
The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
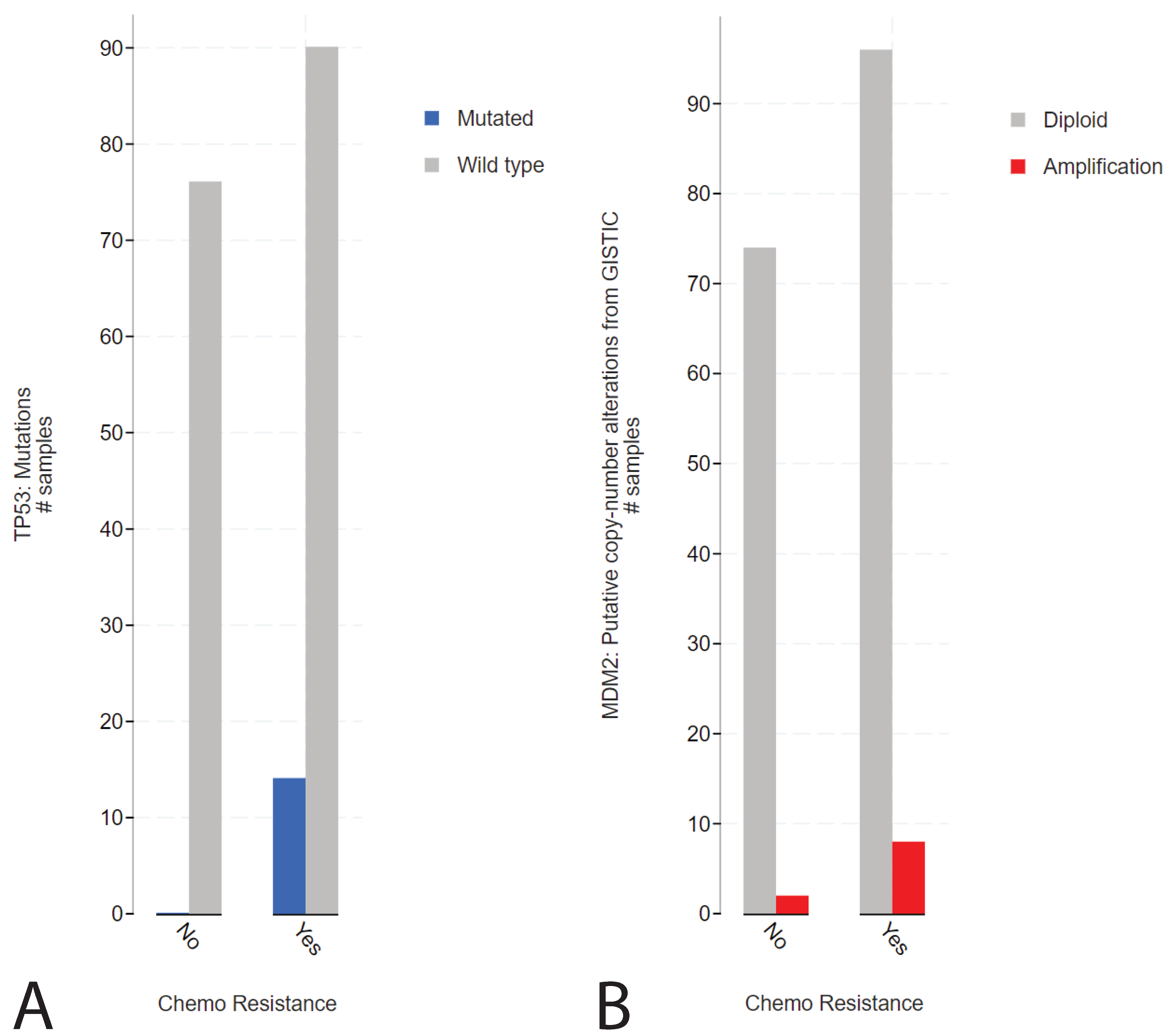
2.1. Presence of TP53 Mutations in Refractory Cisplatin-Resistant GCTs with a Specificity towards Mediastinal Localization
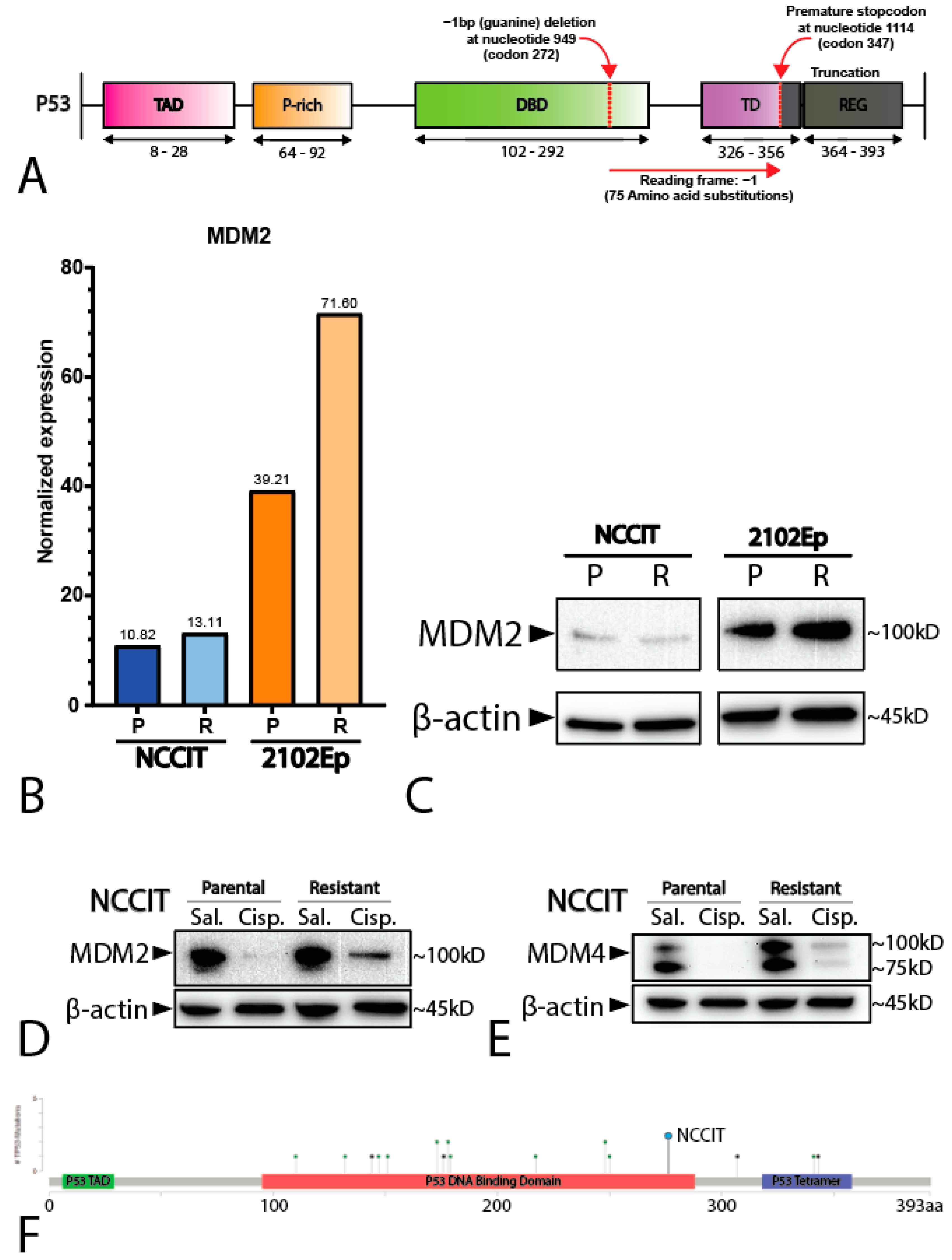
2.2. Mediastinal GCT Cell Line NCCIT Harbors Low Levels of MDM2 and Mutant TP53 whereas Testicular GCT Cell Line 2102Ep Harbors Wild-Type TP53 and High Levels of MDM2
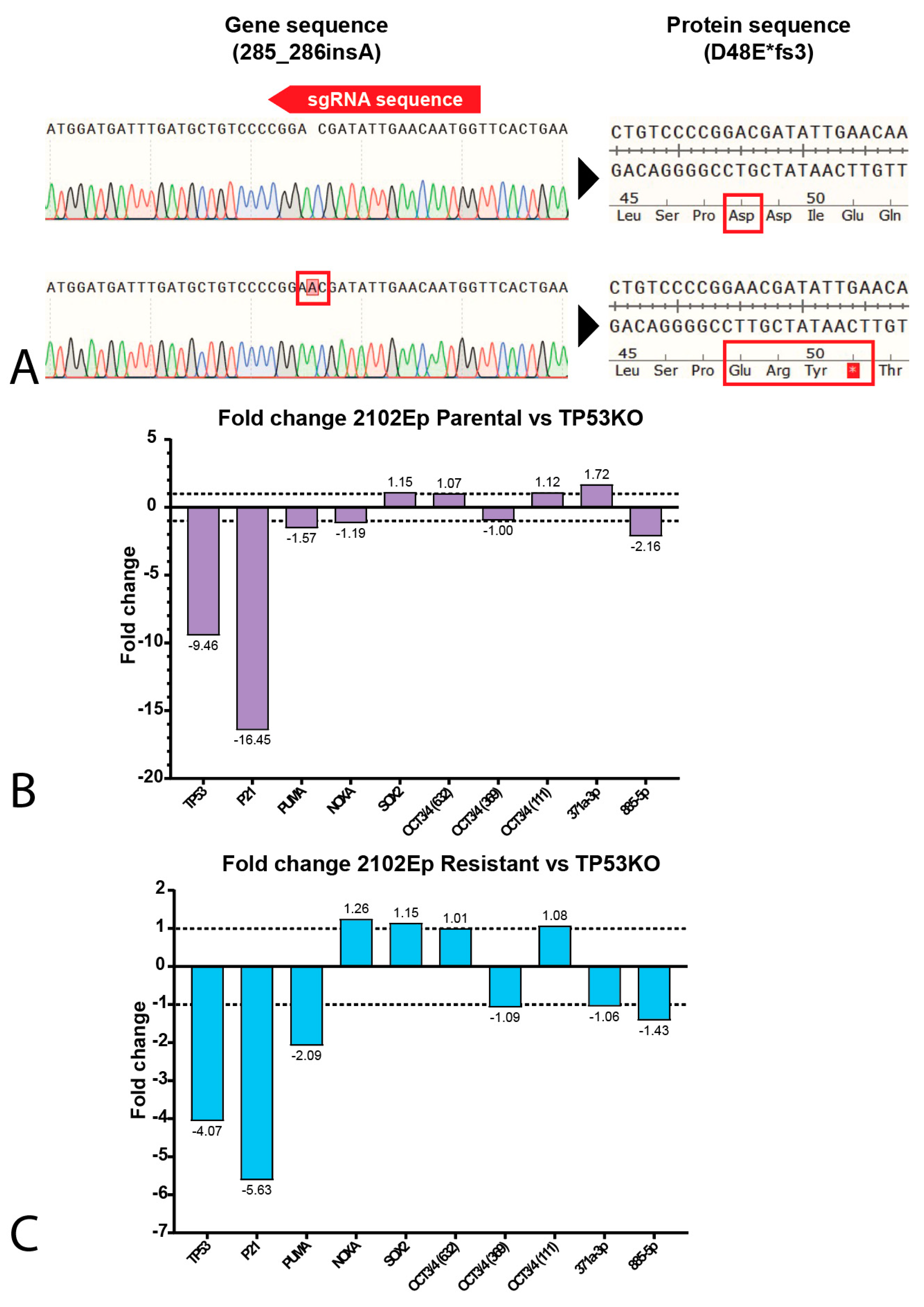
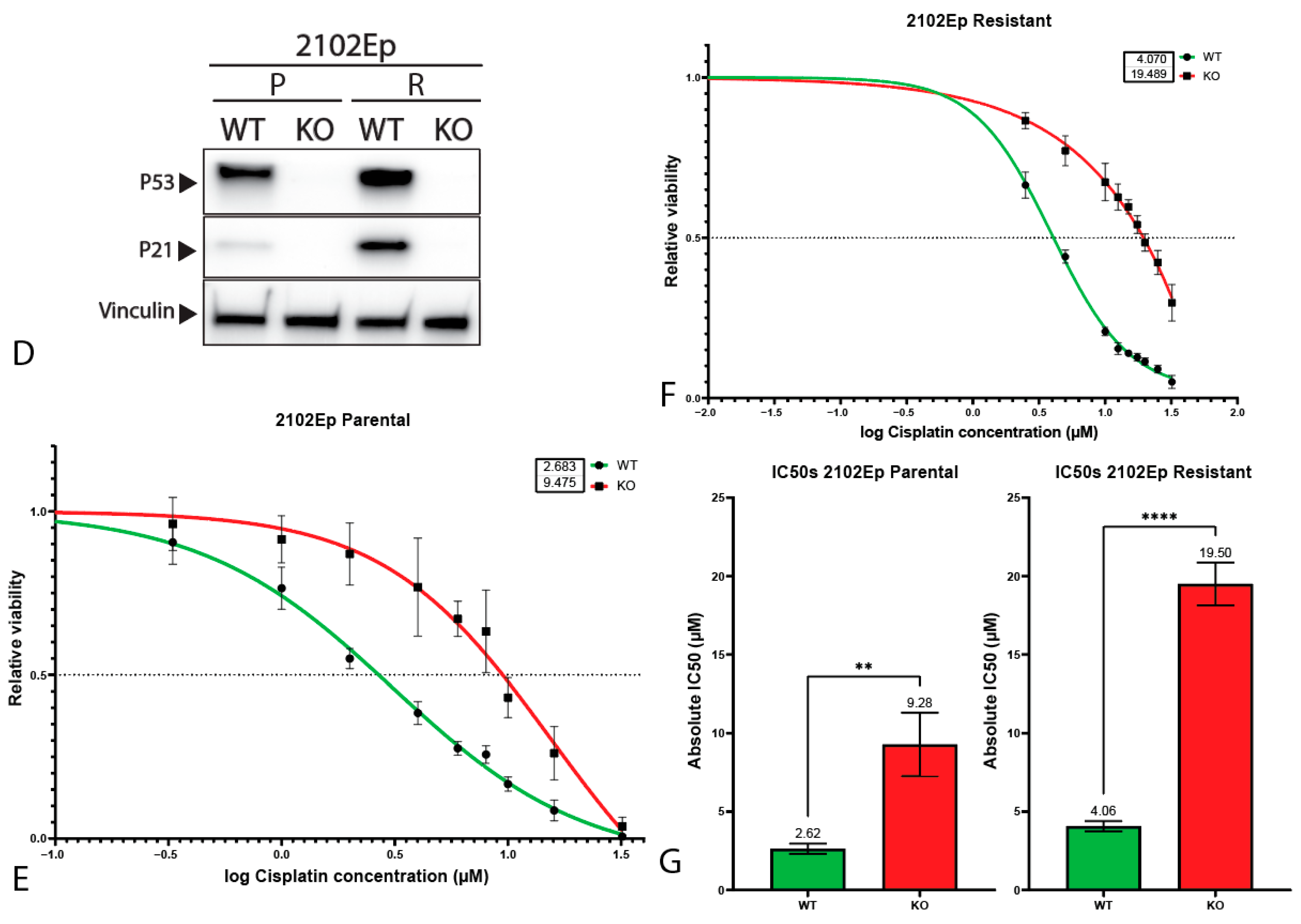
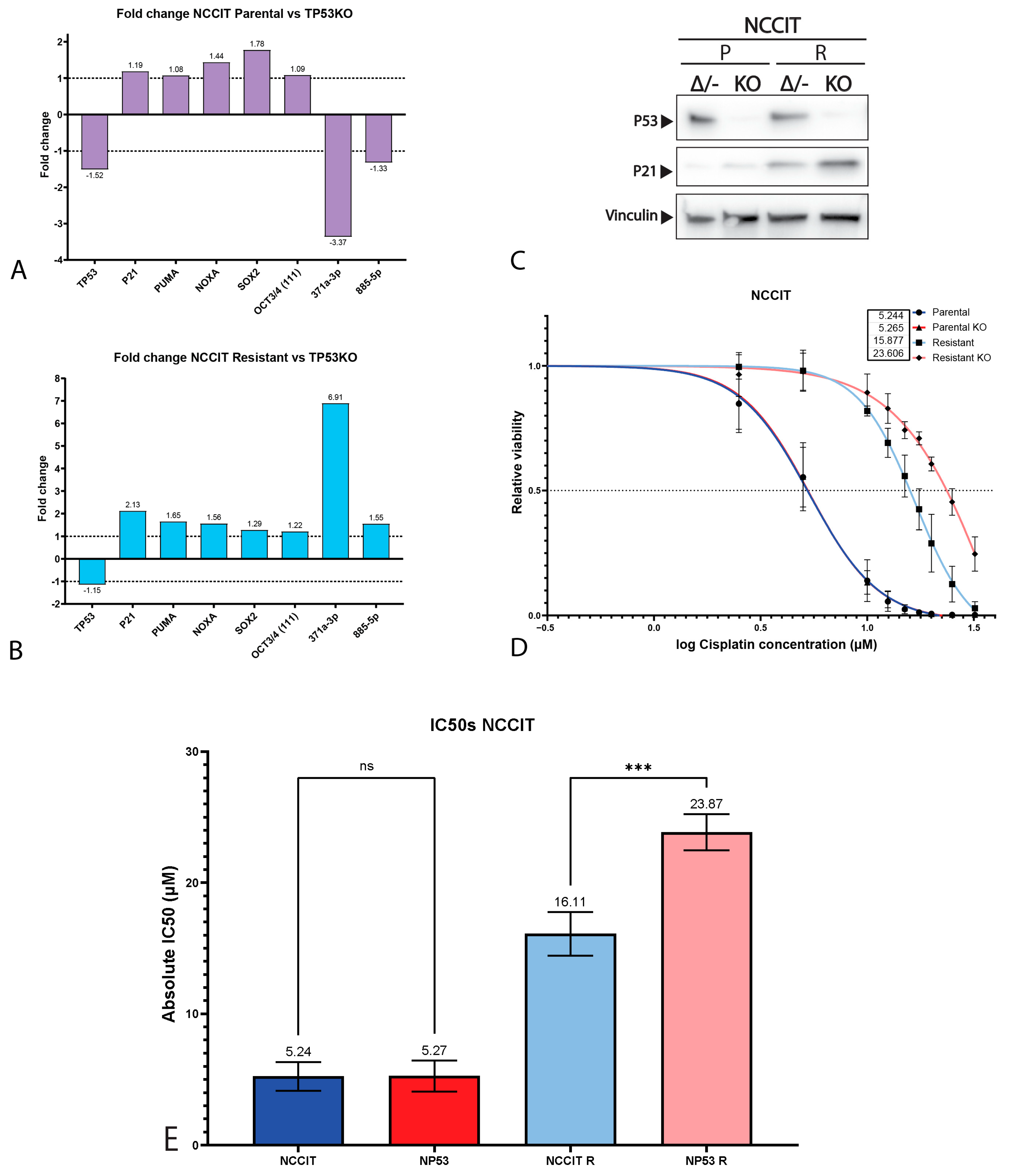
2.3. P53 Is Involved in Cisplatin Resistance in Both Wild-Type (Testicular) and Mutant (Mediastinal) GCT Cell Lines
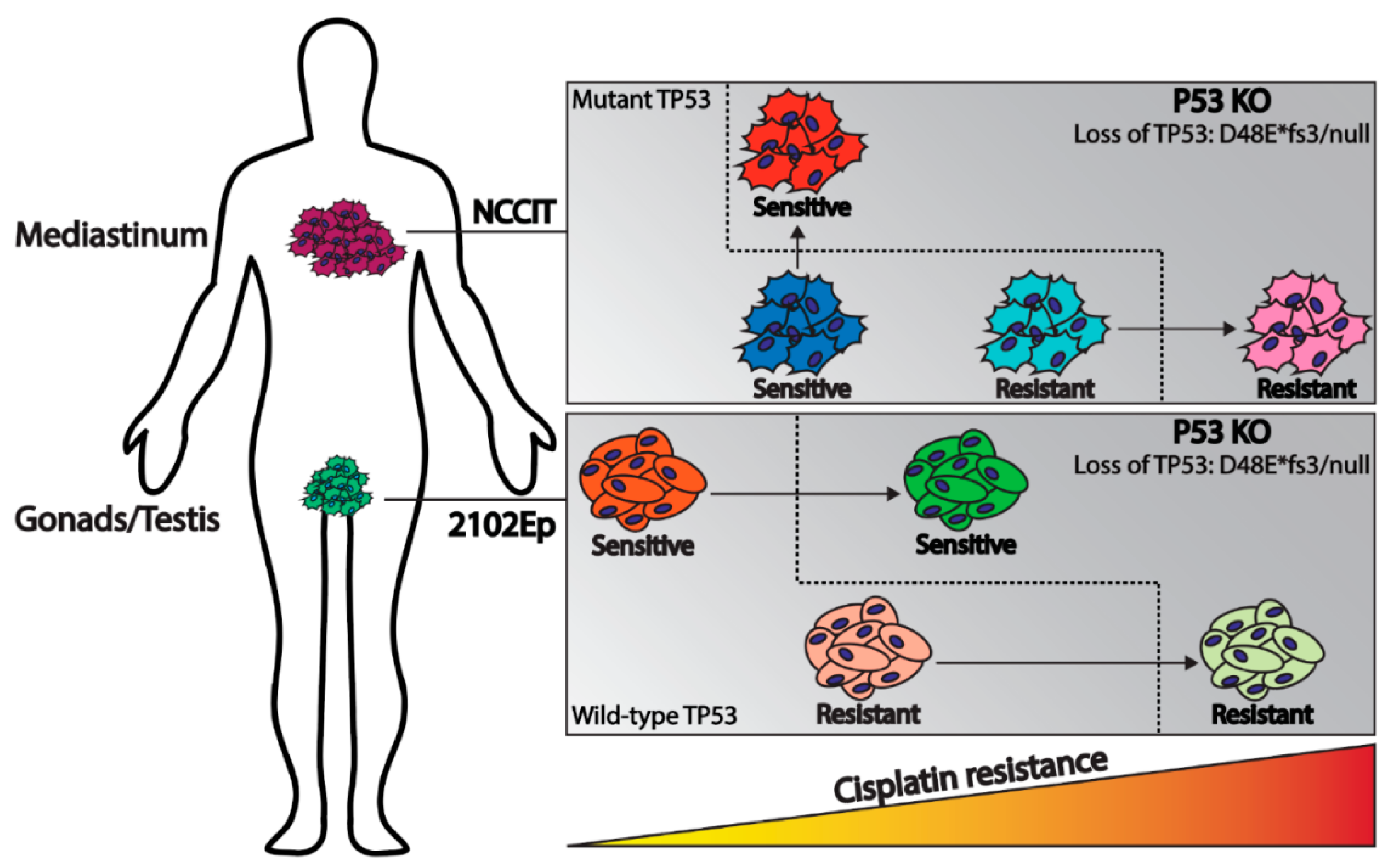
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. cBioPortal
4.3. RNA-seq
4.4. Western Blot
4.5. CRISPR-Mediated Knock-Out Cell Lines
4.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6.1. RNA Isolation
4.6.2. miRNA Profiling
4.6.3. mRNA Gene Expression
4.7. Viability Assays
4.8. Genotyping with GSA Arrays
4.9. Data Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L. Testicular cancer. Nat. Rev. Dis. Prim. 2018, 4, 29. [Google Scholar] [CrossRef]
- Palumbo, C.; Mistretta, F.A.; Mazzone, E.; Knipper, S.; Tian, Z.; Perrotte, P.; Antonelli, A.; Montorsi, F.; Shariat, S.F.; Saad, F.; et al. Contemporary Incidence and Mortality Rates in Patients with Testicular Germ Cell Tumors. Clin. Genitourin. Cancer 2019, 17, e1026–e1035. [Google Scholar] [CrossRef]
- Einhorn, L.H. Cis-Diamminedichloroplatinum, Vinblastine, and Bleomycin Combination Chemotherapy in Disseminated Testicular Cancer. Ann. Intern. Med. 1977, 87, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.C.; Kirschner, A.; Scarpato, K.R.; Morgans, A.K. Current Management of Refractory Germ Cell Tumors and Future Directions. Curr. Oncol. Rep. 2017, 19, 8. [Google Scholar] [CrossRef]
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer Statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Oosterhuis, J.W.; Looijenga, L. Testicular Germ-Cell Tumours in a Broader Perspective. Nat. Rev. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human Germ Cell Tumours from a Developmental Perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef]
- Timmerman, D.; Remmers, T.; Hillenius, S.; Looijenga, L. Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 5377. [Google Scholar] [CrossRef] [PubMed]
- Bagrodia, A.; Lee, B.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic Determinants of Cisplatin Resistance in Patients with Advanced Germ Cell Tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Kersemaekers, A.-M.F.; Mayer, F.; Molier, M.; Van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Role of P53 and MDM2 in Treatment Response of Human Germ Cell Tumors. J. Clin. Oncol. 2002, 20, 1551–1561. [Google Scholar] [CrossRef]
- Ulbright, T.M.; Orazi, A.; De Riese, W.; De Riese, C.; Messemer, E.J.; Foster, R.S.; Donohue, J.P.; Eble, J.N. The Correlation of P53 Protein Expression with Proliferative Activity and Occult Metastases in Clinical Stage I Non-Seminomatous Germ Cell Tumors of the Testis. Mod. Pathol. 1994, 7, 64–68. [Google Scholar]
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival Responses of Human Embryonic Stem Cells to DNA Damage. J. Cell. Physiol. 2009, 220, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; Van Der Kuip, H. p53 Hypersensitivity is the Predominant Mechanism of the Unique Responsiveness of Testicular Germ Cell Tumor (TGCT) Cells to Cisplatin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef]
- Jacobsen, C.; Honecker, F. Cisplatin Resistance in Germ Cell Tumours: Models and Mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline Genome Protection: Implications for Gamete Quality and Germ Cell Tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Mühlenberg, T.; Leahy, M.; Hoiczyk, M.; Gauler, T.; Schuler, M.; Looijenga, L. Therapeutic Potential of Mdm2 Inhibition in Malignant Germ Cell Tumours. Eur. Urol. 2010, 57, 679–687. [Google Scholar] [CrossRef]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.; De Jong, S. Disruption of the MDM2–p53 Interaction Strongly Potentiates p53-Dependent Apoptosis in Cisplatin-Resistant Human Testicular Carcinoma Cells via the Fas/FasL Pathway. Cell Death Dis. 2011, 2, e148. [Google Scholar] [CrossRef] [Green Version]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; Van Der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef]
- Mego, M.; Van Agthoven, T.; Gronesova, P.; Chovanec, M.; Miskovska, V.; Mardiak, J.; Looijenga, L.H.J. Clinical Utility of Plasma miR-371a-3p in Germ Cell Tumors. J. Cell. Mol. Med. 2019, 23, 1128–1136. [Google Scholar] [CrossRef]
- Fåhraeus, R.; Olivares-Illana, V. MDM2’s Social Network. Oncogene 2013, 33, 4365–4376. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-T.; Wang, J.-W.; Yang, L.; Wang, J.; Zhang, W. Primary Germ Cell Tumor in the Mediastinum-Report of 47 Cases. Zhonghua Zhong Liu Za Zhi 2006, 28, 863–866. [Google Scholar] [PubMed]
- Burger, H.; Nooter, K.; Boersma, A.W.; Kortland, C.J.; Stoter, G. Lack of Correlation Between Cisplatin-Induced Apoptosis, p53 Status and Expression of Bcl-2 Family Proteins in Testicular Germ Cell Tumour Cell Lines. Int. J. Cancer 1997, 73, 592–599. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [Green Version]
- de Jong, J.; Stoop, H.; Gillis, A.J.M.; Hersmus, R.; van Gurp, R.J.H.L.M.; van de Geijn, G.-J.M.; van Drunen, E.; Beverloo, H.B.; Schneider, D.T.; Sherlock, J.K.; et al. Further Characterization of the First Seminoma Cell Line TCam-2. Genes Chromosom. Cancer 2008, 47, 185–196. [Google Scholar] [CrossRef]
- Li, J.; Kurokawa, M. Regulation of MDM2 Stability After DNA Damage. J. Cell. Physiol. 2015, 230, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Spierings, D.; De Vries, E.E.G.; Vellenga, E.; De Jong, S. The Attractive Achilles Heel of Germ Cell Tumours: An Inherent Sensitivity to Apoptosis-Inducing Stimuli. J. Pathol. 2003, 200, 137–148. [Google Scholar] [CrossRef]
- Kerley-Hamilton, J.S.; Pike, A.M.; Li, N.; DiRenzo, J.; Spinella, M.J. A p53-Dominant Transcriptional Response to Cisplatin in Testicular Germ Cell Tumor-Derived Human Embyronal Carcinoma. Oncogene 2005, 24, 6090–6100. [Google Scholar] [CrossRef] [Green Version]
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA Repair Machinery and P53 in the Testicular Germ Cell Cancer: A Review. Oncotarget 2016, 7, 85641–85649. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, A.R.; Chan, C.S. Why are there Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 as Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, J.; Gillis, A.J.; Berg, A.V.D.; Dorssers, L.C.J.; Belge, G.; Dieckmann, K.-P.; Roest, H.P.; Van Der Laan, L.J.W.; Gietema, J.; Hamilton, R.J.; et al. Identification and Validation Model for Informative Liquid Biopsy-Based microRNA Biomarkers: Insights from Germ Cell Tumor In Vitro, In Vivo and Patient-Derived Data. Cells 2019, 8, 1637. [Google Scholar] [CrossRef] [Green Version]
- Almstrup, K.; Lobo, J.; Mørup, N.; Belge, G.; Meyts, E.R.-D.; Looijenga, L.H.J.; Dieckmann, K.-P. Application of miRNAs in the Diagnosis and Monitoring of Testicular Germ Cell Tumours. Nat. Rev. Urol. 2020, 17, 201–213. [Google Scholar] [CrossRef]
- Murray, M.J.; Bell, E.; Raby, K.L.; Rijlaarsdam, M.; Gillis, A.J.M.; Looijenga, L.; Brown, H.; Destenaves, B.; Nicholson, J.C.; Coleman, N.S. A pipeline to Quantify Serum and Cerebrospinal Fluid Micrornas for Diagnosis and Detection of Relapse in Paediatric Malignant Germ-Cell Tumours. Br. J. Cancer 2016, 114, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Murray, M.J.; Halsall, D.J.; Hook, C.E.; Williams, D.M.; Nicholson, J.C.; Coleman, N.; Sweet, W.; Duh, Y.-J.; Greenfield, L.; Tarco, E.; et al. Identification of MicroRNAs from the miR-371∼373 and miR-302 Clusters as Potential Serum Biomarkers of Malignant Germ Cell Tumors. Am. J. Clin. Pathol. 2011, 135, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Syring, I.; Bartels, J.; Holdenrieder, S.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Circulating Serum miRNA (miR-367-3p, miR-371a-3p, miR-372-3p and miR-373-3p) as Biomarkers in Patients with Testicular Germ Cell Cancer. J. Urol. 2015, 193, 331–337. [Google Scholar] [CrossRef]
- Leão, R.; Albersen, M.; Looijenga, L.H.; Tandstad, T.; Kollmannsberger, C.; Murray, M.J.; Culine, S.; Coleman, N.; Belge, G.; Hamilton, R.J.; et al. Circulating MicroRNAs, the Next-Generation Serum Biomarkers in Testicular Germ Cell Tumours: A Systematic Review. Eur. Urol. 2021, 80, 456–466. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 Regulators as Targets in Anticancer Therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [Green Version]
- Kollmannsberger, C.; Nichols, C.; Meisner, C.; Mayer, F.; Kanz, L.; Bokemeyer, C. Identification of Prognostic Subgroups Among Patients with Metastatic ‘IGCCCG Poor-Prognosis’ Germ-Cell Cancer: An Explorative Analysis Using Cart Modeling. Ann. Oncol. 2000, 11, 1115–1120. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H. Mediastinal Germ Cell Tumors: Many Questions and Perhaps an Answer. J. Clin. Investig. 2020, 130, 6238–6241. [Google Scholar] [CrossRef] [PubMed]
- Saiki, A.Y.; Caenepeel, S.; Yu, D.; Lofgren, J.A.; Osgood, T.; Robertson, R.; Canon, J.; Su, C.; Jones, A.; Zhao, X.; et al. MDM2 Antagonists Synergize Broadly and Robustly with Compounds Targeting Fundamental Oncogenic Signaling Pathways. Oncotarget 2014, 5, 2030–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-P.; Liu, F.; Wang, W. Two-Phase Dynamics of p53 in the DNA Damage Response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beishline, K.; Azizkhan-Clifford, J. Interplay between the Cell Cycle and Double-Strand Break Response in Mammalian Cells. Cell Cycle Control 2014, 1170, 41–59. [Google Scholar] [CrossRef]
- Aubrey, B.; Kelly, G.L.; Janic, A.; Herold, M.; Strasser, A. How does p53 Induce Apoptosis and how does this Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.; Stoop, H.; Scheffer, G.L.; Scheper, R.; Oosterhuis, J.W.; Looijenga, L.; Bokemeyer, C. Molecular Determinants of Treatment Response in Human Germ Cell Tumors. Clin. Cancer Res. 2003, 9, 767–773. [Google Scholar]
- Dieckmann, K.-P.; Spiekermann, M.; Balks, T.; Flor, I.; Löning, T.; Bullerdiek, J.; Belge, G. MicroRNAs miR-371-3 in Serum as Diagnostic Tools in the Management of Testicular Germ Cell Tumours. Br. J. Cancer 2012, 107, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, K.-P.; Radtke, A.; Spiekermann, M.; Balks, T.; Matthies, C.; Becker, P.; Ruf, C.; Oing, C.; Oechsle, K.; Bokemeyer, C.; et al. Serum Levels of MicroRNA miR-371a-3p: A Sensitive and Specific New Biomarker for Germ Cell Tumours. Eur. Urol. 2017, 71, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Dieckmann, K.-P.; Radtke, A.; Geczi, L.; Matthies, C.; Anheuser, P.; Eckardt, U.; Sommer, J.; Zengerling, F.; Trenti, E.; Pichler, R.; et al. Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J. Clin. Oncol. 2019, 37, 1412–1423. [Google Scholar] [CrossRef]
- Koster, R.; Di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; Berg, A.V.D.; Suurmeijer, A.; Bischoff, R.; Gietema, J.; De Jong, S. Cytoplasmic p21 Expression Levels Determine Cisplatin Resistance in Human Testicular Cancer. J. Clin. Investig. 2010, 120, 3594–3605. [Google Scholar] [CrossRef] [Green Version]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-Independent p21 Expression Causes Genomic Instability by Deregulating Replication Licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational Signatures Reveal the role of RAD52 in p53-Independent p21-Driven Genomic Instability. Genome Biol. 2018, 19, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreis, N.-N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Teshima, S.; Shimosato, Y.; Hirohashi, S.; Tome, Y.; Hayashi, I.; Kanazawa, H.; Kakizoe, T. Four new human germ cell tumor cell lines. Lab. Investig. 1988, 59, 328–336. [Google Scholar]
- Wang, N.; Trend, B.; Bronson, D.L.; Fraley, E.E. Nonrandom abnormalities in chromosome 1 in human testicular cancers. Cancer Res. 1980, 40, 796–802. [Google Scholar] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmerman, D.M.; Eleveld, T.F.; Gillis, A.J.M.; Friedrichs, C.C.; Hillenius, S.; Remmers, T.L.; Sriram, S.; Looijenga, L.H.J. The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors. Int. J. Mol. Sci. 2021, 22, 11774. https://doi.org/10.3390/ijms222111774
Timmerman DM, Eleveld TF, Gillis AJM, Friedrichs CC, Hillenius S, Remmers TL, Sriram S, Looijenga LHJ. The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors. International Journal of Molecular Sciences. 2021; 22(21):11774. https://doi.org/10.3390/ijms222111774
Chicago/Turabian StyleTimmerman, Dennis M., Thomas F. Eleveld, Ad J. M. Gillis, Carlijn C. Friedrichs, Sanne Hillenius, Tessa L. Remmers, Sruthi Sriram, and Leendert H. J. Looijenga. 2021. "The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors" International Journal of Molecular Sciences 22, no. 21: 11774. https://doi.org/10.3390/ijms222111774
APA StyleTimmerman, D. M., Eleveld, T. F., Gillis, A. J. M., Friedrichs, C. C., Hillenius, S., Remmers, T. L., Sriram, S., & Looijenga, L. H. J. (2021). The Role of TP53 in Cisplatin Resistance in Mediastinal and Testicular Germ Cell Tumors. International Journal of Molecular Sciences, 22(21), 11774. https://doi.org/10.3390/ijms222111774