
Sulfonylurea Receptor 1 in Central Nervous System Injury: An Updated Review

 ,
,
Abstract
1. Introduction
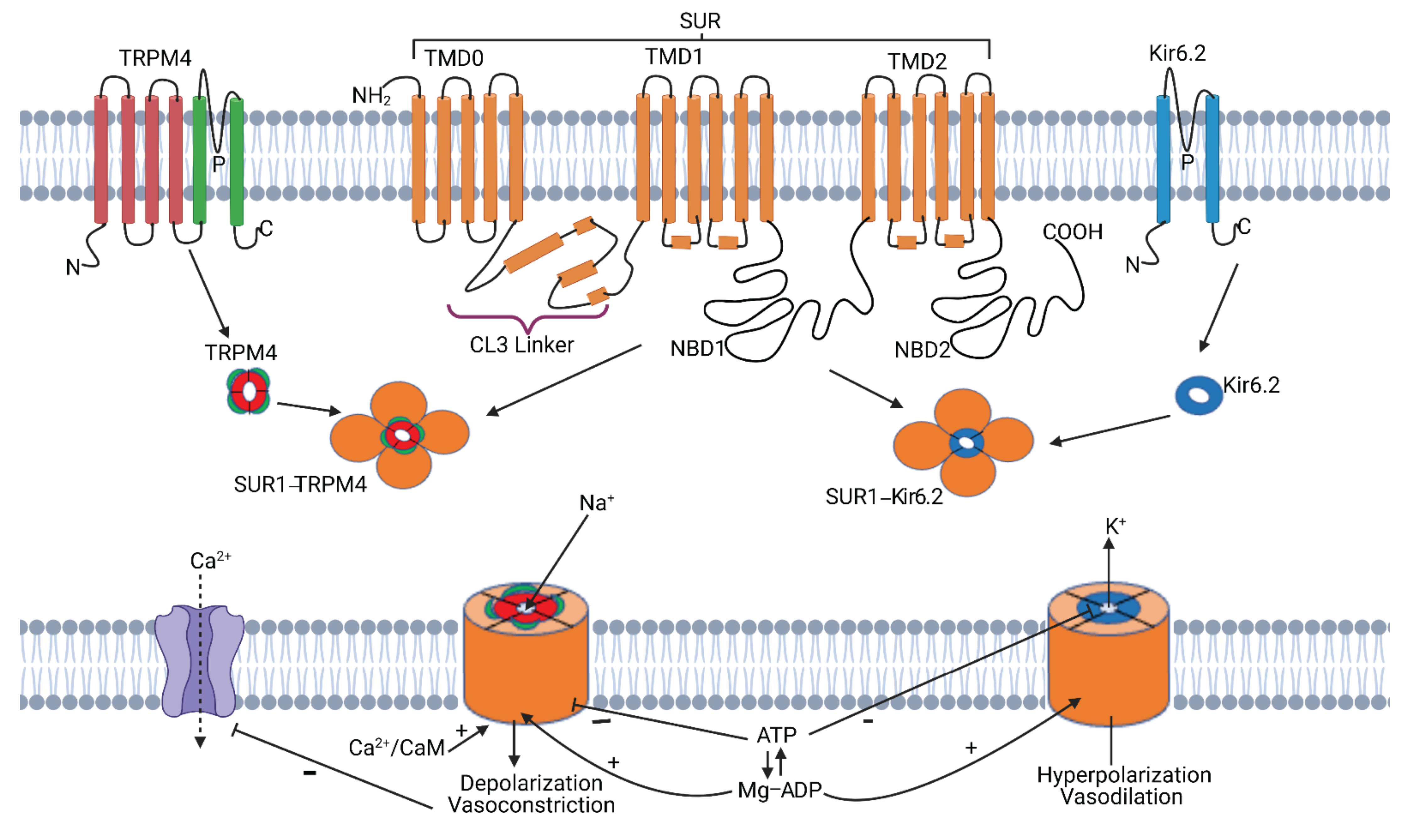
2. SUR1-TRPM4
2.1. SUR1-TRPM4 Structure
2.2. SUR1-TRPM4—Discovery and Function
2.3. SUR1-TRPM4: Biophysical and Pharmacological Properties
2.4. SUR1-TRPM4: Transcriptional Regulation
2.4.1. Hif1a and Sp1
2.4.2. TNFa and NF-κB
2.4.3. Toll-like Receptor (TLR)-4
2.5. SUR1 Pathways
2.5.1. Cerebral Edema Pathways
2.5.2. BBB Permeability Pathways
2.5.3. Neuroinflammation Pathways
2.5.4. Cell Death Pathways
3. SUR1-TRPM4 Expression and Inhibition in CNS injury
3.1. Ischemic Stroke
3.1.1. Ischemic Stroke—Preclinical Studies
Ischemic Stroke—In Vitro Studies
Ischemic Stroke—In Vivo Expression Patterns
Ischemic Stroke—In Vivo Channel Blockade
3.1.2. Ischemic Stroke—Human Studies
Ischemic Stroke—Human Expression Patterns
Ischemic Stroke—Clinical Retrospective Studies
Ischemic Stroke—Clinical Trials
3.2. TBI
3.2.1. TBI—Preclinical Studies
TBI—In Vivo Expression Patterns
TBI—In Vivo Channel Blockade
3.2.2. TBI—Clinical Studies
TBI—Human Expression Patterns
TBI—Human Genetic Variation
TBI—Clinical Trials
3.3. SCI
3.3.1. SCI—Preclinical Studies
SCI—In Vivo Expression Patterns
SCI—In Vivo Channel Blockade
3.3.2. SCI—Clinical Studies
SCI—Human Expression Patterns
SCI—Clinical Trials
3.4. SAH
3.4.1. SAH—Preclinical Studies
SAH—In Vivo Expression Patterns
SAH—In Vivo Channel Blockade
3.4.2. SAH—Clinical Studies
SAH—Human Expression Patterns
SAH—Clinical Trials
3.5. Cardiac Arrest
3.5.1. Cardiac Arrest—Preclinical Studies
Cardiac Arrest—In Vivo Expression Patterns
Cardiac Arrest—In Vivo Channel Blockade
3.5.2. Cardiac Arrest—Clinical Studies
Cardiac Arrest—Human Expression Patterns
Cardiac Arrest—Clinical Trials
3.6. ICH
3.6.1. ICH—Preclinical Studies
ICH—In Vivo Expression Patterns
ICH—In Vivo Channel Blockade
3.6.2. ICH—Clinical Studies
ICH—Human Expression Patterns
ICH—Clinical Trials
3.7. Multiple Sclerosis (MS) and Experimental Autoimmune Encephalitis (EAE)
3.7.1. MS and EAE—Preclinical Studies
EAE—In Vivo Expression Patterns
EAE—In Vivo Channel Blockade
3.7.2. MS and EAE—Clinical Studies
MS—Human Expression Patterns
MS—Clinical Trials
3.8. Neuro-Oncology
3.8.1. Neuro-Oncology—Preclinical Studies
Neuro-Oncology—In Vivo Expression Patterns
Neuro-Oncology—In Vivo Channel Blockade
3.8.2. Neuro-Oncology—Clinical Studies
Neuro-Oncology—Human Expression Patterns
Neuro-Oncology—Clinical Trials
3.9. Acute Liver Failure (ALF)
3.9.1. ALF—Preclinical Studies
ALF—In Vitro Studies
ALF—In Vivo Expression Pattern
ALF—In Vivo Channel Blockade
3.9.2. ALF—Clinical Studies
3.10. Status Epilepticus
3.10.1. Status Epilepticus–Preclinical Studies
Status Epilepticus—In Vivo Expression Patterns
Status Epilepticus—In Vivo Channel Blockade
3.10.2. Status Epilepticus—Clinical Studies
4. SUR1-TRPM4 Expression and Inhibition in Other Neurological Conditions
4.1. Neuropathic Pain
4.1.1. Neuropathic Pain—Preclinical Studies
Neuropathic Pain—In Vivo Expression Patterns
Neuropathic Pain—In Vivo Channel Blockade
4.1.2. Neuropathic Pain—Clinical Studies
Neuropathic Pain—Human Expression Patterns
Neuropathic Pain—Clinical Trials
4.2. HIV-Associated Neurocognitive Disorders (HAND)
4.2.1. HAND—Preclinical Studies
HAND—In Vivo Expression Patterns
HAND—In Vitro Channel Blockade
4.2.2. HAND—Clinical Studies
HAND—Human Expression Patterns
HAND—Clinical Trials
4.3. Retinopathy
4.3.1. Retinopathy—Preclinical Studies
Retinal Preclinical SUR1 Expression Patterns
Retinopathy—In Vivo Channel Blockade
4.3.2. Retinal Expression—Clinical Studies
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADC | Apparent Diffusion Coefficient |
| ALF | Acute Liver Failure |
| AQP4 | Aquaporin 4 |
| ASPECTS | Alberta Stroke Program Early CT Score |
| ASTRAL | Antagonizing SUR1-TRPM4 To Reduce the progression of intracerebral Hematoma and edema surrounding Lesions |
| ATP | Adenosine Triphosphate |
| BAX | Bcl-Associated X protein |
| BBB | Blood–Brain Barrier |
| BSCB | Blood–Spinal Cord Barrier |
| BTB | Brain–Tumor Barrier |
| CAMKII | Calmodulin-Dependent Protein Kinase II |
| CCI | Controlled Cortical Impact |
| CHARM | Ciara in Large Hemispheric Infarction Analyzing Modified Rankin and Mortality |
| CNS | Central Nervous System |
| COX2 | Cyclooxygenase 2 |
| CSF | Cerebrospinal Fluid |
| DC | Decompressive Craniectomy |
| FLAIR | Fluid-Attenuated Inversion Recovery |
| FPI | Fluid Percussion Injury |
| FRET | Förster Resonance Energy Transfer |
| GAIN | Genetic Association in Neurotrauma |
| GAMES | Glyburide Advantage in Malignant Edema and Stroke |
| GASH | Glibenclamide in Aneurysmatic Subarachnoid Hemorrhage |
| GCS | Glasgow Coma Scale |
| GFAP | Glial Fibrillary Acidic Protein |
| GOS | Glasgow Outcome Scale |
| HAND | HIV Associated Neurocognitive Disorder |
| HIF1α | Hypoxia inducible factor 1α |
| ICH | Intracerebral Hemorrhage |
| IFNγ | Interferon-γ |
| IL-17 | Interleukin-17 |
| iNOS | inducible Nitric Oxide Synthase |
| LHI | Large Hemispheric Infarction |
| MA | Mithramycin-A |
| MCAO | Middle Cerebral Artery Occlusion |
| MMP-9 | Matrix Metalloproteinase-9 |
| MRI | Magnetic Resonance Imaging |
| NeuN | Neuronal Nuclear Protein |
| NF-κB | Nuclear Factor κ- Light-Chain Enhancer of Activated B cells |
| NO | Nitric Oxide |
| NOS2 | Nitric Oxide Synthase 2 |
| NBS | Nucleotide Binding Site |
| NIHSS | National Institute of Health Stroke Scale |
| OBTT | Operation Brain Trauma Therapy |
| PACAP | Pituitary Adenylate Cyclase-Activating Polypeptide |
| PHN | Progressive Hemorrhagic Necrosis |
| SAH | Subarachnoid Hemorrhage |
| SCI | Spinal Cord Injury |
| SCING | Spinal Cord Injury Neuroprotection with Glyburide |
| Sp1 | Specificity Protein 1 |
| SUR1 | Sulfonylurea Receptor 1 |
| TAA | Thioacetamide |
| TBI | Traumatic Brain Injury |
| TLR4 | Toll-like Receptor 4 |
| tPA | Tissue Plasminogen Activator |
| TNFα | Tumor Necrosis factor α |
| TRPM4 | Transient Receptor Potential Melastatin 4 |
| VISTA | Virtual International Stroke Trial Archive |
| vWF | Von Willebrand Factor |
| ZO-1 | Zona Occludens 1 |
References
- Shi, N.-Q.; Ye, B.; Makielski, J.C. Function and distribution of the SUR isoforms and splice variants. J. Mol. Cell. Cardiol. 2005, 39, 51–60. [Google Scholar] [CrossRef]
- Aittoniemi, J.; Fotinou, C.; Craig, T.J.; de Wet, H.; Proks, P.; Ashcroft, F.M. SUR1: A unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. B Biol. Sci. 2008, 364, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Deeley, R.G.; Westlake, C.; Cole, S.P.C. Transmembrane Transport of Endo- and Xenobiotics by Mammalian ATP-Binding Cassette Multidrug Resistance Proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Miki, T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 2003, 81, 133–176. [Google Scholar] [CrossRef]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The sulfonylurea receptor 1 (Sur1)-transient receptor potential me-lastatin 4 (Trpm4) channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Schwartzbauer, G.T.; Gerzanich, V. Sulfonylurea Receptor 1 in Central Nervous System Injury: A Focused Review. Br. J. Pharmacol. 2012, 32, 1699–1717. [Google Scholar] [CrossRef]
- Chen, M.; B, J.M. Cell Swelling and a Nonselective Cation Channel Regulated by Internal Ca2+ and ATP in Native Reactive Astrocytes from Adult Rat Brain. J. Neurosci. 2001, 21, 6512–6521. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Dong, Y.; Simard, J.M. Functional Coupling between Sulfonylurea Receptor Type 1 and a Nonselective Cation Channel in Reactive Astrocytes from Adult Rat Brain. J. Neurosci. 2003, 23, 8568–8577. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Aarabi, B.; Gerzanich, V. The Sur1-Trpm4 Channel in Spinal Cord Injury. J. Spine 2013. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Bell, J.; Citerio, G.; Hemphill, J.C.; Kimberly, W.T.; Narayan, R.K.; Sahuquillo, J.; Sheth, K.N.; Simard, J.M. Role of Sulfonylurea Receptor 1 and Glibenclamide in Traumatic Brain Injury: A Review of the Evidence. Int. J. Mol. Sci. 2020, 21, 409. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. Br. J. Pharmacol. 2015, 36, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood–Brain Barrier Breakdown in Acute and Chronic Cerebrovascular Disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef]
- Griepp, D.W.; Lee, J.; Moawad, C.M.; Davati, C.; Runnels, J.; Fiani, B. BIIB093 (intravenous glibenclamide) for the prevention of severe cerebral edema. Surg. Neurol. Int. 2021, 12, 80. [Google Scholar] [CrossRef]
- Zusman, B.E.; Kochanek, P.M.; Jha, R.M. Cerebral Edema in Traumatic Brain Injury: A Historical Framework for Current Therapy. Curr. Treat. Options Neurol. 2020, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Kent, T.; Chen, M.; Tarasov, K.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef]
- Cooper, D.J.; Rosenfeld, J.V.; Murray, L.; Arabi, Y.; Davies, A.R.; D’Urso, P.; Kossmann, T.; Ponsford, J.; Seppelt, I.; Reilly, P.; et al. Decompressive Craniectomy in Diffuse Traumatic Brain Injury. N. Engl. J. Med. 2011, 364, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef]
- Kamel, H.; Navi, B.B.; Nakagawa, K.; Hemphill, J.C.; Ko, N.U. Hypertonic saline versus mannitol for the treatment of elevated in-tracranial pressure: A meta-analysis of randomized clinical trials. Crit. Care Med. 2011, 39, 554–559. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M. A Precision Medicine Approach to Cerebral Edema and Intracranial Hypertension after Severe Traumatic Brain Injury: Quo Vadis? Curr. Neurol. Neurosci. Rep. 2018, 18, 105. [Google Scholar] [CrossRef]
- Nielson, J.L.; Cooper, S.R.; Yue, J.K.; Sorani, M.D.; Inoue, T.; Yuh, E.L.; Mukherjee, P.; Petrossian, T.C.; Paquette, J.; Lum, P.Y.; et al. Uncovering Precision Phenotype-Biomarker Associations in Traumatic Brain Injury Using Topological Data Analysis. PLoS ONE 2017, 12, e0169490. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M. Adding insight to injury: A new era in neurotrauma. Lancet Neurol. 2017, 16, 578–580. [Google Scholar] [CrossRef]
- Stocchetti, N.; Carbonara, M.; Citerio, G.; Ercole, A.; Skrifvars, M.B.; Smielewski, P.; Zoerle, T.; Menon, D.K. Severe Traumatic Brain Injury: Targeted Management in the Intensive Care Unit. Lancet Neurol. 2017, 16, 452–464. [Google Scholar] [CrossRef]
- Jha, R.M.; Puccio, A.M.; Chou, S.H.-Y.; Chang, C.-C.H.; Wallisch, J.S.; Molyneaux, B.J.; Zusman, B.E.; Shutter, L.A.; Poloyac, S.M.; Janesko-Feldman, K.L.; et al. Sulfonylurea Receptor-1. Crit. Care Med. 2017, 45, e255–e264. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Battey, T.W.K.; Pham, L.; Wu, O.; Yoo, A.J.; Furie, K.L.; Singhal, A.B.; Elm, J.J.; Stern, B.J.; Sheth, K.N. Glyburide Is Associated with Attenuated Vasogenic Edema in Stroke Patients. Neurocrit. Care 2013, 20, 193–201. [Google Scholar] [CrossRef] [PubMed]
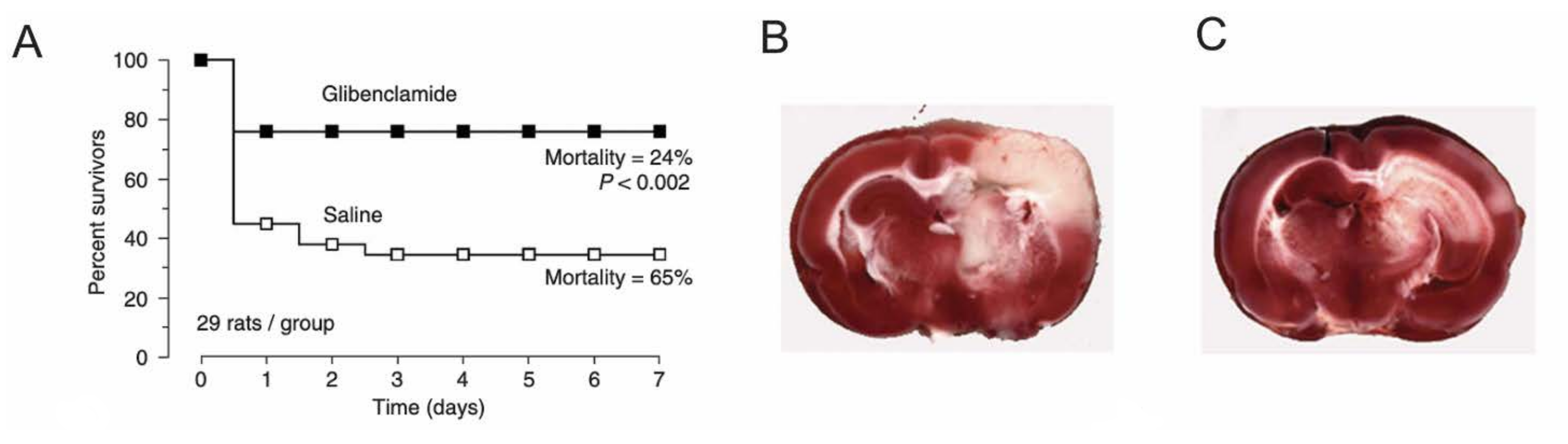
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key Role of Sulfonylurea Receptor 1 in Progressive Secondary Hemorrhage after Brain Contusion. J. Neurotrauma 2009, 26, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Norenberg, M.D.; Tosun, C.; Chen, Z.; Ivanova, S.; Tsymbalyuk, O.; Bryan, J.; Landsman, D.; Gerzanich, V. Brief Suppression of Abcc8 Prevents Autodestruction of Spinal Cord After Trauma. Sci. Transl. Med. 2010, 2, 28–29. [Google Scholar] [CrossRef]
- Simard, J.M.; Tsymbalyuk, O.; Ivanov, A.; Ivanova, S.; Bhatta, S.; Geng, Z.; Woo, S.K.; Gerzanich, V. Endothelial Sulfonylurea Receptor 1–Regulated NCCa-ATP Channels Mediate Progressive Hemorrhagic Necrosis Following Spinal Cord Injury. J. Clin. Invest. 2007, 117, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Gerzanich, V.; Stokum, J.A.; Ivanova, S.; Woo, S.K.; Tsymbalyuk, O.; Sharma, A.; Akkentli, F.; Imran, Z.; Aarabi, B.; Sahuquillo, J.; et al. Sulfonylurea Receptor 1, Transient Receptor Potential Cation Channel Subfamily M Member 4, and KIR6.2: Role in Hemorrhagic Progression of Contusion. J. Neurotrauma 2019, 36, 1060–1079. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.D.; Gerzanich, V.; Geng, Z.; Simard, J.M. Glibenclamide Reduces Hippocampal Injury and Preserves Rapid Spatial Learning in a Model of Traumatic Brain Injury. J. Neuropathol. Exp. Neurol. 2010, 69, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, H.M.; Shenton, M.E.; Pasternak, O.; Simard, J.M.; Okonkwo, D.O.; Aldrich, C.; He, F.; Jain, S.; Hayman, E.G. Magnetic Resonance Imaging Pilot Study of Intravenous Glyburide in Traumatic Brain Injury. J. Neurotrauma 2020, 37, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Molyneaux, B.J.; Jackson, T.C.; Wallisch, J.; Park, S.-Y.; Poloyac, S.; Vagni, V.A.; Janesko-Feldman, K.L.; Hoshitsuki, K.; Minnigh, M.B.; et al. Glibenclamide Produces Region-Dependent Effects on Cerebral Edema in a Combined Injury Model of Traumatic Brain Injury and Hemorrhagic Shock in Mice. J. Neurotrauma 2018, 35, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Mondello, S.; Bramlett, H.M.; Dixon, C.E.; Shear, D.A.; Dietrich, W.D.; Wang, K.K.; Yang, Z.; Hayes, R.L.; Poloyac, S.M.; et al. Glibenclamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2021, 38, 628–645. [Google Scholar] [CrossRef] [PubMed]
- Zusman, B.E.; Kochanek, P.M.; Bell, M.J.; Adelson, P.D.; Wisniewski, S.R.; Au, A.K.; Clark, R.S.; Bayır, H.; Janesko-Feldman, K.; Jha, R.M. Cerebrospinal Fluid Sulfonylurea Receptor-1 is Associated with Intracranial Pressure and Outcome after Pediatric TBI: An Exploratory Analysis of the Cool Kids Trial. J. Neurotrauma 2021, 38, 1615–1619. [Google Scholar] [CrossRef] [PubMed]
- Tata, S.; Zusman, B.E.; Kochanek, P.M.; Gerzanich, V.; Kwon, M.S.; Woo, S.K.; Clark, R.S.B.; Janesko-Feldman, K.; Vagni, V.A.; Simard, J.M.; et al. Abcc8 (Sulfonylurea Receptor-1) Impact on Brain Atrophy after Traumatic Brain Injury Varies by Sex. J. Neurotrauma 2021, 38, 2473–2485. [Google Scholar] [CrossRef] [PubMed]
- Tosun, C.; Kurland, D.B.; Mehta, R.; Castellani, R.J.; Dejong, J.; Kwon, M.S.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Inhibition of the Sur1-Trpm4 Channel Reduces Neuroinflammation and Cognitive Impairment in Subarachnoid Hemorrhage. Stroke 2013, 44, 3522–3528. [Google Scholar] [CrossRef]
- Dundar, T.T.; Abdallah, A.; Yurtsever, I.; Guler, E.M.; Ozer, O.F.; Uysal, O. Serum SUR1 and TRPM4 in patients with subarachnoid hemorrhage. Neurosurg. Rev. 2019, 43, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Valverde, T.; Vidal-Jorge, M.; Martínez-Saez, E.; Castro, L.; Arikan, F.; Cordero, E.; Rădoi, A.; Poca, M.-A.; Simard, J.M.; Sahuquillo, J. Sulfonylurea Receptor 1 in Humans with Post-Traumatic Brain Contusions. J. Neurotrauma 2015, 32, 1478–1487. [Google Scholar] [CrossRef]
- Thompson, E.M.; Halvorson, K.; McLendon, R. Sulfonylurea receptor 1 expression is variable in adult and pediatric brain tumors. Clin. Neuropathol. 2018, 37, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Geng, Z.; Woo, S.K.; Ivanova, S.; Tosun, C.; Melnichenko, L.; Gerzanich, V. Glibenclamide Reduces Inflammation, Vasogenic Edema, and Caspase-3 Activation after Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2008, 29, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Pishko, G.L.; Muldoon, L.L.; Neuwelt, E.A. Inhibition of SUR1 decreases the vascular permeability of cerebral me-tastases. Neoplasia 2013, 15, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Zafardoost, P.; Ghasemi, A.A.; Salehpour, F.; Piroti, C.; Ziaeii, E. Evaluation of the Effect of Glibenclamide in Patients With Diffuse Axonal Injury Due to Moderate to Severe Head Trauma. Trauma Mon. 2016, 21, e25113. [Google Scholar] [CrossRef] [PubMed]
- Zweckberger, K.; Hackenberg, K.; Jung, C.; Hertle, D.; Kiening, K.; Unterberg, A.; Sakowitz, O. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience 2014, 272, 199–206. [Google Scholar] [CrossRef]
- Xu, Z.-M.; Yuan, F.; Liu, Y.-L.; Ding, J.; Tian, H.-L. Glibenclamide Attenuates Blood–Brain Barrier Disruption in Adult Mice after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Keledjian, K.; Hayman, E.; Karimy, J.K.; Pampori, A.; Imran, Z.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Glibenclamide Pretreatment Protects against Chronic Memory Dysfunction and Glial Activation in Rat Cranial Blast Traumatic Brain Injury. Behav. Brain Res. 2017, 333, 43–53. [Google Scholar] [CrossRef]
- Pergakis, M.; Badjatia, N.; Chaturvedi, S.; Cronin, C.A.; Kimberly, W.T.; Sheth, K.N.; Simard, J.M. BIIB093 (IV glibenclamide): An investigational compound for the prevention and treatment of severe cerebral edema. Expert Opin. Investig. Drugs 2019, 28, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.N.; Elm, J.J.; Molyneaux, B.J.; Hinson, H.; Beslow, L.A.; Sze, G.K.; Ostwaldt, A.-C.; del Zoppo, G.J.; Simard, J.M.; Jacobson, S.; et al. Safety and Efficacy of Intravenous Glyburide on Brain Swelling after Large Hemispheric Infarction (GAMES-RP): A Randomised, Double-Blind, Placebo-Controlled Phase 2 Trial. Lancet Neurol. 2016, 15, 1160–1169. [Google Scholar] [CrossRef]
- Sheth, K.N.; Petersen, N.H.; Cheung, K.; Elm, J.J.; Hinson, H.E.; Molyneaux, B.J.; Beslow, L.A.; Sze, G.K.; Simard, J.M.; Kimberly, W.T. Long-Term Outcomes in Patients Aged ≤70 Years With Intravenous Glyburide From the Phase II GAMES-RP Study of Large Hemispheric Infarction. Stroke 2018, 49, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.N.; Kimberly, W.T.; Elm, J.J.; Kent, T.A.; Yoo, A.J.; Thomalla, G.; Campbell, B.; Donnan, G.A.; Davis, S.M.; Albers, G.W.; et al. Exploratory analysis of glyburide as a novel therapy for preventing brain swelling. Neurocrit. Care 2014, 21, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Patel, A.P.; Kimberly, W.T.; Sheth, K.N.; Simard, J.M. Cerebral edema after cardiopulmonary resuscitation: A thera-peutic target following cardiac arrest? Neurocrit. Care 2018, 28, 276–287. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P., IV; Boyd, A.E., III; González, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the β Cell High-Affinity Sulfonylurea Receptor: A Regulator of Insulin Secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Tampé, R. Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr. Opin. Struct. Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef]
- Tusnády, G.E.; Bakos, É.; Váradi, A.; Sarkadi, B. Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett. 1997, 402, 1–3. [Google Scholar] [CrossRef]
- Matsuo, M.; Kioka, N.; Amachi, T.; Ueda, K. ATP Binding Properties of the Nucleotide-binding Folds of SUR1. J. Biol. Chem. 1999, 274, 37479–37482. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. TRPM4 Is a Ca2+-Activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Nilius, B.; Prenen, J.; Tang, J.; Wang, C.; Owsianik, G.; Janssens, A.; Voets, T.; Zhu, M.X. Regulation of the Ca2+ Sensitivity of the Nonselective Cation Channel TRPM4. J. Biol. Chem. 2005, 280, 6423–6433. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Yuan, J.X.-J. Introduction to TRP Channels: Structure, Function, and Regulation. In Membrane Receptors, Channels and Transporters in Pulmonary Circulation; Springer: Berlin/Heidelberg, Germany, 2009; pp. 99–108. [Google Scholar] [CrossRef]
- Vennekens, R.; Nilius, B. Insights into TRPM4 Function, Regulation and Physiological Role. In Handbook of Experimental Pharmacology; Transient Receptor Potential (TRP) Channels; Humana Press: Totowa, NJ, USA, 2007; pp. 269–285. [Google Scholar] [CrossRef]
- Zerangue, N.; Schwappach, B.; Jan, Y.N.; Jan, L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 1999, 22, 537–548. [Google Scholar] [CrossRef]
- Luo, Z.-W.; Ovcjak, A.; Wong, R.; Yang, B.-X.; Feng, Z.-P.; Sun, H.-S. Drug development in targeting ion channels for brain edema. Acta Pharmacol. Sin. 2020, 41, 1272–1288. [Google Scholar] [CrossRef]
- Bryan, J.; Muñoz, A.; Zhang, X.; Düfer, M.; Drews, G.; Krippeit-Drews, P.; Aguilar-Bryan, L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflügers Arch.-Eur. J. Physiol. 2007, 453, 703–718. [Google Scholar] [CrossRef]
- Burke, M.A.; Mutharasan, R.K.; Ardehali, H. The Sulfonylurea Receptor, an Atypical ATP-Binding Cassette Protein, and Its Regulation of the KATPChannel. Circ. Res. 2008, 102, 164–176. [Google Scholar] [CrossRef]
- Yamada, K.; Inagaki, N. Neuroprotection by KATP channels. J. Mol. Cell. Cardiol. 2005, 38, 945–949. [Google Scholar] [CrossRef]
- Haider, S.; Antcliff, J.F.; Proks, P.; Sansom, M.S.; Ashcroft, F.M. Focus on Kir6.2: A key component of the ATP-sensitive potassium channel. J. Mol. Cell. Cardiol. 2005, 38, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Chen, M.; Tarasov, K.V.; Bhatta, S.; Ivanova, S.; Melnitchenko, L.; Tsymbalyuk, N.; West, G.A.; Gerzanich, V. Newly Expressed SUR1-Regulated NCCa-ATP Channel Mediates Cerebral Edema after Ischemic Stroke. Nat. Med. 2006, 12, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ämmälä, C.; Moorhouse, A.; Gribble, F.; Ashfleld, R.; Proks, P.; Smith, P.A.; Sakura, H.; Coles, B.; Ashcroft, S.J.H.; Ashcroft, F.M.; et al. Promiscuous coupling between the sulphonylurea receptor and inwardly rectifying potassium channels. Nature 1996, 379, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P., IV; Kunjilwar, K.; Gonzalez, G.; Schwanstecher, M.; Panten, U.; Aguilar-Bryan, L.; Bryan, J. Association and Stoichiometry of KATP Channel Subunits. Neuron 1997, 18, 827–838. [Google Scholar] [CrossRef]
- Mehta, R.I.; Tosun, C.; Ivanova, S.; Tsymbalyuk, N.; Famakin, B.M.; Kwon, M.S.; Castellani, R.J.; Gerzanich, V.; Simard, J.M. Sur1-Trpm4 Cation Channel Expression in Human Cerebral Infarcts. J. Neuropathol. Exp. Neurol. 2015, 74, 835–849. [Google Scholar] [CrossRef]
- Tsymbalyuk, O.; Gerzanich, V.; Mumtaz, A.; Andhavarapu, S.; Ivanova, S.; Makar, T.K.; Sansur, C.; Keller, A.; Nakamura, Y.; Bryan, J.; et al. SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol. Pain 2021, 17, 17448069211006603. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Wessell, A.; Gerzanich, V.; Sheth, K.N.; Simard, J.M. Mechanisms of Global Cerebral Edema Formation in Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care 2016, 26, 301–310. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2018, 145, 230–246. [Google Scholar] [CrossRef]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; Del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in cerebral ischemia and stroke. Neurocrit. Care 2013, 20, 319–333. [Google Scholar] [CrossRef]
- Simard, J.M.; Kahle, K.T.; Gerzanich, V. Molecular mechanisms of microvascular failure in central nervous system injury--synergistic roles of NKCC1 and SUR1/TRPM4. J. Neurosurg. 2010, 113, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Cartier, E.A.; Conti, L.R.; Vandenberg, C.A.; Shyng, S.L. Defective trafficking and function of KATP channels caused by a sulfonyl-urea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc. Natl. Acad. Sci. USA 2001, 98, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.-F.; Casey, J.; Shyng, S.-L. Sulfonylureas Correct Trafficking Defects of Disease-causing ATP-sensitive Potassium Channels by Binding to the Channel Complex. J. Biol. Chem. 2006, 281, 33403–33413. [Google Scholar] [CrossRef] [PubMed]
- Partridge, C.J.; Beech, D.; Sivaprasadarao, A. Identification and Pharmacological Correction of a Membrane Trafficking Defect Associated with a Mutation in the Sulfonylurea Receptor Causing Familial Hyperinsulinism. J. Biol. Chem. 2001, 276, 35947–35952. [Google Scholar] [CrossRef]
- Findlay, I. Effects of pH upon the inhibition by sulphonylurea drugs of ATP-sensitive K+ channels in cardiac muscle. J. Pharmacol. Exp. Ther. 1992, 262, 71–79. [Google Scholar]
- Nelson, D.A.; Bryan, J.; Wechsler, S.; Clement, J.P.; Aguilar-Bryan, L. The High-Affinity Sulfonylurea Receptor: Distribution, Glycosylation, Purification, and Immunoprecipitation of Two Forms from Endocrine and Neuroendocrine Cell Lines. Biochemistry 1996, 35, 14793–14799. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Kraig, R.P.; Tanabe, J.; Pulsinelli, W.A. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. Integr. Comp. Physiol. 1991, 260, R581–R588. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Woo, S.K.; Bhatta, S.; Gerzanich, V. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr. Opin. Pharmacol. 2008, 8, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ashfield, R.; Ashcroft, S.J. Cloning of the promoters for the beta-cell ATP-sensitive K-channel subunits Kir6.2 and SUR1. Diabetes 1998, 47, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Sánchez, C.; Ito, Y.; Ferrer, J.; Reitman, M.; LeRoith, D. Characterization of the Mouse Sulfonylurea Receptor 1 Promoter and Its Regulation. J. Biol. Chem. 1999, 274, 18261–18270. [Google Scholar] [CrossRef]
- Kim, J.-W.; Seghers, V.; Cho, J.-H.; Kang, Y.; Kim, S.; Ryu, Y.; Baek, K.; Aguilar-Bryan, L.; Lee, Y.-D.; Bryan, J.; et al. Transactivation of the Mouse Sulfonylurea Receptor I Gene by BETA2/NeuroD. Mol. Endocrinol. 2002, 16, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Gorogawa, S.; Fujitani, Y.; Kaneto, H.; Hazama, Y.; Watada, H.; Miyamoto, Y.; Takeda, K.; Akira, S.; Magnuson, M.A.; Yamasaki, Y.; et al. Insulin Secretory Defects and Impaired Islet Architecture in Pancreatic β-Cell-Specific STAT3 Knockout Mice. Biochem. Biophys. Res. Commun. 2004, 319, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Vatamaniuk, M.Z.; Brestelli, J.E.; Friedman, J.R.; Matschinsky, F.M.; Kaestner, K.H. Foxa2 regulates multiple pathways of insulin secretion. J. Clin. Investig. 2004, 114, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Geng, Z.; Chen, Z.; Ivanov, A.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Sequential Activation of Hypoxia-Inducible Factor 1 and Specificity Protein 1 Is Required for Hypoxia-Induced Transcriptional Stimulation of Abcc8. J. Cereb. Blood Flow Metab. 2011, 32, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Helton, R.; Cui, J.; Scheel, J.; Ellison, J.A.; Ames, C.; Gibson, C.; Blouw, B.; Ouyang, L.; Dragatsis, I.; Zeitlin, S.; et al. Brain-Specific Knock-Out of Hypoxia-Inducible Factor-1 Reduces Rather Than Increases Hypoxic-Ischemic Damage. J. Neurosci. 2005, 25, 4099–4107. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hu, Q.; Yan, J.; Lei, J.; Qin, L.; Shi, X.; Luan, L.; Yang, L.; Wang, K.; Han, J.; et al. Multiple Effects of 2ME2 and D609 on the Cortical Expression of HIF-1α and Apoptotic Genes in a Middle Cerebral Artery Occlusion-Induced Focal Ischemia Rat Model. J. Neurochem. 2007, 102, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ostrowski, R.P.; Zhou, C.; Tang, J.; Zhang, J.H. Suppression of hypoxia-inducible factor-1α and its downstream genes reduces acute hyperglycemia-enhanced hemorrhagic transformation in a rat model of cerebral ischemia. J. Neurosci. Res. 2010, 88, 2046–2055. [Google Scholar] [CrossRef]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1α inhibition ameliorates neonatal brain injury in a rat pup hypoxic–ischemic model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF-1 alpha inhibition ameliorates neonatal brain damage after hypoxic-ischemic injury. In Handbook of Experimental Pharmacology; Springer: Vienna, Austria, 2008; Volume 102, pp. 395–399. [Google Scholar] [CrossRef]
- Kurland, D.B.; Gerzanich, V.; Karimy, J.K.; Woo, S.K.; Vennekens, R.; Freichel, M.; Nilius, B.; Bryan, J.; Simard, J.M. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J. Neuroinflamm. 2016, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2013, 62, 26–38. [Google Scholar] [CrossRef]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of Toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef]
- Stokum, J.A.; Kwon, M.S.; Woo, S.K.; Tsymbalyuk, O.; Vennekens, R.; Gerzanich, V.; Simard, J.M. SUR1-TRPM4 and AQP4 Form a Heteromultimeric Complex That Amplifies Ion/Water Osmotic Coupling and Drives Astrocyte Swelling. Glia 2017, 66, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Gerzanich, V.; Kwon, M.S.; Woo, S.K.; Ivanov, A.; Simard, J.M. SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS ONE 2018, 13, e0195526. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Gerzanich, V.; Nimmagadda, V.K.C.; Jain, R.; Lam, K.; Mubariz, F.; Trisler, D.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; et al. Silencing of Abcc8 or Inhibition of Newly Upregulated Sur1-Trpm4 Reduce Inflammation and Disease Progression in Experimental Autoimmune Encephalomyelitis. J. Neuroinflamm. 2015, 12. [Google Scholar] [CrossRef]
- Gerzanich, V.; Makar, T.K.; Guda, P.R.; Kwon, M.S.; Stokum, J.A.; Woo, S.K.; Ivanova, S.; Ivanov, A.; Mehta, R.I.; Morris, A.B.; et al. Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2017, 14, 177. [Google Scholar] [CrossRef]
- Clément, T.; Rodriguez-Grande, B.; Badaut, J. Aquaporins in brain edema. J. Neurosci. Res. 2018, 98, 9–18. [Google Scholar] [CrossRef]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Mestre, H.; Mori, Y.; Nedergaard, M. The Brain’s Glymphatic System: Current Controversies. Trends Neurosci. 2020, 43, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Stæger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367, 6483. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Tornroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021. [Google Scholar] [CrossRef]
- MacAulay, N. Molecular mechanisms of brain water transport. Nat. Rev. Neurosci. 2021, 22, 326–344. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Iliff, J.J.; Bill, R.M. Aquaporin 4 and glymphatic flow have central roles in brain fluid homeostasis. Nat. Rev. Neurosci. 2021, 22, 650–651. [Google Scholar] [CrossRef]
- Fang, Y.; Shi, H.; Ren, R.; Huang, L.; Okada, T.; Lenahan, C.; Gamdzyk, M.; Travis, Z.D.; Lu, Q.; Tang, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide Attenuates Brain Edema by Protecting Blood–Brain Barrier and Glymphatic System After Subarachnoid Hemorrhage in Rats. Neurotherapeutics 2020, 17, 1954–1972. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Geng, Z.; Silver, F.L.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; Colucci, M.; Gerzanich, V. Does Inhibiting Sur1 Complement Rt-PA in Cerebral Ischemia? Ann. N. Y. Acad. Sci. 2012, 1268, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Yurovsky, V.; Tsymbalyuk, N.; Melnichenko, L.; Ivanova, S.; Gerzanich, V. Protective Effect of Delayed Treatment With Low-Dose Glibenclamide in Three Models of Ischemic Stroke. Stroke 2009, 40, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Tsymbalyuk, N.; Tsymbalyuk, O.; Ivanova, S.; Yurovsky, V.; Gerzanich, V. Glibenclamide Is Superior to Decompressive Craniectomy in a Rat Model of Malignant Stroke. Stroke 2010, 41, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, D.M.; Nassar, N.N.; El Salam, R.A. Glibenclamide ameliorates ischemia–reperfusion injury via modulating oxidative stress and inflammatory mediators in the rat hippocampus. Brain Res. 2011, 1385, 257–262. [Google Scholar] [CrossRef]
- Simard, J.M.; Woo, S.K.; Tsymbalyuk, N.; Voloshyn, O.; Yurovsky, V.; Ivanova, S.; Lee, R.; Gerzanich, V. Glibenclamide—10-h Treatment Window in a Clinically Relevant Model of Stroke. Transl. Stroke Res. 2012, 3, 286–295. [Google Scholar] [CrossRef]
- Ortega, F.J.; Gimeno-Bayon, J.; Espinosa-Parrilla, J.F.; Carrasco, J.; Batlle, M.; Pugliese, M.; Mahy, N.; Rodriguez, M.J. ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia–ischemia in rats. Exp. Neurol. 2012, 235, 282–296. [Google Scholar] [CrossRef]
- Wali, B.; Ishrat, T.; Atif, F.; Hua, F.; Stein, N.G.; Sayeed, I. Glibenclamide Administration Attenuates Infarct Volume, Hemispheric Swelling, and Functional Impairments following Permanent Focal Cerebral Ischemia in Rats. Stroke Res. Treat. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Jolkkonen, J.; Mahy, N.; Rodriguez, M.J. Glibenclamide Enhances Neurogenesis and Improves Long-Term Functional Recovery after Transient Focal Cerebral Ischemia. Br. J. Pharmacol. 2012, 33, 356–364. [Google Scholar] [CrossRef]
- Arikan Abello, F.; Martínez Valverde, T.; Sánchez Guerrero, Á.; Campos Martorell, M.; Esteves Coelho, M.; Gándara Sabatini, D.F.; Torne Torne, R.; Castro González, L.; Sahuquillo Barris, J. Malignant infarction of the middle cerebral artery in a porcine model. A pilot study. PLoS ONE 2017, 12, e0172637. [Google Scholar] [CrossRef] [PubMed]
- Burgos, I.M.A.; Ortiz-Plata, A.; Franco-Pérez, J.; Millán, A.; Aguilera, P. Resveratrol reduces cerebral edema through inhibition of de novo SUR1 expression induced after focal ischemia. Exp. Neurol. 2020, 330, 113353. [Google Scholar] [CrossRef]
- Luu, W.; Bjork, J.; Salo, E.; Entenmann, N.; Jurgenson, T.; Fisher, C.; Klein, A.H. Modulation of SUR1 KATP Channel Subunit Activity in the Peripheral Nervous System Reduces Mechanical Hyperalgesia after Nerve Injury in Mice. Int. J. Mol. Sci. 2019, 20, 2251. [Google Scholar] [CrossRef]
- Mehta, R.I.; Ivanova, S.; Tosun, B.C.; Castellani, R.J.; Gerzanich, V.; Simard, J.M. Sulfonylurea Receptor 1 Expression in Human Cerebral Infarcts. J. Neuropathol. Exp. Neurol. 2013, 72, 871–883. [Google Scholar] [CrossRef]
- Kunte, H.; Schmidt, S.; Eliasziw, M.; del Zoppo, G.J.; Simard, J.M.; Masuhr, F.; Weih, M.; Dirnagl, U. Sulfonylureas Improve Outcome in Patients With Type 2 Diabetes and Acute Ischemic Stroke. Stroke 2007, 38, 2526–2530. [Google Scholar] [CrossRef] [PubMed]
- Favilla, C.G.; Mullen, M.T.; Ali, M.; Higgins, P.; Kasner, S.E. Virtual International Stroke Trials Archive (VISTA) Collaboration. Sul-fonylurea use before stroke does not influence outcome. Stroke 2011, 42, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Kunte, H.; Busch, M.; Trostdorf, K.; Vollnberg, B.; Harms, L.; Mehta, R.I.; Castellani, R.J.; Mandava, P.; Kent, T.; Simard, J.M. Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas. Ann. Neurol. 2012, 72, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Horsdal, H.T.; Mehnert, F.; Rungby, J.; Johnsen, S.P. Type of Preadmission Antidiabetic Treatment and Outcome among Patients with Ischemic Stroke: A Nationwide Follow-up Study. J. Stroke Cerebrovasc. Dis. 2012, 21, 717–725. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, F.; Song, C.; Li, L.; Yang, X.; Wang, X.; Yu, L.; Guo, J.; Wang, K.; Fu, F.; et al. Glibenclamide Advantage in Treating Edema After Intracerebral Hemorrhage (GATE-ICH): Study Protocol for a Multicenter Randomized, Controlled, Assessor-Blinded Trial. Front. Neurol. 2021, 12, 579. [Google Scholar] [CrossRef]
- da Costa, B.B.S.; Windlin, I.C.; Koterba, E.; Yamaki, V.N.; Rabelo, N.N.; Solla, D.J.F.; Teixeira, M.J.; Figueiredo, E.G. Glibenclamide in Aneurysmatic Subarachnoid Hemorrhage (GASH): Study Protocol for a Randomized Controlled Trial. Trials 2019, 20, 413. [Google Scholar] [CrossRef]
- Simard, J.M.; Chen, M. Regulation by Sulfanylurea Receptor Type 1 of a Non-selective Cation Channel Involved in Cytotoxic Edema of Reactive Astrocytes. J. Neurosurg. Anesthesiol. 2004, 16, 98–99. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, X.; Huang, K.; Gu, Y.; Hu, Y.; Wu, Y.; Ji, Z.; Wang, Q.; Pan, S. Glibenclamide Enhances the Therapeutic Benefits of Early Hypothermia after Severe Stroke in Rats. Aging Dis. 2018, 9, 685–695. [Google Scholar] [CrossRef] [PubMed]
- King, Z.; Sheth, K.N.; Kimberly, W.T.; Simard, J.M. Profile of intravenous glyburide for the prevention of cerebral edema following large hemispheric infarction: Evidence to date. Drug Des. Dev. Ther. 2018, 12, 2539–2552. [Google Scholar] [CrossRef]
- Sheth, K.N.; Kimberly, W.T.; Elm, J.J.; Kent, T.A.; Mandava, P.; Yoo, A.J.; Thomalla, G.; Campbell, B.; Donnan, G.A.; Davis, S.M.; et al. Pilot Study of Intravenous Glyburide in Patients with a Large Ischemic Stroke. Stroke 2014, 45, 281–283. [Google Scholar] [CrossRef]
- Jha, R.; Battey, T.W.K.; Pham, L.; Lorenzano, S.; Furie, K.L.; Sheth, K.N.; Kimberly, W.T. Fluid-Attenuated Inversion Recovery Hyperintensity Correlates With Matrix Metalloproteinase-9 Level and Hemorrhagic Transformation in Acute Ischemic Stroke. Stroke 2014, 45, 1040–1045. [Google Scholar] [CrossRef]
- Kimberly, W.T.; Bevers, M.B.; von Kummer, R.; Demchuk, A.M.; Romero, J.M.; Elm, J.J.; Hinson, H.E.; Molyneaux, B.J.; Simard, J.M.; Sheth, K.N. Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP Trial. Neurology 2018, 91, e2163–e2169. [Google Scholar] [CrossRef]
- Jha, R.M.; Koleck, T.; Puccio, A.M.; Okonkwo, D.; Park, S.-Y.; Zusman, B.; Clark, R.S.B.; Shutter, L.; Wallisch, J.; Empey, P.; et al. Regionally clustered ABCC8 polymorphisms in a prospective cohort predict cerebral oedema and outcome in severe traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1152–1162. [Google Scholar] [CrossRef]
- Jha, R.M.; Desai, S.M.; Zusman, B.E.; Koleck, T.A.; Puccio, A.M.; Okonkwo, D.O.; Park, S.-Y.; Shutter, L.A.; Kochanek, P.M.; Conley, Y.P. DownstreamTRPM4Polymorphisms Are Associated with Intracranial Hypertension and Statistically Interact WithABCC8Polymorphisms in a Prospective Cohort of Severe Traumatic Brain Injury. J. Neurotrauma 2019, 36, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Puccio, A.M.; Okonkwo, D.O.; Zusman, B.E.; Park, S.-Y.; Wallisch, J.; Empey, P.; Shutter, L.; Clark, R.S.B.; Kochanek, P.M.; et al. ABCC8 Single Nucleotide Polymorphisms are Associated with Cerebral Edema in Severe TBI. Neurocrit. Care 2016, 26, 213–224. [Google Scholar] [CrossRef]
- Khalili, H.; Derakhshan, N.; Niakan, A.; Ghaffarpasand, F.; Salehi, M.; Eshraghian, H.; Shakibafard, A.; Zahabi, B. Effects of Oral Glibenclamide on Brain Contusion Volume and Functional Outcome of Patients with Moderate and Severe Traumatic Brain Injuries: A Randomized Double-Blind Placebo-Controlled Clinical Trial. World Neurosurg. 2017, 101, 130–136. [Google Scholar] [CrossRef]
- Castro, L.; Noelia, M.; Vidal-Jorge, M.; Sánchez-Ortiz, D.; Gándara, D.; Martínez-Saez, E.; Cicuéndez, M.; Poca, M.A.; Simard, J.M.; Sahuquillo, J. Kir6.2, the Pore-Forming Subunit of ATP-Sensitive K+ Channels, Is Overexpressed in Human Posttraumatic Brain Contusions. J. Neurotrauma 2019, 36, 165–175. [Google Scholar] [CrossRef]
- Gorse, K.M.; Lantzy, M.K.; Lee, E.D.; Lafrenaye, A.D. Transient Receptor Potential Melastatin 4 Induces Astrocyte Swelling But Not Death after Diffuse Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Bramlett, H.M.; Dixon, C.E.; Shear, D.A.; Dietrich, W.D.; Schmid, K.E.; Mondello, S.; Wang, K.; Hayes, R.L.; Povlishock, J.T.; et al. Approach to Modeling, Therapy Evaluation, Drug Selection, and Biomarker Assessments for a Multicenter Pre-Clinical Drug Screening Consortium for Acute Therapies in Severe Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2016, 33, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Bramlett, H.M.; Dixon, C.E.; Dietrich, W.D.; Mondello, S.; Wang, K.K.W.; Hayes, R.L.; Lafrenaye, A.; Povlishock, J.T.; Tortella, F.C.; et al. Operation Brain Trauma Therapy: 2016 Update. Mil. Med. 2018, 183, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Shen, G.; Su, Z.; He, Z.; Yuan, L. Glibenclamide ameliorates the disrupted blood–brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome. Brain Behav. 2019, 9, e01254. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Elmer, J.; Zusman, B.E.; Desai, S.; Puccio, A.M.; Okonkwo, D.O.; Park, S.Y.; Shutter, L.A.; Wallisch, J.S.; Conley, Y.P.; et al. Intracranial Pressure Trajectories. Crit. Care Med. 2018, 46, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Zusman, B.E.; Puccio, A.M.; Okonkwo, D.O.; Pease, M.; Desai, S.M.; Leach, M.; Conley, Y.P.; Kochanek, P.M. Genetic Variants Associated With Intraparenchymal Hemorrhage Progression After Traumatic Brain Injury. JAMA Netw. Open 2021, 4, e2116839. [Google Scholar] [CrossRef]
- Kurland, D.B.; Tosun, C.; Pampori, A.; Karimy, J.K.; Caffes, N.M.; Gerzanich, V.; Simard, J.M. Glibenclamide for the Treatment of Acute CNS Injury. Pharmaceuticals 2013, 6, 1287–1303. [Google Scholar] [CrossRef] [PubMed]
- Hosier, H.; Peterson, D.; Tsymbalyuk, O.; Keledjian, K.; Smith, B.R.; Ivanova, S.; Gerzanich, V.; Popovich, P.G.; Simard, J.M. A Direct Comparison of Three Clinically Relevant Treatments in a Rat Model of Cervical Spinal Cord Injury. J. Neurotrauma 2015, 32, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Popovich, P.G.; Tsymbalyuk, O.; Gerzanich, V. Spinal cord injury with unilateral versus bilateral primary hemor-rhage—Effects of glibenclamide. Exp. Neurol. 2012, 233, 829–835. [Google Scholar] [CrossRef]
- Simard, J.M.; Tsymbalyuk, O.; Keledjian, K.; Ivanov, A.; Ivanova, S.; Gerzanich, V. Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp. Neurol. 2012, 233, 566–574. [Google Scholar] [CrossRef]
- Popovich, P.G.; Lemeshow, S.; Gensel, J.C.; Tovar, C.A. Independent evaluation of the effects of glibenclamide on reducing pro-gressive hemorrhagic necrosis after cervical spinal cord injury. Exp. Neurol. 2012, 233, 615–622. [Google Scholar] [CrossRef]
- Minnema, A.J.; Mehta, A.; Boling, W.W.; Schwab, J.; Simard, J.M.; Farhadi, H.F. SCING—Spinal Cord Injury Neuroprotection with Glyburide: A pilot, open-label, multicentre, prospective evaluation of oral glyburide in patients with acute traumatic spinal cord injury in the USA. BMJ Open 2019, 9, e031329. [Google Scholar] [CrossRef]
- Gerzanich, V.; Woo, S.K.; Vennekens, R.; Tsymbalyuk, O.; Ivanova, S.; Ivanov, A.; Geng, Z.; Chen, Z.; Nilius, B.; Flockerzi, V.; et al. De Novo Expression of Trpm4 Initiates Secondary Hemorrhage in Spinal Cord Injury. Nat. Med. 2009, 15, 185–191. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, J.; Yu, T.; Chen, Z.; Xiao, Z.; Wang, J.; Hu, Y.; Wu, Y.; Zhu, D. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics 2018, 8, 4181–4198. [Google Scholar] [CrossRef]
- Simard, J.M.; Popovich, P.G.; Tsymbalyuk, O.; Caridi, J.; Gullapalli, R.P.; Kilbourne, M.J.; Gerzanich, V. MRI evidence that glibenclamide reduces acute lesion expansion in a rat model of spinal cord injury. Spinal Cord 2013, 51, 823–827. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schwartz, G.; Fehlings, M.G. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: Im-proved behavioral and neuroanatomical recovery with riluzole. J. Neurosurg. 2001, 94 (Suppl. 2), 245–256. [Google Scholar] [CrossRef]
- Tetreault, L.A.; Zhu, M.P.; Wilson, J.R.; Karadimas, S.K.; Fehlings, M.G. The impact of riluzole on neurobehavioral outcomes in pre-clinical models of traumatic and nontraumatic spinal cord injury: Results from a systematic review of the literature. Glob. Spine J. 2020, 10, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, H.Y.; Park, C.S.; Ju, B.G.; Yune, T.Y. Mithramycin A Improves Functional Recovery by Inhibiting BSCB Disruption and Hemorrhage after Spinal Cord Injury. J. Neurotrauma 2018, 35, 508–520. [Google Scholar] [CrossRef]
- Lee, J.Y.; Choi, H.Y.; Na, W.H.; Ju, B.G.; Yune, T.Y. Ghrelin inhibits BSCB disruption/hemorrhage by attenuating MMP-9 and SUR1/TrpM4 expression and activation after spinal cord injury. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, H.Y.; Na, W.H.; Ju, B.G.; Yune, T.Y. 17β-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology 2015, 156, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Tosun, C.; Koltz, M.T.; Kurland, D.B.; Ijaz, H.; Gurakar, M.; Schwartzbauer, G.; Coksaygan, T.; Ivanova, S.; Gerzanich, V.; Simard, J.M. The Protective Effect of Glibenclamide in a Model of Hemorrhagic Encephalopathy of Prematurity. Brain Sci. 2013, 3, 215–238. [Google Scholar] [CrossRef]
- Dankiewicz, J.; Cronberg, T.; Lilja, G.; Jakobsen, J.C.; Levin, H.; Ullén, S.; Rylander, C.; Wise, M.P.; Oddo, M.; Cariou, A.; et al. Hypothermia versus Normothermia after Out-of-Hospital Cardiac Arrest. N. Engl. J. Med. 2021, 384, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Thoma, B. Translating Targeted Temperature Management Trials into Postarrest Care. N. Engl. J. Med. 2021, 384, 2344–2345. [Google Scholar] [CrossRef]
- Jackson, T.C.; Kochanek, P.M. A New Vision for Therapeutic Hypothermia in the Era of Targeted Temperature Management: A Speculative Synthesis. Ther. Hypothermia Temp. Manag. 2019, 9, 13–47. [Google Scholar] [CrossRef]
- Lascarrou, J.-B.; Merdji, H.; Le Gouge, A.; Colin, G.; Grillet, G.; Girardie, P.; Coupez, E.; Dequin, P.-F.; Cariou, A.; Boulain, T.; et al. Targeted Temperature Management for Cardiac Arrest with Nonshockable Rhythm. N. Engl. J. Med. 2019, 381, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Wetterslev, J.; Cronberg, T.; Erlinge, D.; Gasche, Y.; Hassager, C.; Horn, J.; Hovdenes, J.; Kjaergaard, J.; Kuiper, M.; et al. Targeted Temperature Management at 33 °C versus 36°C after Cardiac Arrest. N. Engl. J. Med. 2013, 369, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 2002, 346, 549–556. [Google Scholar] [CrossRef]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. N. Engl. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Taguchi, N.; Isaka, Y.; Nakamura, T.; Tanaka, M. Glibenclamide and Therapeutic Hypothermia Have Comparable Effect on Attenuating Global Cerebral Edema Following Experimental Cardiac Arrest. Neurocrit. Care 2017, 29, 119–127. [Google Scholar] [CrossRef]
- Huang, K.; Gu, Y.; Hu, Y.; Ji, Z.; Wang, S.; Lin, Z.; Li, X.; Xie, Z.; Pan, S. Glibenclamide Improves Survival and Neurologic Outcome After Cardiac Arrest in Rats*. Crit. Care Med. 2015, 43, e341–e349. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Z.; Gu, Y.; Hu, Y.; Ji, Z.; Wang, S.; Lin, Z.; Li, X.; Xie, Z.; Pan, S. Glibenclamide Is Comparable to Target Temperature Management in Improving Survival and Neurological Outcome After Asphyxial Cardiac Arrest in Rats. JAHA 2016, 5, e003465. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Z.; Gu, Y.; Ji, Z.; Lin, Z.; Wang, S.; Pan, S.; Wu, Y. Glibenclamide Prevents Water Diffusion Abnormality in the Brain After Cardiac Arrest in Rats. Neurocrit. Care 2018, 29, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Li, L.; Chen, Q.; Tao, Y.; Yang, L.; Zhang, B.; Zhang, J.H.; Feng, H.; Chen, Z.; Tang, J.; et al. Role of Glibenclamide in Brain Injury After Intracerebral Hemorrhage. Transl. Stroke Res. 2016, 8, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kung, T.F.C.; Wilkinson, C.M.; Dirks, C.A.; Jickling, G.C.; Colbourne, F. Glibenclamide does not improve outcome following severe collagenase-induced intracerebral hemorrhage in rats. PLoS ONE 2021, 16, e0252584. [Google Scholar] [CrossRef]
- Wilkinson, C.M.; Brar, P.S.; Balay, C.J.; Colbourne, F. Glibenclamide, a Sur1-Trpm4 antagonist, does not improve outcome after collagenase-induced intracerebral hemorrhage. PLoS ONE 2019, 14, e0215952. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.; Yang, B.; Hu, Z. Neuroprotective potential of glibenclamide is mediated by antioxidant and anti-apoptotic pathways in intracerebral hemorrhage. Brain Res. Bull. 2018, 142, 18–24. [Google Scholar] [CrossRef]
- Chang, J.J.; Khorchid, Y.; Kerro, A.; Burgess, L.G.; Goyal, N.; Alexandrov, A.W.; Alexandrov, A.V.; Tsivgoulis, G. Sulfonylurea drug pretreatment and functional outcome in diabetic patients with acute intracerebral hemorrhage. J. Neurol. Sci. 2017, 381, 182–187. [Google Scholar] [CrossRef]
- Simard, J.M.; Castellani, R.J.; Ivanova, S.; Koltz, M.T.; Gerzanich, V. Sulfonylurea Receptor 1 in the Germinal Matrix of Premature Infants. Pediatr. Res. 2008, 64, 648–652. [Google Scholar] [CrossRef]
- Schattling, B.; Steinbach, K.; Thies, E.; Kruse, M.; Menigoz, A.; Ufer, F.; Flockerzi, V.; Brück, W.; Pongs, O.; Vennekens, R.; et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2012, 18, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Trapp, B.D. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol. 2011, 93, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, C.; Volckaert, T.; Wolke, U.; Sze, M.; De Rycke, R.; Waisman, A.; Prinz, M.; Beyaert, R.; Pasparakis, M.; Van Loo, G. Oligodendrocyte-Specific FADD Deletion Protects Mice from Autoimmune-Mediated Demyelination. J. Immunol. 2010, 185, 7646–7653. [Google Scholar] [CrossRef]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Experientia 2008, 65, 2702–2720. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, C.F.; Raine, C.S. The astrocyte in multiple sclerosis revisited. Glia 2013, 61, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Farez, M.F. The role of astrocytes in multiple sclerosis progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Ludwin, S.K.; Rao, V.T.; Moore, C.S.; Antel, J. Astrocytes in multiple sclerosis. Mult. Scler. J. 2016, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Valdes, V.; Tong, X.Y.; Shamaladevi, N.; Gonzalez, W.; Norenberg, M.D. Sulfonylurea Receptor 1 Contributes to the Astrocyte Swelling and Brain Edema in Acute Liver Failure. Transl. Stroke Res. 2014, 5, 28–37. [Google Scholar] [CrossRef]
- Betjemann, J.P.; Lowenstein, D.H. Status epilepticus in adults. Lancet Neurol. 2015, 14, 615–624. [Google Scholar] [CrossRef]
- Lin, Z.; Huang, H.; Gu, Y.; Huang, K.; Hu, Y.; Ji, Z.; Wu, Y.; Wang, S.; Yang, T.; Pan, S. Glibenclamide ameliorates cerebral edema and improves outcomes in a rat model of status epilepticus. Neuropharmacology 2017, 121, 1–11. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic Pain: A Maladaptive Response of the Nervous System to Damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Jiang, B.-C.; Gao, Y.-J. Chemokines in neuron–glial cell interaction and pathogenesis of neuropathic pain. Cell. Mol. Life Sci. 2017, 74, 3275–3291. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, R.; Tilley, D.M.; Vogel, L.; Benyamin, R. The Role of Glia and the Immune System in the Development and Maintenance of Neuropathic Pain. Pain Pract. 2010, 10, 167–184. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef]
- Song, J.-G.; Hahm, K.D.; Kim, Y.K.; Gil Leem, J.; Lee, C.; Jeong, S.M.; Park, P.H.; Shin, J.W. Adenosine Triphosphate–Sensitive Potassium Channel Blockers Attenuate the Antiallodynic Effect of R-PIA in Neuropathic Rats. Anesth. Analg. 2011, 112, 1494–1499. [Google Scholar] [CrossRef]
- de Los Monteros-Zuñiga, A.E.; Izquierdo, T.; Quiñonez-Bastidas, G.N.; Rocha-González, H.I.; Godínez-Chaparro, B. Anti-allodynic effect of mangiferin in neuropathic rats: Involvement of nitric oxide-cyclic GMP-ATP sensitive K+ channels pathway and serotoninergic system. Pharmacol. Biochem. Behav. 2016, 150–151, 190–197. [Google Scholar] [CrossRef]
- Longhi-Balbinot, D.T.; Rossaneis, A.C.; Ribeiro, F.P.; Bertozzi, M.M.; Cunha, F.Q.; Alves-Filho, J.C.; Cunha, T.; Peron, J.P.; Miranda, K.M.; Casagrande, R.; et al. The nitroxyl donor, Angeli’s salt, reduces chronic constriction injury-induced neuropathic pain. Chem. Interact. 2016, 256, 1–8. [Google Scholar] [CrossRef]
- Koh, W.U.; Shin, J.W.; Bang, J.-Y.; Kim, S.G.; Song, J.-G. The Antiallodynic Effects of Nefopam Are Mediated by the Adenosine Tri-phosphate-Sensitive Potassium Channel in a Neuropathic Pain Model. Anesth. Analg. 2016, 123, 762–770. [Google Scholar] [CrossRef]
- Bertozzi, M.M.; Rossaneis, A.C.; Fattori, V.; Longhi-Balbinot, D.T.; Freitas, A.; Cunha, F.Q.; Alves-Filho, J.C.; Cunha, T.M.; Casagrande, R.; Verri, W.A. Diosmin reduces chronic constriction injury-induced neuropathic pain in mice. Chem. Interact. 2017, 273, 180–189. [Google Scholar] [CrossRef]
- Zulazmi, N.A.; Gopalsamy, B.; Min, J.C.S.; Farouk, A.A.O.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Zerumbone Alleviates Neuropathic Pain through the Involvement of l-Arginine-Nitric Oxide-cGMP-K+ ATP Channel Pathways in Chronic Constriction Injury in Mice Model. Molecules 2017, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Mata-Bermudez, A.; Izquierdo, T.; Monteros-Zuñiga, E.D.L.; Coen, A.; Godínez-Chaparro, B. Antiallodynic effect induced by 6-gingerol in neuropathic rats is mediated by activation of the serotoninergic system and the nitric oxide-cyclic guanosine monophosphate-adenosine triphosphate-sensitive K+ channel pathway. Phytother. Res. 2018, 32, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Pastrana-Quintos, T.; Salgado-Moreno, G.; Pérez-Ramos, J.; Coen, A.; Godínez-Chaparro, B. Anti-allodynic effect induced by cur-cumin in neuropathic rat is mediated through the NO-cyclic-GMP-ATP sensitive K+ channels pathway. BMC Complement. Med. Ther. 2020, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Makar, T.; Gerzanich, V.; Kalakonda, S.; Ivanova, S.; Pereira, E.F.R.; Andharvarapu, S.; Zhang, J.; Simard, J.M.; Zhao, R.Y. HIV-1 Vpr-Induced Proinflammatory Response and Apoptosis Are Mediated through the Sur1-Trpm4 Channel in Astrocytes. mBio 2020, 11, e02939-20. [Google Scholar] [CrossRef] [PubMed]
- Berdugo, M.; Delaunay, K.; Naud, M.-C.; Guegan, J.; Moulin, A.; Savoldelli, M.; Picard, E.; Radet, L.; Jonet, L.; Djerada, Z.; et al. The antidiabetic drug glibenclamide exerts direct retinal neuroprotection. Transl. Res. 2020, 229, 83–99. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Title | Disease and Model Details | Regulation/Inhibition/Interaction | Signaling Pathway Proteins | Authors/Year |
|---|---|---|---|---|
| Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage | SAH (rat, filament puncture) | Inhibition of SUR1 by GLI |
| Simard et al., 2009 [40] |
| Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage | SAH (rat, filament puncture and entorhinal injection) | Gene suppression of Abcc8/Inhibition of SUR1 by GLI |
| Tosun et al., 2013 [36] |
| Glibenclamide Attenuates Blood–Brain Barrier Disruption in Adult Mice after Traumatic Brain Injury | TBI (mouse, CCI) | Inhibition of SUR1 by GLI |
| Xu et al., 2017 [44] |
| SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells | Cerebral ischemia/ reperfusion (in vitro, and rat ischemia-reperfusion) | NF-κB activation/ SUR1-TRPM4 channel opening by tPA |
| Gerzanich et al., 2018 [96] |
| Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury | TBI (rat, cortical impact) | Inhibition of SUR1 by GLI |
| Patel et al., 2010 [30] |
| Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis | EAE (mouse EAE, MOG33–55 peptide) | Abcc8−/− mice/Inhibition of SUR1 by GLI |
| Makar et al., 2015 [97] |
| Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis | EAE (mouse EAE, MOG33–55 peptide) | Inhibition of SUR1 by GLI |
| Gerzanich et al., 2017 [98] |
| Glibenclamide pretreatment protects against chronic memory dysfunction and glial activation in rat cranial blast traumatic brain injury | TBI (rat, direct cranial blast) | Inhibition of SUR1 by GLI |
| Stokum et al., 2017 [45] |
| The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia | Activation of TLR4 by LPS (primary culture, adult rat microglia) | Abcc8−/− mice/Inhibition of SUR1 by GLI |
| Kurland et al., 2016 [92] |
| Authors, Year | Study Title | Model | Species | Drug Details | Results |
|---|---|---|---|---|---|
| In vitro studies: ischemic stroke | |||||
| Chen et al., 2001 [7] | Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain | Chemically induced hypoxia in non-reactive isolated astrocytes using 1 nM NaN3 | NA | NA |
|
| Chen et al., 2003 [8] | Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain | Chemically induced hypoxia in non-reactive astrocytes using 1 nM NaN3 | NA | GLI |
|
| In vivo studies: ischemic stroke | |||||
| Simard et al., 2006 [65] | Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke | Massive middle cerebral artery (MCA) infarction associated with malignant cerebral edema | Rat | GLI Infusion rate: 75 ng/h Infusion time: 2–3 min after MCAo |
|
| Simard et al., 2009 [112] | Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke | 3 models Thromboembolic MCA occlusion (MCAo), Transient MCAo and Permanent MCAo | Rat | GLI Time: variable Loading dose: 3.3–10 mg/kg Infusion rate: 75–200 ng/h |
|
| Simard et al., 2010 [113] | Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke | MCAo severe ischemia reperfusion | Rat | GLI Time: 15 min before MCAo Loading dose: 10 mg/kg i.p. Infusion rate: 200 ng/h |
|
| Abdallah et al., 2011 [114] | Glibenclamide ameliorates ischemia-reperfusion injury via modulating oxidative stress and inflammatory mediators in the rat hippocampus | Ischemia reperfusion | Rat | GLI Time: 10 min before ischemia/reperfusion Dose: 1 mg/kg i.p. |
|
| Simard et al., 2012 [115] | Glibenclamide 10 h treatment window in a clinically relevant model of stroke. | Intra-arterial occluder MCAo | Rat | GLI Time: 4.5 vs. 10 h post MCAo Loading dose: 10 μg/kg, Infusion rate: 200 ng/h |
|
| Ortega et al., 2012 [116] | ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia-ischemia in rats | Transient MCAo | Rat | GLI Time: 6, 12, 24 h after MCAo Doses: 0.06 μg, 0.6 μg, 6 μg |
|
| Wali et al., 2012 [117] | Glibenclamide administration attenuates infarct volume, hemispheric swelling, and functional impairments following permanent focal cerebral ischemia in rats | Permanent MCAo | Rat | GLI Time: 5 min after MCAo Loading dose: 10 mg/kg, Infusion rate: 200 ng/h |
|
| Ortega et al., 2013 [118] | Glibenclamide enhances neurogenesis and improves long-term functional recovery after transient focal cerebral ischemia | Transient MCAo | Rat | GLI Time: 6 h, 12 h, 24 h after reperfusion Dose: 0.6 mg |
|
| Arikan et al., 2017 [119] | Malignant infarction of the middle cerebral artery in a porcine model. A pilot study | Malignant infarction of the middle cerebral artery | Pig | NA |
|
| Alquisiras et al., 2020 [120] | Resveratrol reduces cerebral edema through inhibition of de novo SUR1 expression induced after focal ischemia | MCAo | Rat | NA |
|
| Woo et al., 2020 [121] | SUR1-TRPM4 channels, not KATP, mediate brain swelling following cerebral ischemia | Permanent MCAo | Rat | Abcc8 antisense ODN Trpm4 antisense ODN |
|
| Human expression studies: ischemic stroke | |||||
| Mehta et al., 2013 [122] | Sulfonylurea receptor 1 expression in human cerebral infarcts | Post-mortem brain specimen within 31 d after ischemic stroke | 13 patients | NA |
|
| Mehta et al., 2015 [68] | Sur1-Trpm4 cation channel expression in human cerebral infarcts | Post-mortem brain specimen | 14 patients | NA |
|
| Clinical retrospective studies: ischemic stroke | |||||
| Kunte et al., 2007 [123] | Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke | Acute ischemic stroke | 61 patients | Oral sulfonylurea |
|
| Favilla et al., 2011 [124] | Sulfonylurea use before stroke does not influence outcome | Patients enrolled in non-reperfusion ischemic stroke trials | 1050 patients | Oral sulfonylurea |
|
| Kunte et al., 2012 [125] | Hemorrhagic transformation of ischemic stroke in diabetics on sulfonylureas | Diabetic patients with acute ischemic stroke | 220 patients | Oral sulfonylurea |
|
| Horsdal et al., 2012 [126] | Type of preadmission antidiabetic treatment and outcome among patients with ischemic stroke: a nationwide follow-up study | Diabetic patient with acute ischemic stroke | 4817 patients | Oral sulfonylurea |
|
| Clinical trials: ischemic stroke | |||||
| Sheth et al., 2014 [117] | Pilot study of intravenous glyburide in patients with a large ischemic stroke | Large hemispheric infarction | 10 patients | Intravenous glyburide (RP-1127) |
|
| Sheth et al., 2016 [47] | Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomized, double-blind, placebo-controlled phase 2 trial | Large hemispheric infarction | 86 patients | Intravenous glyburide (RP-1127) Phase II double-blind (GAMES-RP) |
|
| NCT Identifier | Phase | Drug | Studied Population | Outcome/References |
|---|---|---|---|---|
| NCT03741530 | Phase I (GATE-ICH) | Oral glyburide (1.25 mg) | ICH | Completed, results not yet reported [127] |
| NCT02864953 | Phase III (CHARM) | BIIB093 (newest name for RP-1127) | Large hemispheric infarction including thrombectomy | Recruiting [46] |
| NCT03954041 | Phase II (ASTRAL) | Intravenous glyburide (RP-1127) | Brain contusion | Recruiting [46] |
| NCT02524379 | Phase I (SCING) | Oral glyburide (3.125–2.5 mg on days 1–3) | Acute cervical traumatic SCI | Active, not recruiting [46] |
| NCT03569540 | Phase IV (GASH) | Oral glyburide (0.5 mg) | SAH | Unknown [128] |
| Authors, Year | Study Title | Model | Species | Drug Details | Results |
|---|---|---|---|---|---|
| In vivo studies: TBI | |||||
| Simard et al., 2009 [26] | Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion | Weight-drop model of focal cortical contusion (10 g dropped from 5 cm, velocity = 1 m/s) | Rat | GLI Time: 10 min of injury Loading dose: 10 mg/kg i.p, Infusion rate: 200 ng/h |
|
| Patel et al., 2010 [30] | Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. | Milder injury weight drop model (10 g dropped from 3 cm, velocity = 0.77 m/s) | Rat | GLI Time: 10 min of injury Loading dose: 10 mg/kg i.p, Infusion rate: 200 ng/h |
|
| Zweckberger et al., 2014 [43] | Glibenclamide reduces secondary brain damage after experimental traumatic brain injury | CCI (1.5mm tissue displacement, velocity = 7.5 m/s, dwell time = 300 ms) | Rat | GLI Time: 15 min after CCI Loading dose 10 mg/kg i.p, Infusion rate 10 mL/h |
|
| Xu et al., 2017 [44] | Glibenclamide attenuates blood–brain barrier disruption in adult mice after traumatic brain injury | CCI (1.5 mm tissue displacement, velocity = 1.5 m/s, dwell time = 100 ms) | Mouse | GLI Time: immediately after CCI Dose:10 mg i.p. |
|
| Jha et al., 2018 [24] | Glibenclamide produces region-dependent effects on cerebral edema in a combined injury model of traumatic brain injury and hemorrhagic shock in mice | CCI (5 m/s, 1 mm depth) + hemorrhagic shock (HS) | Mouse | GLI Time: 10 min after CCI Loading dose: 20 mg/kg i.v. Infusion rate: 0.4 mg/h |
|
| Gerzanich et al., 2019 [29] | Sulfonylurea receptor 1, transient receptor potential cation channel subfamily M member 4, and kir6.2: role in hemorrhagic progression of contusion | CCI (4.5 mm tissue displacement, velocity = 1 m/s, dwell time = 200 ms) | Rat | GLI Time: 10 min after CCI Loading dose: 10 mg/kg i.p., Infusion rate: 400 ng/h |
|
| Jha et al., 2021 [33] | Glibenclamide treatment in traumatic brain injury: operation brain trauma therapy | Fluid percussion injury (FPI), CCI (4 m/s; 2.6 mm depth), penetrating ballistic like brain injury | Rat | GLI Time: 10 min after injury Loading dose: 10 mg/kg i.p., Infusion rate: 0.20 μg/h |
|
| Tata et al., 2021 [35] | Abcc8 (Sulfonylurea receptor-1) impact on brain atrophy after TBI varies by sex | CCI (1.2 mm displacement, velocity = 5 m/s, dwell time = 50–60 ms) | Mouse | NA (Abcc8−/−) |
|
| Human expression and genetics studies: TBI | |||||
| Martinez et al., 2015 [38] | Sulfonylurea receptor 1 in humans with post-traumatic brain contusions | Contusional TBI | 26 patient samples | NA |
|
| Jha et al., 2017 [24] | Sulfonylurea receptor-1: a novel biomarker for cerebral edema in severe traumatic brain injury | CSF samples from severe TBI | 28 patients 15 controls | NA |
|
| Jha et al., 2017 [123] | ABCC8 single nucleotide polymorphisms are associated with cerebral edema in severe TBI | Candidate gene study in severe TBI (ABCC8) | 385 patients analyzed | NA |
|
| Jha et al., 2018 [120] | Regionally clustered ABCC8 polymorphisms in a prospective cohort predict cerebral oedema and outcome in severe traumatic brain injury | Tag-SNP study in severe TBI (ABCC8) | 410 patients analyzed | NA |
|
| Castro et al., 2019 [139] | Kir6.2, the pore-forming subunit of ATP-sensitive K+ channels, is overexpressed in human posttraumatic brain contusions | Contusional TBI | 32 patients | NA |
|
| Gerzanich et al., 2019 [29] | Sulfonylurea receptor 1, transient receptor potential cation channel subfamily M member 4, and kir6.2: role in hemorrhagic progression of contusion | Specimens from patients with non-ballistic, closed head injury or contusion-TBI who underwent decompressive craniectomy | 16 patients | NA |
|
| Jha et al., 2019 [122] | Downstream TRPM4 polymorphisms are associated with intracranial hypertension and statistically interact with ABCC8 polymorphisms in a prospective cohort of severe traumatic brain injury | Candidate gene study in severe TBI (TRPM4) | 385 patients analyzed | NA |
|
| Zusman et al., 2021 [34] | Cerebrospinal fluid sulfonylurea receptor-1 is associated with intracranial pressure and outcome after pediatric TBI: an exploratory analysis of the cool kids trial | CSF samples from pediatric patients with severe TBI | 16 patients, 7 controls | NA |
|
| Jha et al., 2021 [131] | Genetic variants associated with intraparenchymal hemorrhage progression after traumatic brain injury. | Candidate gene study of hemorrhage progression in severe TBI (ABCC8, TRPM4) | 321 patients analyzed | NA |
|
| Clinical trials: TBI | |||||
| Zafardoost et al., 2016 [42] | Evaluation of the effect of glibenclamide in patients with diffuse axonal injury due to moderate to severe head trauma | Diffuse axonal injury Randomized trial | 40 patients | Oral GLI 1.25 mg every 12 h for 1 week |
|
| Khalili et al., 2017 [138] | Effects of oral glibenclamide on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injuries: a randomized double-blind placebo-controlled clinical trial | Moderate to severe contusional TBI Randomized trial | 66 patients | Oral GLI 10 mg daily for 10 d |
|
| Eisenberg et al., 2020 [31] | Magnetic resonance imaging pilot study of intravenous glyburide in traumatic brain injury | TBI with GCS 4-14 Randomized Trial | 28 patients | GLI total daily dose on D1 was 3.12 mg, on D2 and D3 was 2.67 mg/day |
|
| Authors, Year | Study Title | Model | Species | GLI Dose | Results |
|---|---|---|---|---|---|
| In vivo studies: SCI | |||||
| Simard et al., 2007 [28] | Endothelial sulfonylurea receptor 1-regulated NC Ca-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury | Severe unilateral SCI (10 g weight dropped from 2.5 cm) | Rat | GLI Time: 2-3 min after SCI Dose: 200 ng/h Others: repaglinide, Abcc8 antisense oligo deoxynucleotide ODN |
|
| Gerzanich et al., 2009 [139] | De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury | Unilateral SCI | Mouse Rat | Trpm4−/− Trpm4 antisense ODN |
|
| Simard et al., 2010 [27] | Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma | Unilateral SCI | Mouse Rat | GLI Time: 15 min after SCI Loading dose: 10 mg/kg Infusion rate: 200 ng/h Abcc8−/− Abcc8 antisense ODN |
|
| Simard et al., 2012 [148] | Spinal cord injury with unilateral versus bilateral primary hemorrhage–effects of glibenclamide | Unilateral/Bilateral primary SCI | Rat | GLI Time: min after SCI Loading dose: 10 mg/kg Infusion rate: 200 ng/h |
|
| Simard et al., 2012 [149] | Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury | Unilateral cervical SCI | Rat | GLI Time: 3 h after SCI Loading dose:10 mg/kg Infusion rate: 200 ng/h Other: riluzole |
|
| Hosier et al., 2015 [147] | A direct comparison of three clinically relevant treatments in a rat model of cervical spinal cord injury | Unilateral impact to the cervical spinal cord at C7 | Rat | GLI Time: 4 h after SCI Loading dose: 10 mg/kg, Infusion rate: 400 ng/h Others: hypothermia, riluzole |
|
| Yao et al., 2018 [153] | Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury | Thoracic SCI | Mouse | Flufenamic acid |
|
| Human studies: SCI | |||||
| Simard et al., 2010 [27] | Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma | Autopsy sample from patients with traumatic SCI | Human | NA |
|
| Authors, Year | Study Title | Model | Species | GLI Dose | Results |
|---|---|---|---|---|---|
| In vivo studies: SAH | |||||
| Simard et al., 2009 [40] | Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage | Mild-moderate SAH using a filament-based endovascular puncture of internal carotid artery | Rat | GLI Time: <15 min after SAH Loading dose: 10 mg/kg Infusion rate: 200 ng/h |
|
| Tosun et al., 2013 [36] | Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage | SAH induced by stereotactic injection of fresh non-heparinized autologous blood into the subarachnoid space of the entorhinal cortex, OR by filament puncture of carotid artery | Rat | GLI Time: <10 min after SAH Loading dose: 10 mg/kg Infusion rate: 200 ng/h |
|
| Fang et al., 2020 [110] | Pituitary adenylate cyclase-activating polypeptide attenuates brain edema by protecting blood-brain barrier and glymphatic system after subarachnoid hemorrhage in rats | SAH | Rat | PACAP38 |
|
| Human studies: SAH | |||||
| Tosun et al., 2013 [36] | Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage | Autopsy sample from SAH patients | 7 patients | NA |
|
| Dundar et al., 2020 [37] | Serum SUR1 and TRPM4 in patients with subarachnoid hemorrhage | Aneurysmal SAH patients | 44 patients | NA |
|
| Authors, Year | Study Title | Model | Species | GLI Dose | Results |
|---|---|---|---|---|---|
| In vivo studies: Cardiac arrest | |||||
| Huang et al., 2015 [169] | Glibenclamide improves survival and neurologic outcome after cardiac arrest in rats | 8 min asphyxia cardiac arrest | Rat | GLI Time: 10 min Loading dose:10 μg/kg Maintenance dose: 1.2 μg at 6, 12, 18, 24 h |
|
| Huang et al., 2016 [170] | Glibenclamide is comparable to target temperature management in improving survival and neurological outcome after asphyxial cardiac arrest in rats | 10 min asphyxia cardiac arrest | Rat | GLI Time: at randomization Loading dose: 10 μg/kg 4 maintenance doses of 1.2 μg per 6 h after ROSC |
|
| Huang et al., 2018 [171] | Glibenclamide prevents water diffusion abnormality in the brain after cardiac arrest in rats | 15 min asphyxia cardiac arrest | Rat | GLI Time: 15 min post-ROSC Loading dose 10 μg/kg Maintenance: four maintenance doses of 1.2 μg at 6, 12, 18, and 24 h after ROSC |
|
| Nakayama et al., 2018 [168] | Glibenclamide and therapeutic hypothermia have comparable effect on attenuating global cerebral edema following experimental cardiac arrest | CA induced by IV injection of 0.05 mL cold 0.5 mol/L KCL | Mouse | GLI Loading dose: 10 mg/kg Maintenance dose: 4 mg/kg 6 h and 18 h post ROSC |
|
| In vivo studies: Intracerebral hemorrhage (ICH) | |||||
| Tosun et al., 2013 [160] | The protective effect of glibenclamide in a model of hemorrhagic encephalopathy of prematurity | Model of hemorrhagic encephalopathy induced by 20 min of intrauterine ischemia, followed by an intraperitoneal injection of glycerol | Rat | GLI Time: immediately before closing the laparotomy Loading dose:10 μg/kg Infusion rate 400 ng/h |
|
| Jiang et al., 2017 [172] | Role of glibenclamide in brain injury after intracerebral hemorrhage | Autologous blood infusion ICH model | Rat | GLI Time: end of surgery Loading dose: 10 μg/kg, Infusion rate: 200 ng/h |
|
| Zhou et al., 2018 [175] | Neuroprotective potential of glibenclamide is mediated by antioxidant and anti-apoptotic pathways in intracerebral hemorrhage | Collagenase induced ICH model | Rat | GLI Time: 30 min before surgery Loading dose:10 μg/kg i.p Maintenance: 1 mg/kg daily |
|
| Xu et al., 2019 [143] | Glibenclamide ameliorates the disrupted blood–brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome | Autologous blood infusion ICH model | Mouse | GLI Time: immediately after ICH Dose:10 μg/kg i.p. |
|
| Wilkinson et al., 2019 [174] | Glibenclamide, a Sur1-Trpm4 antagonist, does not improve outcome after collagenase-induced intracerebral hemorrhage | Collagenase induced ICH model | Rat | GLI Time: 2 h post-ICH Loading dose: 10 μg/kg Infusion rate: 200 ng/h |
|
| Kung et al., 2021 [173] | Glibenclamide does not improve outcome following severe collagenase-induced intracerebral hemorrhage in rats | Collagenase induced ICH model | Rat | GLI Time: 2 h post-ICH Loading dose 10 μg/kg Infusion rate: 200 ng/h |
|
| Human studies: ICH | |||||
| Simard et al., 2008 [177] | Sulfonylurea receptor 1 in the germinal matrix of premature infants | Brain specimen from pre-mature infants with germinal matrix hemorrhage | 12 patients | NA |
|
| In vivo studies: Multiple sclerosis (MS) and experimental autoimmune encephalitis (EAE) | |||||
| Schattling et al., 2012 [178] | TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis | EAE was induced using myelin oligodendrocyte glycoprotein 35–55 | Mouse | GLI Dose: 10 μg daily i.p |
|
| Makar et al., 2015 [97] | Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis | EAE was induced in wild-type (WT) and Abcc8−/− mice using myelin oligodendrocyte glycoprotein 35–55 | Mouse | GLI Dose: 10 μg daily i.p |
|
| Gerzanich et al., 2017 [98] | Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. | EAE was induced using myelin oligodendrocyte glycoprotein 35–55 | Mouse | GLI Dose: 10 μg glibenclamide daily i.p |
|
| Human studies: MS and EAE | |||||
| Gerzanich et al., 2017 [98] | Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. | Demyelinating lesions from MS patients | 9 patients | NA |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, R.M.; Rani, A.; Desai, S.M.; Raikwar, S.; Mihaljevic, S.; Munoz-Casabella, A.; Kochanek, P.M.; Catapano, J.; Winkler, E.; Citerio, G.; et al. Sulfonylurea Receptor 1 in Central Nervous System Injury: An Updated Review. Int. J. Mol. Sci. 2021, 22, 11899. https://doi.org/10.3390/ijms222111899
Jha RM, Rani A, Desai SM, Raikwar S, Mihaljevic S, Munoz-Casabella A, Kochanek PM, Catapano J, Winkler E, Citerio G, et al. Sulfonylurea Receptor 1 in Central Nervous System Injury: An Updated Review. International Journal of Molecular Sciences. 2021; 22(21):11899. https://doi.org/10.3390/ijms222111899
Chicago/Turabian StyleJha, Ruchira M., Anupama Rani, Shashvat M. Desai, Sudhanshu Raikwar, Sandra Mihaljevic, Amanda Munoz-Casabella, Patrick M. Kochanek, Joshua Catapano, Ethan Winkler, Giuseppe Citerio, and et al. 2021. "Sulfonylurea Receptor 1 in Central Nervous System Injury: An Updated Review" International Journal of Molecular Sciences 22, no. 21: 11899. https://doi.org/10.3390/ijms222111899
APA StyleJha, R. M., Rani, A., Desai, S. M., Raikwar, S., Mihaljevic, S., Munoz-Casabella, A., Kochanek, P. M., Catapano, J., Winkler, E., Citerio, G., Hemphill, J. C., Kimberly, W. T., Narayan, R., Sahuquillo, J., Sheth, K. N., & Simard, J. M. (2021). Sulfonylurea Receptor 1 in Central Nervous System Injury: An Updated Review. International Journal of Molecular Sciences, 22(21), 11899. https://doi.org/10.3390/ijms222111899