75–82. [Google Scholar] [CrossRef]
- Gaugler, J.; James, B.; Johnson, T.; Marin, A.; Weuve, J.; Assoc, A.S. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar]
- Huang, Y.D.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericakvance, M.A. Gene Dose of Apolipoprotein-E Type-4 Allele and the Risk of Alzheimers-Disease in Late-Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.Z.; Koger, D.; Delong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE Isoform-Dependent Effects on Brain beta-Amyloid Levels in PDAPP Transgenic Mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Lhotsky, A.; Wang, Y.F.; Dal Forno, G.; An, Y.; Metter, E.J.; Ferrucci, L.; O’brien, R.; Zonderman, A.B. Association of Adiposity Status and Changes in Early to Mid-Adulthood with Incidence of Alzheimer’s Disease. Am. J. Epidemiol. 2008, 168, 1179–1189. [Google Scholar] [CrossRef]
- Mcewen, B.S.; Reagan, L.P. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De La Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’connor, R.; Ravid, R.; O’neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Affholter, J.A.; Fried, V.A.; Roth, R.A. Human insulin-degrading enzyme shares structural and functional homologies with E. coli protease III. Science 1988, 242, 1415–1418. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Hamel, F.G.; Bennett, R.; Ryan, M.P.; Roth, R.A. Human red blood cell insulin-degrading enzyme and rat skeletal muscle insulin protease share antigenic sites and generate identical products from insulin. J. Biol. Chem. 1990, 265, 2984–2987. [Google Scholar] [CrossRef]
- Affholter, J.A.; Hsieh, C.L.; Francke, U.; Roth, R.A. Insulin-Degrading Enzyme—Stable Expression of the Human Complementary-DNA, Characterization of Its Protein Product, and Chromosomal Mapping of the Human and Mouse Genes. Mol. Endocrinol. 1990, 4, 1125–1135. [Google Scholar] [CrossRef]
- Kuo, W.L.; Gehm, B.D.; Rosner, M.R. Regulation of insulin degradation: Expression of an evolutionarily conserved insulin-degrading enzyme increases degradation via an intracellular pathway. Mol. Endocrinol. 1991, 5, 1467–1476. [Google Scholar] [CrossRef][Green Version]
- Roth, R.A.; Mesirow, M.L.; Yokono, K.; Baba, S. Degradation of Insulin-Like Growth Factor-I and Factor-Ii by a Human Insulin Degrading Enzyme. Endocr. Res. 1984, 10, 101–112. [Google Scholar] [CrossRef]
- Ansorge, S.; Bohley, P.; Kirschke, H.; Langner, J.; Wiederanders, B. The Insulin and Glucagon Degrading Proteinase of Rat-Liver—Separation of the Proteinase from the Thiol-Proteindisulfide Oxidoreductases. Biomed. Biochim. Acta 1984, 43, 29–38. [Google Scholar]
- Kurochkin, I.V.; Goto, S. Alzheimers Beta-Amyloid Peptide Specifically Interacts with and Is Degraded by Insulin Degrading Enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef]
- Grasso, G.; Salomone, F.; Tundo, G.R.; Pappalardo, G.; Ciaccio, C.; Spoto, G.; Pietropaolo, A.; Coletta, M.; Rizzarelli, E. Metal ions affect insulin-degrading enzyme activity. J. Inorg. Biochem. 2012, 117, 351–358. [Google Scholar] [CrossRef][Green Version]
- Grasso, G.; Rizzarelli, E.; Spoto, G. How the binding and degrading capabilities of insulin degrading enzyme are-affected by ubiquitin. Biochim. Biophys. Acta-Proteins Proteom. 2008, 1784, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Hamel, F.G.; Upward, J.L.; Bennett, R.G. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology 2003, 144, 2404–2408. [Google Scholar] [CrossRef]
- Song, E.S.; Juliano, M.A.; Juliano, L.; Fried, M.G.; Wagner, S.L.; Hersh, L.B. ATP effects on insulin-degrading enzyme are mediated primarily through its triphosphate moiety. J. Biol. Chem. 2004, 279, 54216–54220. [Google Scholar] [CrossRef] [PubMed]
- Ralat, L.A.; Ren, M.; Schilling, A.B.; Tang, W.J. Protective Role of Cys-178 against the Inactivation and Oligomerization of Human Insulin-degrading Enzyme by Oxidation and Nitrosylation. J. Biol. Chem. 2009, 284, 34005–34018. [Google Scholar] [CrossRef]
- Shen, Y.Q.; Joachimiak, A.; Rosner, M.R.; Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef]
- Sousa, L.; Guarda, M.; Meneses, M.J.; Macedo, M.P.; Miranda, H.V. Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Kang, D.; Mcbride, M.; Li, L.L.; Zhao, J.; Leissring, M.A. Deletion of Insulin-Degrading Enzyme Elicits Antipodal, Age-Dependent Effects on Glucose and Insulin Tolerance. PLoS ONE 2011, 6, e20818. [Google Scholar] [CrossRef] [PubMed]
- Rudovich, N.; Pivovarova, O.; Fisher, E.; Fischer-Rosinsky, A.; Spranger, J.; Mohlig, M.; Schulze, M.B.; Boeing, H.; Pfeiffer, A.F. Polymorphisms within insulin-degrading enzyme (IDE) gene determine insulin metabolism and risk of type 2 diabetes. J. Mol. Med. 2009, 87, 1145–1151. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, D.; Huang, H.; Zhao, Y.; Zhou, H. Characteristics of Insulin-degrading Enzyme in Alzheimer’s Disease: A Meta-Analysis. Curr. Alzheimer Res. 2018, 15, 610–617. [Google Scholar] [CrossRef]
- Morelli, L.; Llovera, R.E.; Mathov, I.; Lue, L.F.; Frangione, B.; Ghiso, J.; Castano, E.M. Insulin-degrading enzyme in brain microvessels: Proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J. Biol. Chem. 2004, 279, 56004–56013. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3 beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef]
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 2002, 81, 1073–1083. [Google Scholar] [CrossRef]
- Wang, Q.M.; Fiol, C.J.; Depaoliroach, A.A.; Roach, P.J. Glycogen-Synthase Kinase-3-Beta Is a Dual-Specificity Kinase Differentially Regulated by Tyrosine and Serine/Threonine Phosphorylation. J. Biol. Chem. 1994, 269, 14566–14574. [Google Scholar] [CrossRef]
- Hughes, K.; Nikolakaki, E.; Plyte, S.E.; Totty, N.F.; Woodgett, J.R. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993, 12, 803–808. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3 beta inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, J.; Liu, Y.J.; Chen, X.; Yu, T.X.; Jia, J.H.; Liu, C.M. PR55 alpha, a Regulatory Subunit of PP2A, Specifically Regulates PP2A-mediated beta-Catenin Dephosphorylation. J. Biol. Chem. 2009, 284, 22649–22656. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Harwood, A.J. Regulation of GSK-3: A cellular multiprocessor. Cell 2001, 105, 821–824. [Google Scholar] [CrossRef]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharmacol. 2009, 156, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Ter Haar, E.; Coll, J.T.; Austen, D.A.; Hsiao, H.M.; Swenson, L.; Jain, J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001, 8, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Bouche, C.; Serdy, S.; Kahn, C.R.; Goldfine, A.B. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 2004, 25, 807–830. [Google Scholar] [CrossRef] [PubMed]
- Rokutanda, S.; Fujita, T.; Kanatani, N.; Yoshida, C.A.; Komori, H.; Liu, W.G.; Mizuno, A.; Komori, T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009, 328, 78–93. [Google Scholar] [PubMed]
- Avila, J.; Leon-Espinosa, G.; Garcia, E.; Garcia-Escudero, V.; Hernandez, F.; Defelipe, J. Tau Phosphorylation by GSK3 in Different Conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef] [PubMed]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Schreyer, S.A.; Shinohara, M.M.; Leboeuf, R.C.; Krebs, E.G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999, 48, 1662–1666. [Google Scholar] [CrossRef]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3 beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; Aoyagi, N.; Ando, K.; Shimohama, S.; Kinoshita, A. GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 2007, 282, 15823–15832. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.T.; Wu, Y.L.; Zou, H.Y.; Wang, R.T.; Zhou, W.H.; Kinoshita, A.; Zhang, M.M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3 beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B.D.; Babu-Khan, S.; Denis, P.; Fan, W.; Kha, H.; Zhang, J.H.; Gong, Y.H.; et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001, 4, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Lee VM, Y. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Perez, M.; Hernandez, F.; Lim, F.; Diaz-Nido, J.; Avila, J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimer’s Dis. 2003, 5, 301–308. [Google Scholar] [CrossRef]
- Hu, S.X.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.S.; Chen, P.P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar] [PubMed]
- Sereno, L.; Coma, M.; Rodriguez, M.; Sanchez-Ferrer, P.; Sanchez, M.B.; Gich, I.; Agullo, J.M.; Perez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.X.; Bai, T.; Sun, Y.L. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Canturk, Z.; Cetinarslan, B.; Tarkun, I.; Canturk, N.Z. Serum ferritin levels in poorly- and well-controlled diabetes mellitus. Endocr. Res. 2003, 29, 299–306. [Google Scholar] [PubMed]
- Altamura, S.; Kopf, S.; Schmidt, J.; Mudder, K.; Da Silva, A.R.; Nawroth, P.; Muckenthaler, M.U. Uncoupled iron homeostasis in type 2 diabetes mellitus. J. Mol. Med. 2017, 95, 1387–1398. [Google Scholar] [PubMed]
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; Mcfarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626. [Google Scholar] [CrossRef]
- Al-Khaldi, A.; Sultan, S. The expression of sirtuins, superoxide dismutase, and lipid peroxidation status in peripheral blood from patients with diabetes and hypothyroidism. BMC Endocr. Disord. 2019, 19, 19. [Google Scholar]
- Shu, T.T.; Lv, Z.G.; Xie, Y.C.; Tang, J.M.; Mao, X.H. Hepcidin as a key iron regulator mediates glucotoxicity-induced pancreatic beta-cell dysfunction. Endocr. Connect. 2019, 8, 150–161. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Paul, S.M.; Demattos, R.B. A beta immunization and anti-A beta antibodies: Potential therapies for the prevention and treatment of Alzheimer’s disease. Adv. Drug Deliv. Rev. 2002, 54, 1603–1613. [Google Scholar]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef]
- Moss, D.W.; Bates, T.E. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur. J. Neurosci. 2001, 13, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Zhu, X.W.; Tabaton, M.; Liu, G.; Mckeel, D.W.; Cohen, M.L.; Wang, X.L.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased Iron and Free Radical Generation in Preclinical Alzheimer Disease and Mild Cognitive Impairment. J. Alzheimer’s Dis. 2010, 19, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased Iron Levels and Decreased Tissue Integrity in Hippocampus of Alzheimer’s Disease Detected in vivo with Magnetic Resonance Imaging. J. Alzheimer’s Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef]
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-Dependent and Tissue-Related Glutathione Redox Status in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 28, 655–666. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.D.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wu, Q.Y.; Chen, C.; Zheng, R.; Chen, Y.; Ni, J.Z.; Song, G.L. Comparison of the effects of selenomethionine and selenium-enriched yeast in the triple-transgenic mouse model of Alzheimer’s disease. Food Funct. 2018, 9, 3965–3973. [Google Scholar] [PubMed]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Rudinskiy, N.; Herrmann, A.G.; Croft, S.; Kim, J.M.; Petrova, V.; Ramos-Rodriguez, J.J.; Pitstick, R.; Wegmann, S.; Garcia-Alloza, M.; et al. Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2016, 44, 3056–3066. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, R.; Kim, H.D.; Maxwell, J.A.; Fukuchi, K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/Delta E9 mice with amyloid plaques. Neurosci. Lett. 2005, 390, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; Laferla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Stover, K.R.; Campbell, M.A.; Van Winssen, C.M.; Brown, R.E. Early detection of cognitive deficits in the 3xTg-AD mouse model of Alzheimer’s disease. Behav. Brain Res. 2015, 289, 29–38. [Google Scholar] [CrossRef]
- He, Z.J.; Han, S.X.; Wu, C.; Liu, L.N.; Zhu, H.Z.; Liu, A.; Lu, Q.Y.; Huang, J.Q.; Du, X.B.; Li, N.; et al. Bis(ethylmaltolato)oxidovanadium(iv) inhibited the pathogenesis of Alzheimer’s disease in triple transgenic model mice. Metallomics 2020, 12, 631. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Li, G.X.; Mu, Y.; Wu, W.X.; Cao, B.X.; Wang, Z.X.; Yu, H.N.; Guan, P.P.; Han, L.; Li, L.Y.; et al. Discovery of Anti-TNBC Agents Targeting PTP1B: Total Synthesis, Structure-Activity Relationship, In Vitro and In Vivo Investigations of Jamunones. J. Med. Chem. 2021, 64, 6008–6020. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A. Insulin resistance, type 2 diabetes, and AD Cerebrovascular disease or neurodegeneration? Neurology 2010, 75, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Bailey, P.; Biswas, C.; Cullinan, C.A.; Doebber, T.W.; Hayes, N.S.; Saperstein, R.; Smith, R.G.; Leibowitz, M.D. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: Binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 1996, 137, 4189–4195. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Davies, P.J.A.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, L.; Liu, S.B.; Zhang, C.; Huang, X.Q.; Li, J.; Zhao, N.M.; Wang, Z. PPAR gamma transcriptionally regulates the expression of insulin-degrading enzyme in primary neurons. Biochem. Biophys. Res. Commun. 2009, 383, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Shioda, N.; Moriguchi, S.; Qin, Z.H.; Fukunaga, K. The vanadium (IV) compound rescues septo-hippocampal cholinergic neurons from neurodegeneration in olfactory bulbectomized mice. Neuroscience 2008, 151, 671–679. [Google Scholar]
- Liu, X.Y.; Zhang, L.J.; Chen, Z.; Liu, L.B. The PTEN inhibitor bpV(pic) promotes neuroprotection against amyloid beta-peptide (25–35)-induced oxidative stress and neurotoxicity. Neurol. Res. 2017, 39, 758–765. [Google Scholar] [CrossRef]
- Prasad, S.; Dubourdieu, D.; Srivastava, A.; Kumar, P.; Lall, R. Metal-Curcumin Complexes in Therapeutics: An Approach to Enhance Pharmacological Effects of Curcumin. Int. J. Mol. Sci. 2021, 22, 7094. [Google Scholar]
- Majithiya, J.B.; Balaraman, R.; Giridhar, R.; Yadav, M.R. Effect of bis[curcumino]oxovanadium complex on non-diabetic and streptozotocin-induced diabetic rats. J. Trace Elem. Med. Biol. 2005, 18, 211–217. [Google Scholar] [CrossRef]
- Nishiyama, T.; Mae, T.; Kishida, H.; Tsukagawa, M.; Mimaki, Y.; Kuroda, M.; Sashida, Y.; Takahashi, K.; Kawada, T.; Nakagawa, K.; et al. Curcuminoids and sesquiterpenoids in turmeric (Curcuma longa L.) suppress an increase in blood glucose level in type 2 diabetic KK-Ay mice. J. Agric. Food Chem. 2005, 53, 959–963. [Google Scholar] [CrossRef]
- Pradhan, A.; Mishra, S.; Surolia, A.; Panda, D. C1 Inhibits Liquid-Liquid Phase Separation and Oligomerization of Tau and Protects Neuroblastoma Cells against Toxic Tau Oligomers. ACS Chem. Neurosci. 2021, 12, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Correia, I.; Chorna, I.; Cavaco, I.; Roy, S.; Kuznetsov, M.L.; Ribeiro, N.; Justino, G.; Marques, F.; Santos-Silva, T.; Santos, M.F.A.; et al. Interaction of [V(IV) O(acac)2] with Human Serum Transferrin and Albumin. Chem. Asian J. 2017, 12, 2062–2084. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name AD Models | AD Pathology in Models | The Effects of Vanadium Compounds |
|---|---|---|
| N2A cell line Swedish mutation of APP | Increased Ab burden | BEOV decreased Ab level [13,15], increased the expression of PPARg |
| SY5Y cell line with Swedish mutation of APP | Increased Ab burden | VAC elevated the levels of PPARg, AMPKa and GSK-3b [12] |
| Tg(APPswe, PSEN1dE9) | Ab senile plaques, spatial learning impaired begin on 12 months of age. | BEOV [13,15] and VAC [12] improved spatial learning activity in Morris water maze, decreased Ab level, increased neuron viability. |
| Tg(APPSwe, TauP301L) | Ab senile plaques and tau filaments tangles, spatial learning activity impaired begin on 6.5 months of age. | BEOV improved spatial learning activity in Morris water maze, decreased Ab level and tau phosphorylation, increased neuron viability [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; You, G.; Liu, Q.; Li, N. Alzheimer’s Disease and Diabetes Mellitus in Comparison: The Therapeutic Efficacy of the Vanadium Compound. Int. J. Mol. Sci. 2021, 22, 11931. https://doi.org/10.3390/ijms222111931
He Z, You G, Liu Q, Li N. Alzheimer’s Disease and Diabetes Mellitus in Comparison: The Therapeutic Efficacy of the Vanadium Compound. International Journal of Molecular Sciences. 2021; 22(21):11931. https://doi.org/10.3390/ijms222111931
Chicago/Turabian StyleHe, Zhijun, Guanying You, Qiong Liu, and Nan Li. 2021. "Alzheimer’s Disease and Diabetes Mellitus in Comparison: The Therapeutic Efficacy of the Vanadium Compound" International Journal of Molecular Sciences 22, no. 21: 11931. https://doi.org/10.3390/ijms222111931
APA StyleHe, Z., You, G., Liu, Q., & Li, N. (2021). Alzheimer’s Disease and Diabetes Mellitus in Comparison: The Therapeutic Efficacy of the Vanadium Compound. International Journal of Molecular Sciences, 22(21), 11931. https://doi.org/10.3390/ijms222111931