
Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
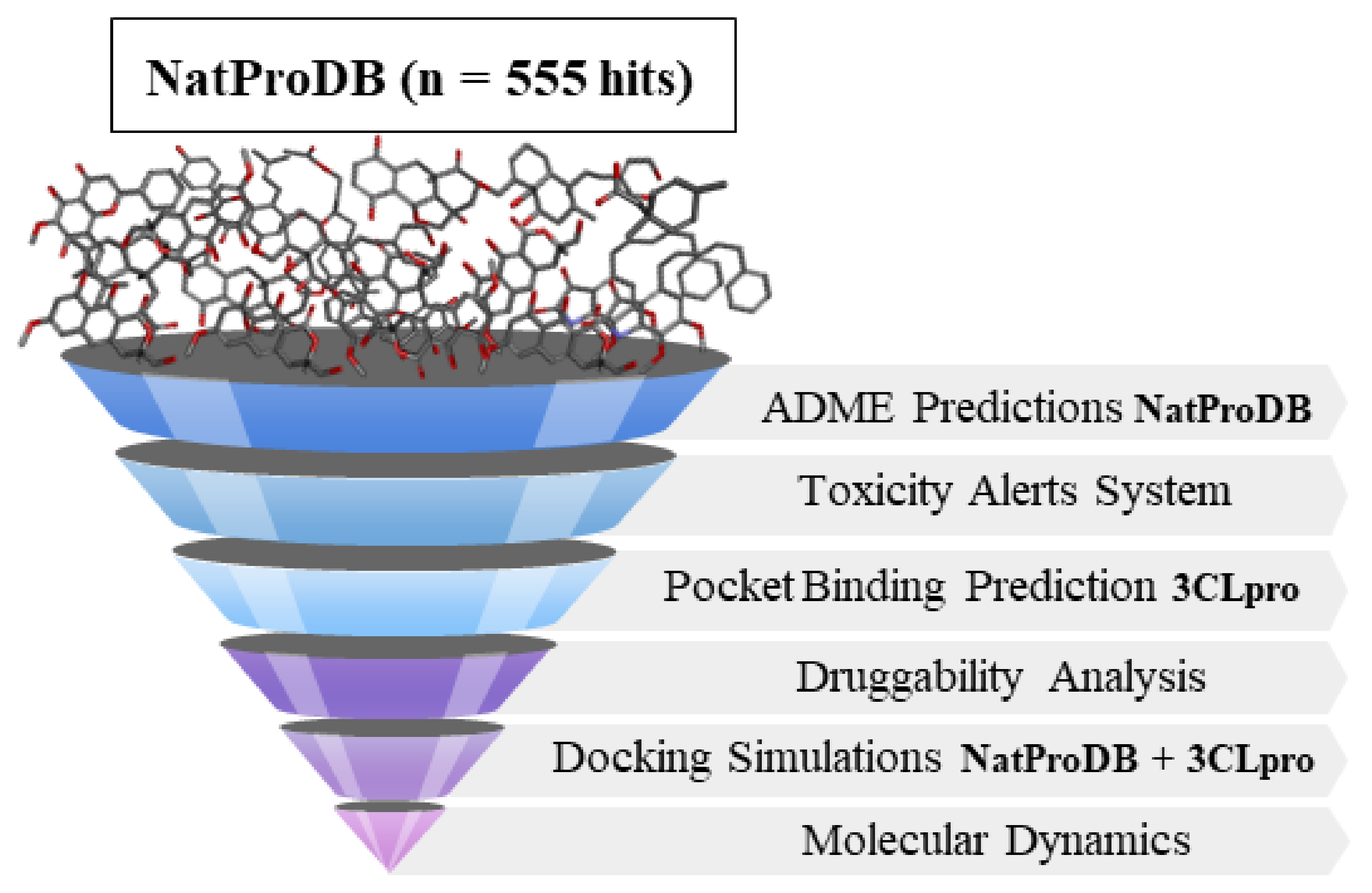
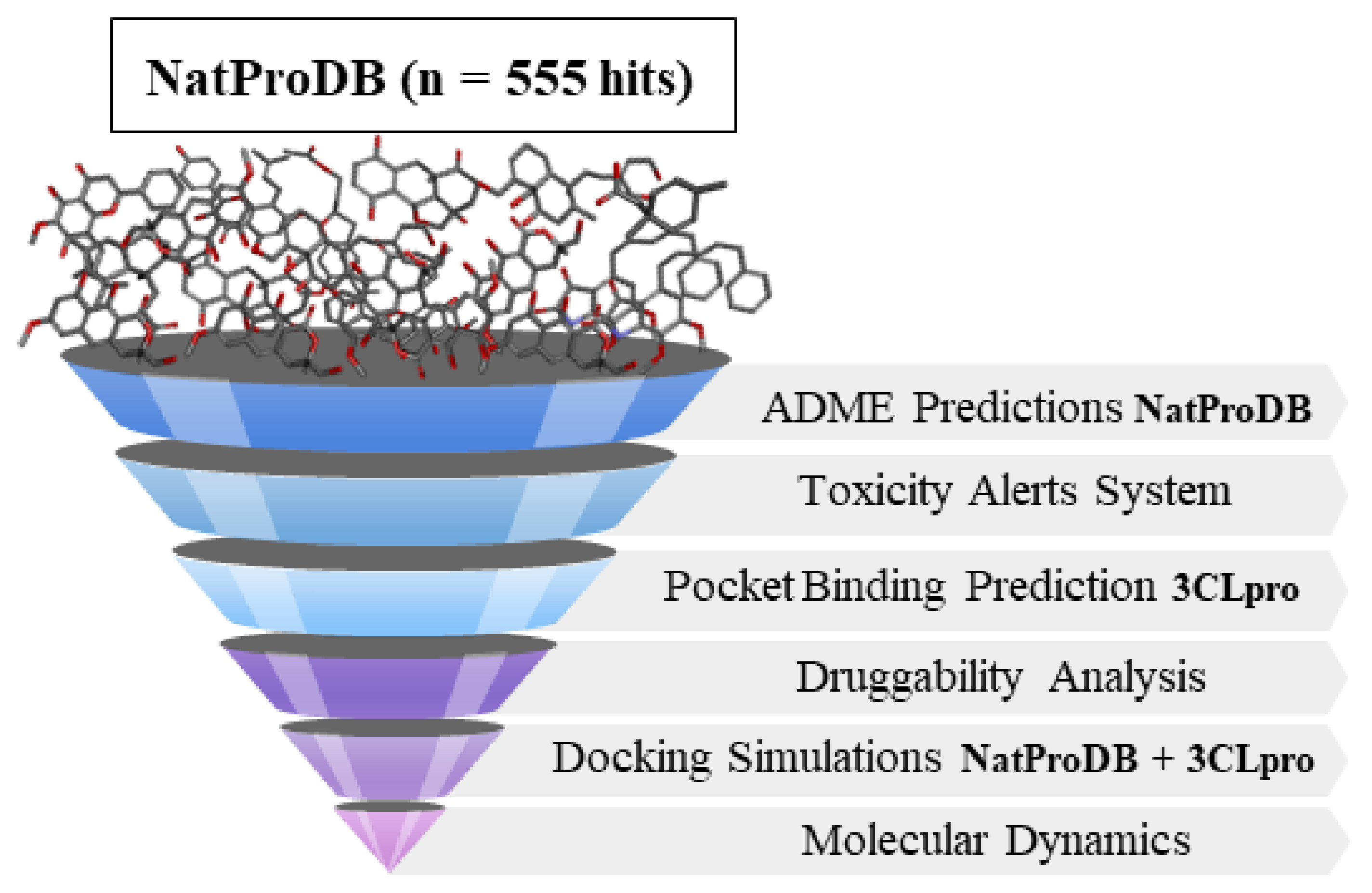
2. Results
2.1. ADMET Analysis by QikPropTM
2.2. Toxicological Analysis by Derek NexusTM
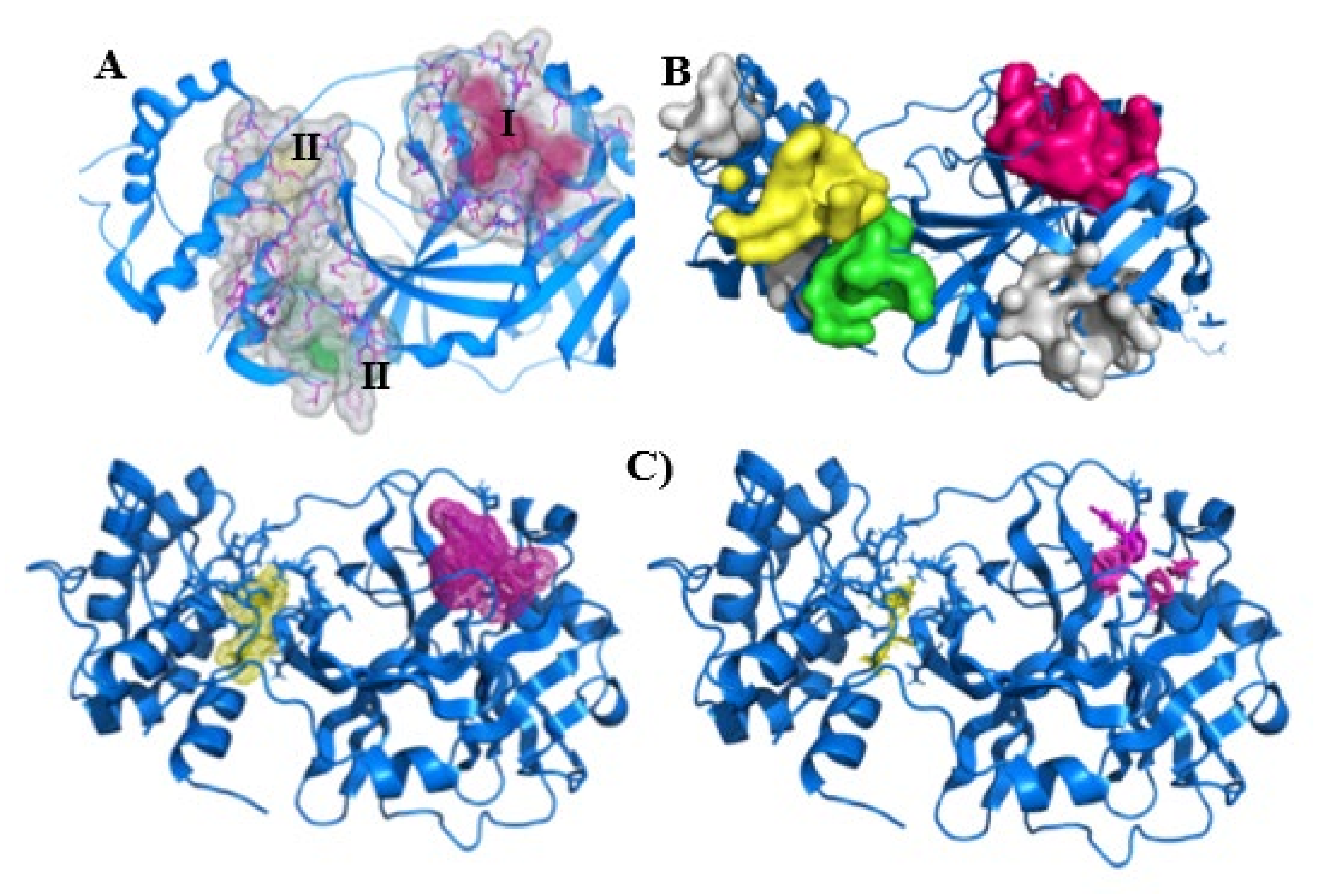
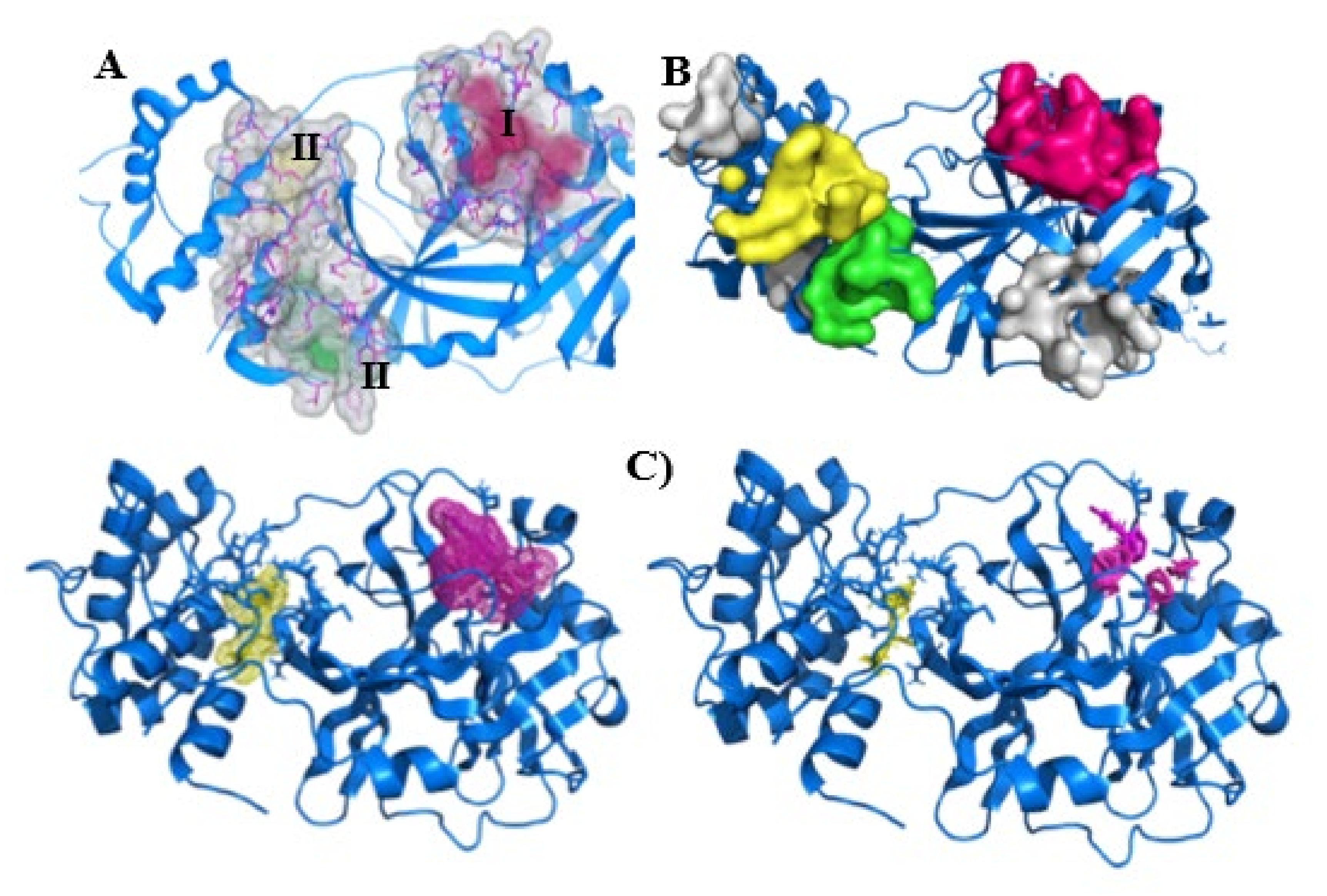
2.3. Site Prediction and Druggability Analysis of SARS-CoV-2 Mpro
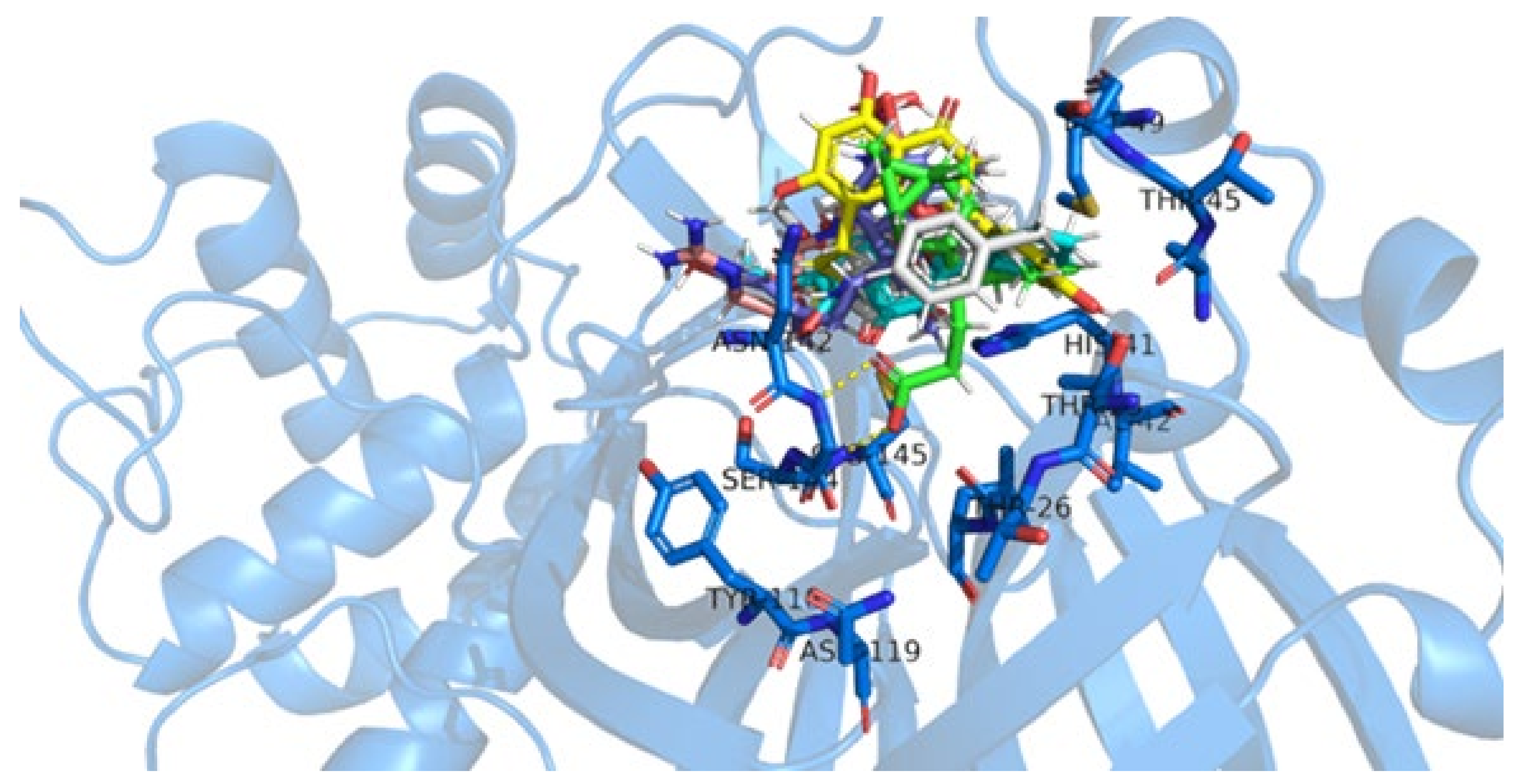
2.4. Molecular Docking Simulations
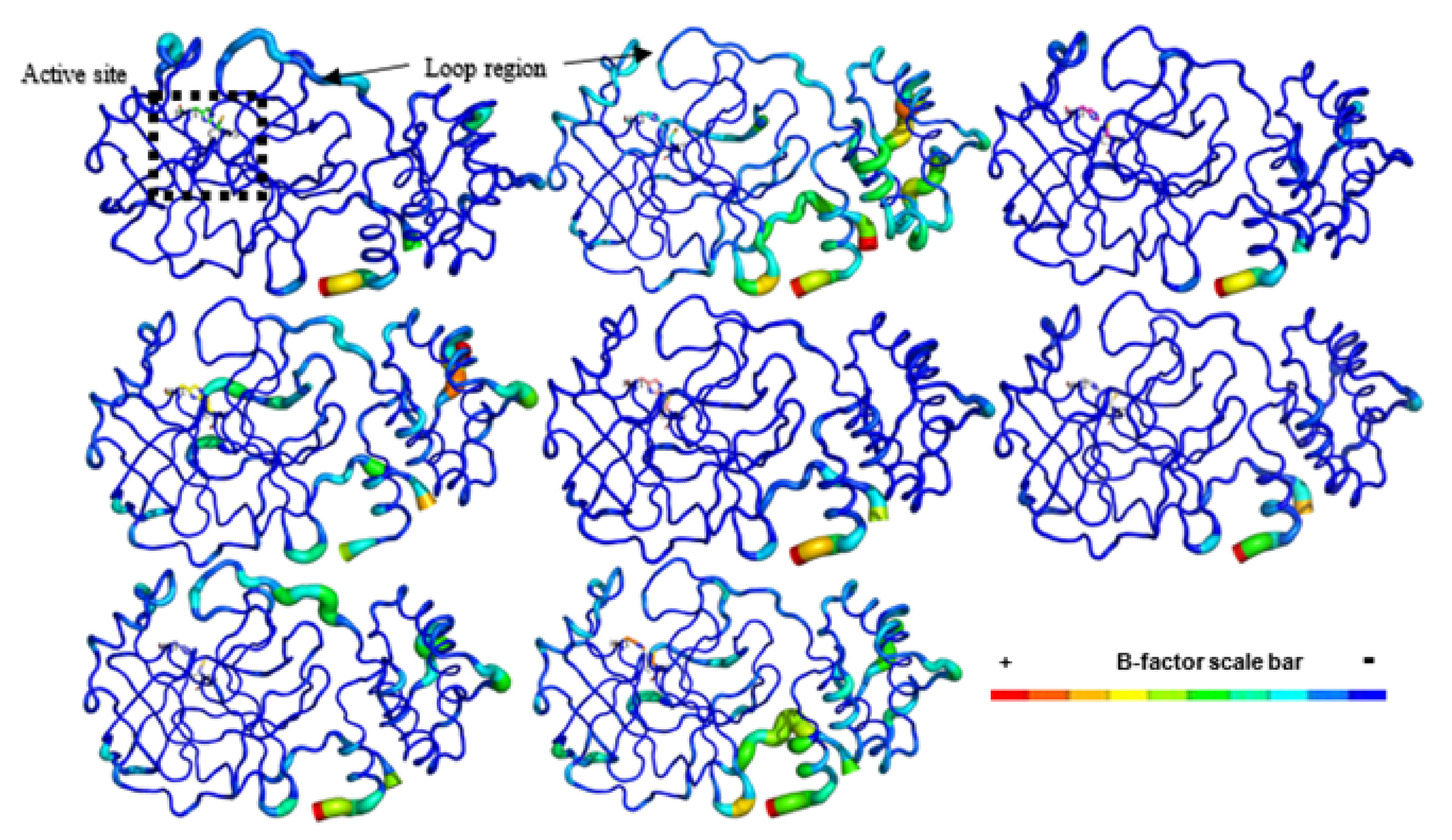
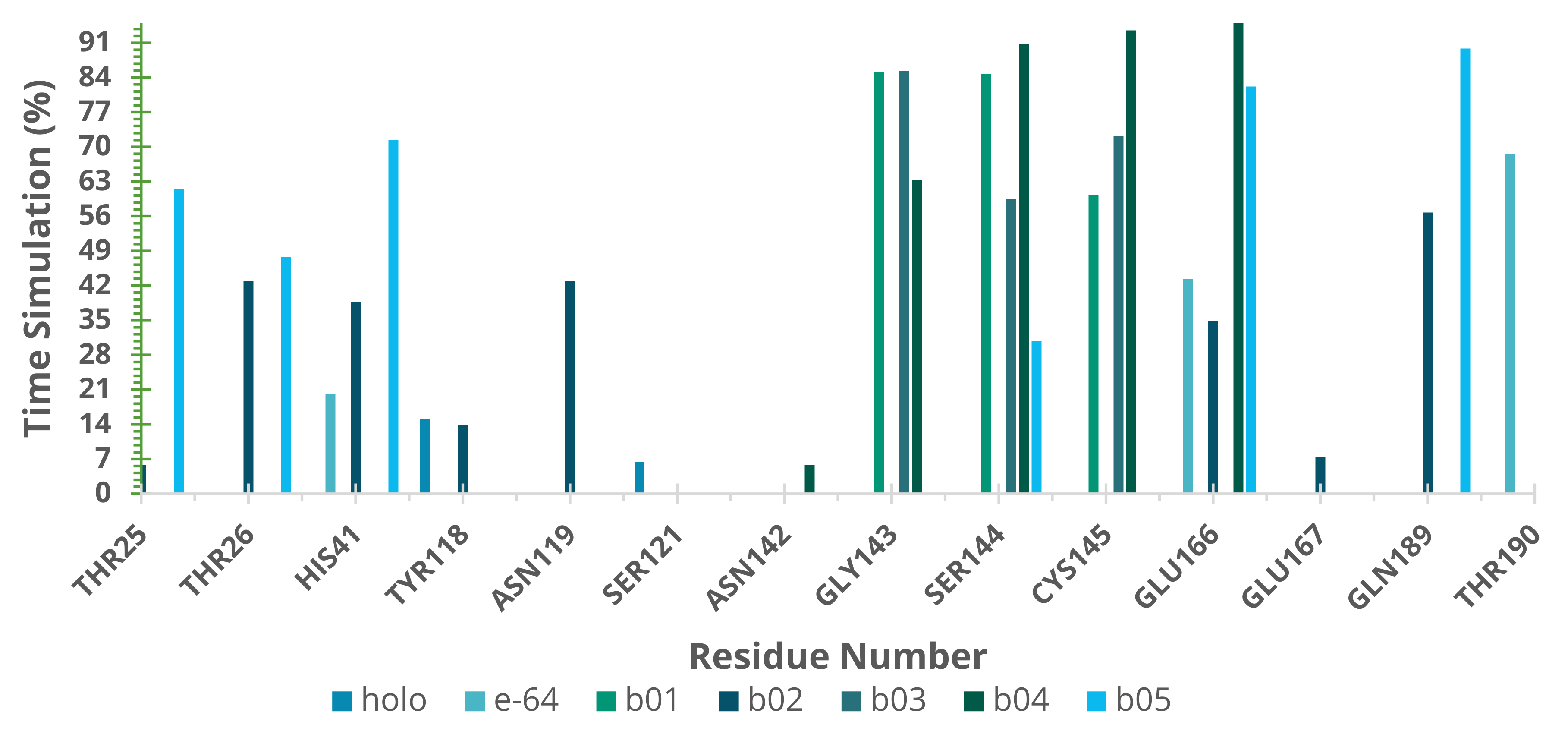
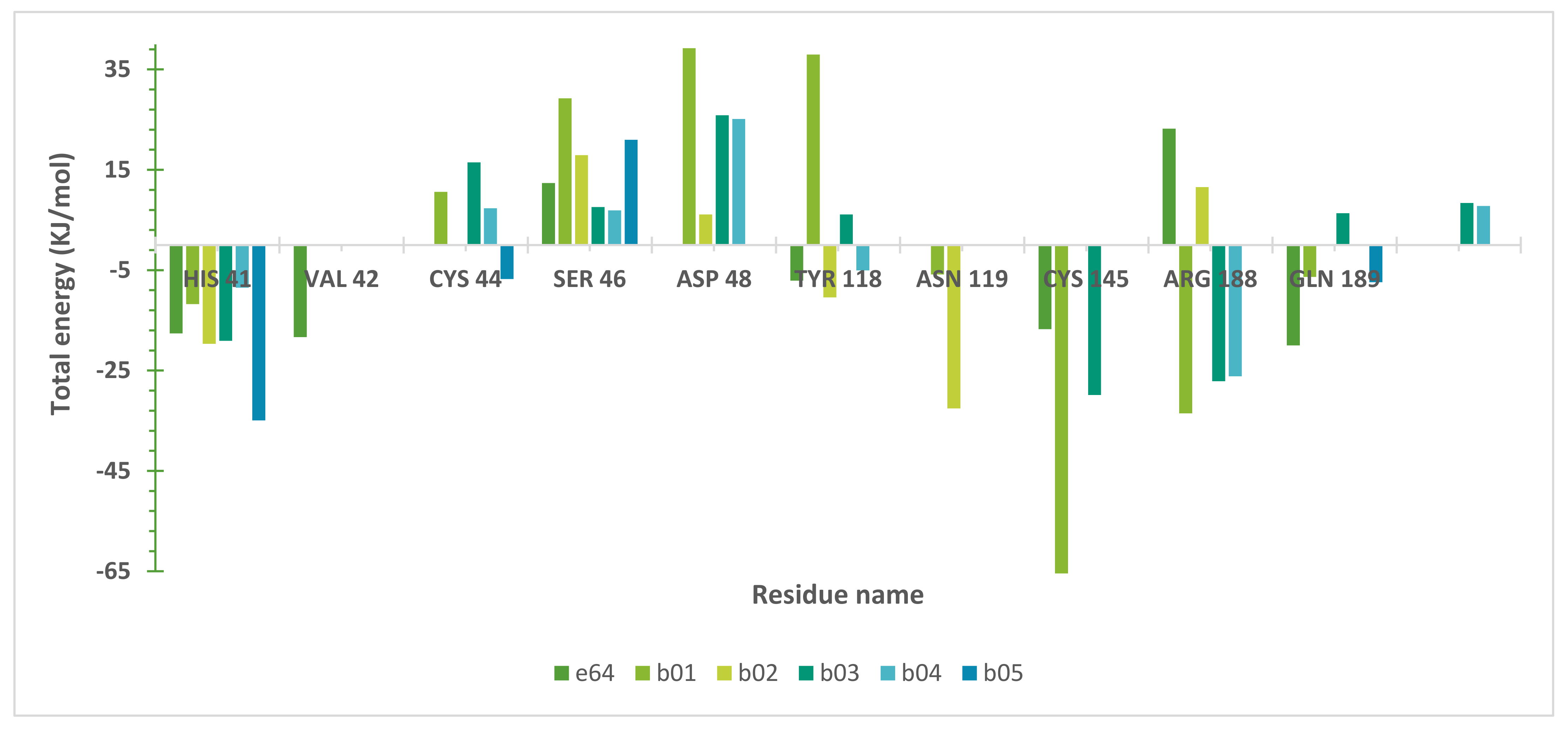
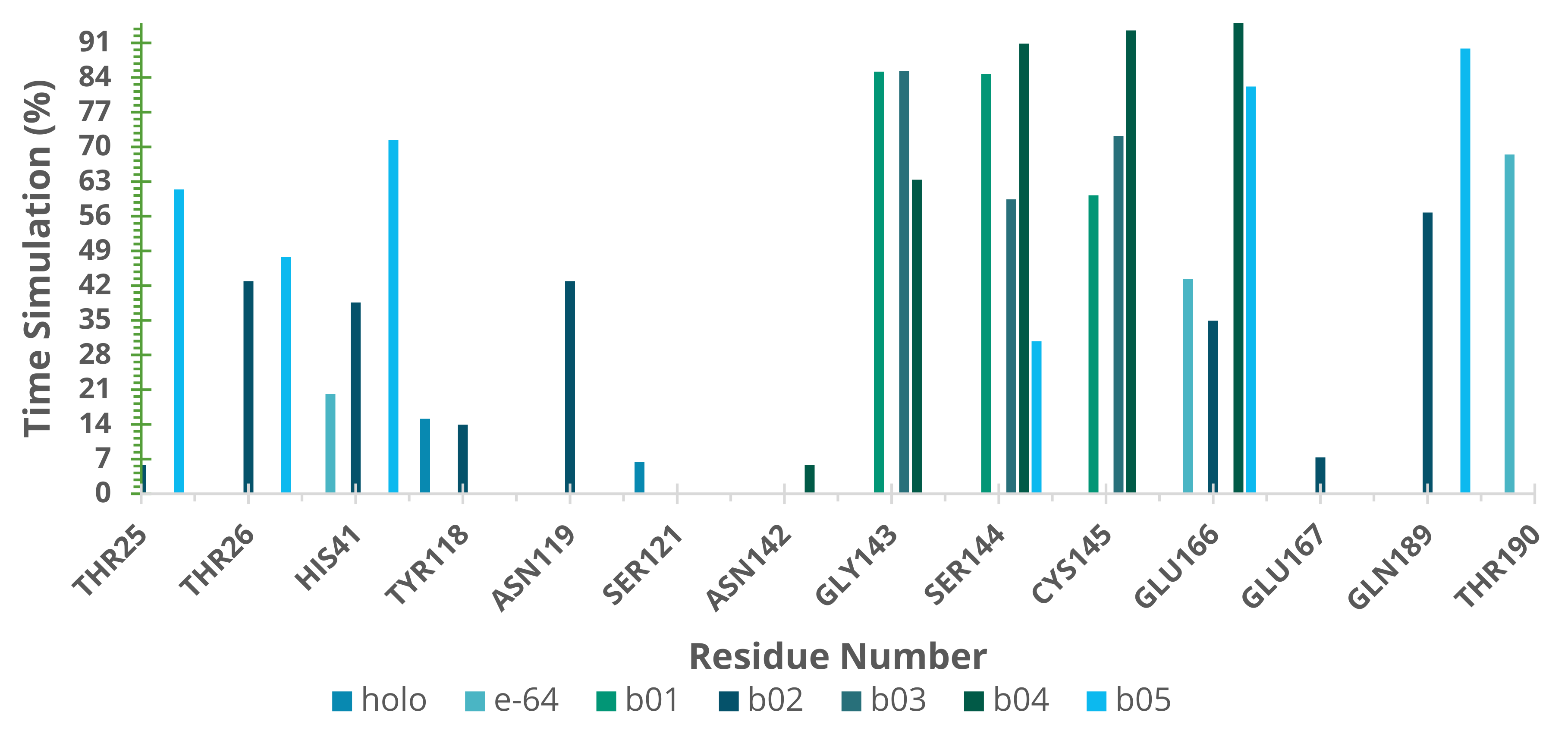
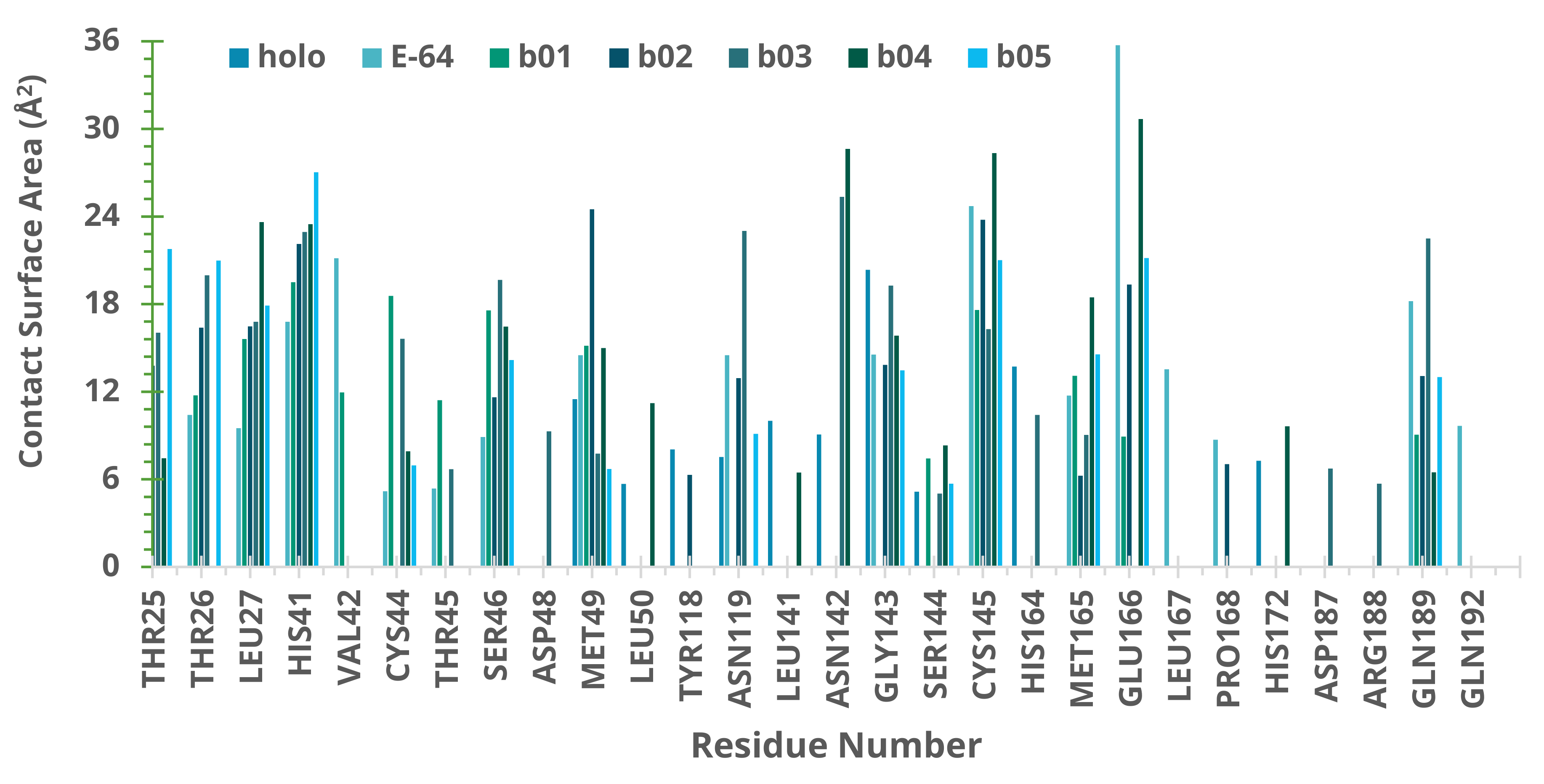
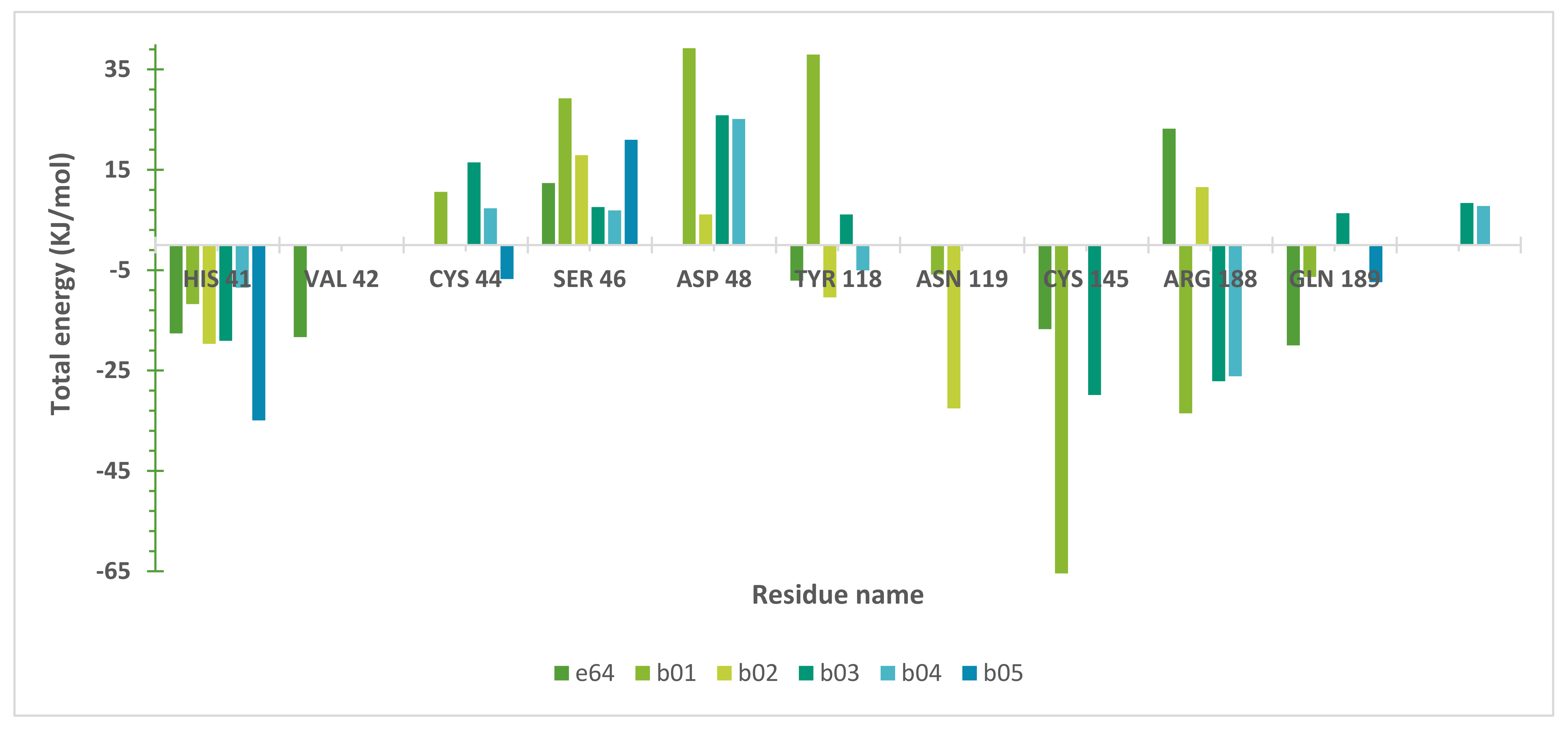
2.5. Molecular Dynamics (MD) Simulations
3. Discussion
4. Materials and Methods
4.1. SARS-CoV-2 Mpro Protein Preparation
4.2. Site Prediction and Druggability Analysis of SARS-CoV-2 Mpro
4.2.1. SeeSAR
4.2.2. PockDrug
4.2.3. FTMap
4.2.4. Molecular Docking on SARS-CoV-2 Mpro
4.2.5. SARS-CoV-2 Ligand Preparation
4.2.6. ADMET Analysis by QikPropTM and DerekTM
4.2.7. Molecular Dynamics (MD) Simulations on SARS-CoV-2 Mpro-NatProDB Complexes
4.2.8. Hydrogen Bond Capacity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| ACE2 | angiotensin-converting enzyme |
| ADMET | absorption, distribution, metabolism, and excretion/toxicity |
| MD | molecular dynamics |
| NatProDB | Natural Products Database of the Bahia Semi-Arid region |
| 3CLpro | SARS-CoV-2 Main Protease |
| CoVs | coronaviruses |
| PDB | Protein Data Bank |
| PSA | polar surface area |
| %HOA | human oral absorption in percentage |
| QPlogPo/w | logarithm of the partition coefficient in 1-octanol/water predicted by QikProp |
| QPLogBB | logarithm of the blood–brain barrier predicted by QikProp |
| QPPCaco | permeability across Caco-2 cells predicted by QikProp |
| CNS | central nervous system |
| RMSF | root-mean square fluctuation |
| LogKhsa | logarithmic human serum albumin-binding predicted by QikProp |
| EA | estimated affinities |
| Hbondcapac. | hydrogen bond capability |
| ΔEbinding | binding energy |
| QPPMDCK | permeability across Madin-Darby Canine Kidney cells predicted by QikProp |
| MW | molecular weight |
| HTS | high-throughput screening |
References
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent Discovery and Development of Inhibitors Targeting Coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2008, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, S.G.V.; Santos, W.C. Clinical Trials on Drug Repositioning for COVID-19 Treatment. Rev. Panam. Salud Públ. 2020, 44, e40. [Google Scholar] [CrossRef]
- Anand, K. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Ziebuhr, J. Molecular Biology of Severe Acute Respiratory Syndrome Coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Snijder, E.J.; Ziebuhr, J. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Bajaj, A.; Purohit, H.J. Understanding SARS-CoV-2: Genetic Diversity, Transmission and Cure in Human. Indian J. Microbiol. 2020, 60, 398–401. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; He, W.-T.; Wang, L.; Lai, A.; Ji, X.; Zhai, X.; Li, G.; Suchard, M.A.; Tian, J.; Zhou, J.; et al. COVID-19: Epidemiology, Evolution, and Cross-Disciplinary Perspectives. Trends Mol. Med. 2020, 26, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Lin, Q.; Jin, S.; You, L. Coronavirus 2019-NCoV: A Brief Perspective from the Front Line. J. Infect. 2020, 80, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.K.S.; Lee, J.-K.; Kalia, V.C. Deploying Biomolecules as Anti-COVID-19 Agents. Indian J. Microbiol. 2020, 60, 263–268. [Google Scholar] [CrossRef]
- Rishi, P.; Thakur, K.; Vij, S.; Rishi, L.; Singh, A.; Kaur, I.P.; Patel, S.K.S.; Lee, J.-K.; Kalia, V.C. Diet, Gut Microbiota and COVID-19. Indian J. Microbiol. 2020, 60, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.; Senthebane, D.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Wink, M. Medicinal Plants: A Source of Anti-Parasitic Secondary Metabolites. Molecules 2012, 17, 2771. [Google Scholar] [CrossRef] [Green Version]
- Fakhar, Z.; Faramarzi, B.; Pacifico, S.; Faramarzi, S. Anthocyanin Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease: An in-Silico Perspective of Therapeutic Targets against COVID-19 Pandemic. J. Biomol. Struct. Dyn. 2020, 39, 6171–6183. [Google Scholar] [CrossRef] [PubMed]
- Mattio, L.M.; Catinella, G.; Pinto, A.; Dallavalle, S. Natural and Nature-Inspired Stilbenoids as Antiviral Agents. Eur. J. Med. Chem. 2020, 202, 112541. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olubiyi, O.O.; Olagunju, M.; Keutmann, M.; Loschwitz, J.; Strodel, B. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Molecules 2020, 25, 3193. [Google Scholar] [CrossRef]
- Sillapachaiyaporn, C.; Chuchawankul, S. HIV-1 Protease and Reverse Transcriptase Inhibition by Tiger Milk Mushroom (Lignosus Rhinocerus) Sclerotium Extracts: In Vitro and in Silico Studies. J. Tradit. Complement. Med. 2020, 10, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, L. Turning the Tide: Natural Products and Natural-Product-Inspired Chemicals as Potential Counters to SARS-CoV-2 Infection. Front. Pharmacol. 2019, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, T.M.; Prado, P.I. Biodiversity of Brazil: A synthesis of the current state of knowledge. In Evaluation of the State of Knowledge on Biological Diversity in Brazil; Baumgarten, L.C., de Andrade, L.A.Z., Cariello, M.O., Eds.; CID Ambiental: Brasilia, Brazil, 2003; pp. 139–144. [Google Scholar]
- Lucchese, A.M.; Vale, A.E. Plantas Da Caatinga: Perfil Botânico, Fitoquímica e Atividade Biológica; Associação Plantas do Nordeste: Recife, Brazil, 2006; Volume 4. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Da Paixão, V.G.; Pita, S.S.D.R. In Silico Identification and Evaluation of New Trypanosoma Cruzi Trypanothione Reductase (TcTR) Inhibitors Obtained from Natural Products Database of the Bahia Semi-Arid Region (NatProDB). Comput. Biol. Chem. 2019, 79, 36–47. [Google Scholar] [CrossRef]
- Macmillan, D.S.; Chilton, M.L. A Defined Approach for Predicting Skin Sensitisation Hazard and Potency Based on the Guided Integration of in Silico, in Chemico and in Vitro Data Using Exclusion Criteria. Regul. Toxicol. Pharmacol. 2019, 101, 35–47. [Google Scholar] [CrossRef]
- Silva, R.; Poiani, J.; Ramos, R.; Costa, J.; Silva, C.; Brasil, D.; Santos, C. Ligand- and Structure-Based Virtual Screening from 16-((Diisobutylamino)Methyl)-6α-Hydroxyivouacapane-7β,17β-Lactone a Compound with Potential Anti-Prostate Cancer Activity. J. Serbian Chem. Soc. 2019, 84, 153–174. [Google Scholar] [CrossRef] [Green Version]
- Duffy, E.M.; Jorgensen, W.L. Prediction of Properties from Simulations: Free Energies of Solvation in Hexadecane, Octanol, and Water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of Drug Solubility from Monte Carlo Simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158. [Google Scholar] [CrossRef]
- Silva, R.C.; Ferreira, I.M.; Federico, L.B.; Hage-Melim, L.I.d.S.; Macêdo, W.J.C.; Porto, A.L.M.; Taft, C.A.; Silva, C.H.T.P.; dos Santos, C.B.R. ADME/Tox Study and Molecular Dynamics Simulations Applied in the Design of New Potential GABA-AT Inhibitors. In Functional Properties of Advanced Engineering Materials and Biomolecules; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Cruz, J.V.; Giuliatti, S.; Alves, L.B.; Silva, R.C.; Ferreira, E.F.B.; Kimani, N.M.; Silva, C.H.T.P.; Souza, J.S.N.d.; Espejo-Román, J.M.; Santos, C.B.R. Identification of Novel Potential Cyclooxygenase-2 Inhibitors Using Ligand- and Structure-Based Virtual Screening Approaches. J. Biomol. Struct. Dyn. 2021, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sutter, A.; Amberg, A.; Boyer, S.; Brigo, A.; Contrera, J.F.; Custer, L.L.; Dobo, K.L.; Gervais, V.; Glowienke, S.; van Gompel, J.; et al. Use of in Silico Systems and Expert Knowledge for Structure-Based Assessment of Potentially Mutagenic Impurities. Regul. Toxicol. Pharmacol. 2013, 67, 39–52. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson1, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Johnson-Davis, K.L.; Dasgupta, A. Special Issues in Therapeutic Drug Monitoring in Patients. In Clinical Challenges in Therapeutic Drug Monitoring; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Braggio, S.; Corsi, M.; Feriani, A.; Fontana, S.; Marocchio, L.; Virginio, C. CHAPTER 15. Discovery Toxicology In Lead Optimisation. In The Handbook of Medicinal Chemistry; Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Hanai, T.; Koseki, A.; Yoshikawa, R.; Ueno, M.; Kinoshita, T.; Homma, H. Prediction of Human Serum Albumin–Drug Binding Affinity without Albumin. Anal. Chim. Acta 2002, 454, 101–108. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of Drug Solubility from Structure. Adv. drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Noctor, T.A.G.; Wainer, I.W.; Hage, D.S. Allosteric and Competitive Displacement of Drugs from Human Serum Albumin by Octanoic Acid, as Revealed by High-Performance Liquid Affinity Chromatography, on a Human Serum Albumin-Based Stationary Phase. J. Chromatogr. B Biomed. Sci. Appl. 1992, 577, 305–315. [Google Scholar] [CrossRef]
- Urien, S.; Bre, F.; Breillout, F.; Bastian, G.; Krikorian, A.; Tillement, J.P. Vinorelbine High-Affinity Binding to Human Platelets and Lymphocytes: Distribution in Human Blood. Cancer Chemother. Pharmacol. 1993, 32, 231–234. [Google Scholar] [CrossRef]
- Haverkamp, W. The Potential for QT Prolongation and Proarrhythmia by Non-Antiarrhythmic Drugs: Clinical and Regulatory Implications. Report on a Policy Conference of the European Society of Cardiology. Eur. Heart J. 2000, 21, 1216–1231. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and Azithromycin as a Treatment of COVID-19: Results of an Open-Label Non-Randomized Clinical Trial. Int. J. Antimicrob. Agents 2020, 56, 105949. [Google Scholar] [CrossRef] [PubMed]
- Malviya, A. Ventricular Arrhythmia Risk Due to Chloroquine/Hydroxychloroquine Treatment for COVID-19: Should It Be Given. Indian Heart J. 2020, 72, 131–132. [Google Scholar] [CrossRef]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.-C. PockDrug-Server: A New Web Server for Predicting Pocket Druggability on Holo and Apo Proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milletti, F.; Vulpetti, A. Predicting Polypharmacology by Binding Site Similarity: From Kinases to the Protein Universe. J. Chem. Inf. Model. 2010, 50, 1418–1431. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Verschueren, K.H.G.; Anand, K.; Shen, J.; Yang, M.; Xu, Y.; Rao, Z.; Bigalke, J.; Heisen, B.; Mesters, J.R.; et al. PH-Dependent Conformational Flexibility of the SARS-CoV Main Proteinase (Mpro) Dimer: Molecular Dynamics Simulations and Multiple X-Ray Structure Analyses. J. Mol. Biol. 2005, 354, 25–40. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the Topology of Active Sites: On the Prediction of Pockets and Subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A Web Server for Automatic Binding Site Prediction, Analysis and Druggability Assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Napoleon, R.L.; Yueh, C.; Whitty, A.; Vajda, S. New Frontiers in Druggability. J. Med. Chem. 2015, 58, 9063–9088. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. The Pymol Molecular Graphics System. 2013. Available online: http://www.pymol.org (accessed on 18 October 2021).
- Wang, T.; Xie, H.-B.; Song, Z.; Niu, J.; Chen, D.-L.; Xia, D.; Chen, J. Role of Hydrogen Bond Capacity of Solvents in Reactions of Amines with CO2: A Computational Study. J. Environ. Sci. 2020, 91, 271–278. [Google Scholar] [CrossRef]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs2: Free ADME/Tox Filtering Tool to Assist Drug Discovery and Chemical Biology Projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- Lagorce, D.; Oliveira, N.; Miteva, M.A.; Villoutreix, B.O. Pan-Assay Interference Compounds (PAINS) That May Not Be Too Painful for Chemical Biology Projects. Drug Discov. Today 2017, 22, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A Web Server for Compound Property Calculation and Chemical Library Design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial Improvements in Large-Scale Redocking and Screening Using the Novel HYDE Scoring Function. J. Comput. Aided. Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef]
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A Consistent Description of HYdrogen Bond and DEhydration Energies in Protein–Ligand Complexes: Methods behind the HYDE Scoring Function. J. Comput. Aided. Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Beglov, D.; Wakefield, A.E.; Egbert, M.; Whitty, A. Cryptic Binding Sites on Proteins: Definition, Detection, and Druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. [Google Scholar] [CrossRef]
- Barrett, A.J.; Kembhavi, A.A.; Brown, M.A.; Kirschke, H.; Knight, C.G.; Tamai, M.; Hanada, K. L-Trans-Epoxysuccinyl-Leucylamido(4-Guanidino)Butane (E-64) and Its Analogues as Inhibitors of Cysteine Proteinases Including Cathepsins B, H and L. Biochem. J. 1982, 201, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Mizoue, K.; Kitamura, K.; Tse, W.-C.; Huber, C.P.; Ishida, T. Structural Basis of Inhibition of Cysteine Proteases by E-64 and Its Derivatives. Biopolymers 1999, 51, 99–107. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully Automated Protein–Ligand Interaction Profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Schneider, R. Database of Homology-Derived Protein Structures and the Structural Meaning of Sequence Alignment. Proteins Struct. Funct. Genet. 1991, 9, 56–68. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A Series of PDB-Related Databanks for Everyday Needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Van Drunen, R.; Van der Spoel, D.; Berendsen, H.; Van der Westhuyzen, D.R. Gromacs-a Parallel Computer for Molecular-Dynamics Simulations. In Proceedings of the 4th International Conference on Computational Physics (PC 92); DeGroot, R.A., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Van der Spoel, D.; van Maaren, P.J.; Larsson, P.; Tîmneanu, N. Thermodynamics of Hydrogen Bonding in Hydrophilic and Hydrophobic Media. J. Phys. Chem. B 2006, 110, 4393–4398. [Google Scholar] [CrossRef]
- Gomes, D.E.B.; Silva, A.W.; Lins, R.D.; Pascutti, P.G.; Soares, T.A. HbMap2Grace 2002. Available online: http://lmdm.biof.ufrj.br/software/hbmap2grace/index.html (accessed on 18 October 2021).
- Gomes, D.E.B.; Sousa, G.L.S.C.; Silva, A.W.S.D.; Pascutti, P.G. SurfinMD 2012. Available online: http://lmdm.biof.ufrj.br/software/surfinmd/index.html (accessed on 18 October 2021).
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized Hydrophobic Interactions and Hydrogen Bonding at the Target-Ligand Interface Leads the Pathways of Drug-Designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Sharma, P.; Vijayan, V.; Pant, P.; Sharma, M.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S. Identification of Potential Drug Candidates to Combat COVID-19: A Structural Study Using the Main Protease (Mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 6649–6659. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinformatics 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, A.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of Nanosystems: Application to Microtubules and the Ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Gbadamosi, I.T. Stay Safe: Helpful Herbal Remedies in Covid-19 Infection. African J. Biomed. Res. 2020, 23, 131–133. [Google Scholar]
- Chou, C.-Y.; Chang, H.-C.; Hsu, W.-C.; Lin, T.-Z.; Lin, C.-H.; Chang, G.-G. Quaternary Structure of the Severe Acute Respiratory Syndrome (SARS) Coronavirus Main Protease. Biochemistry 2004, 43, 14958–14970. [Google Scholar] [CrossRef]
- Zhong, N.; Zhang, S.; Zou, P.; Chen, J.; Kang, X.; Li, Z.; Liang, C.; Jin, C.; Xia, B. Without Its N-Finger, the Main Protease of Severe Acute Respiratory Syndrome Coronavirus Can Form a Novel Dimer through Its C-Terminal Domain. J. Virol. 2008, 82, 4227–4234. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Irschik, H.; Huch, V.; Schummer, D.; Steinmetz, H.; Bock, M.; Schmidt, T.; Kirschning, A.; Müller, R. Carolacton—A Macrolide Ketocarbonic Acid That Reduces Biofilm Formation by the Caries- and Endocarditis-Associated Bacterium Streptococcus Mutans. Eur. J. Org. Chem. 2010, 2010, 1284–1289. [Google Scholar] [CrossRef]
- Anderson, D.E.; Cui, J.; Ye, Q.; Huang, B.; Zu, W.; Gong, J.; Liu, W.; Young Kim, S.; Guo Yan, B.; Sigmundsson, K.; et al. Orthogonal Genome-Wide Screenings in Bat Cells Identify MTHFD1 as a Target of 1 Broad Antiviral Therapy 2 3. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Wiegrebe, W.; Kramer, W.J.; Shamma, M. The Emetine Alkaloids. J. Nat. Prod. 1984, 47, 397–408. [Google Scholar] [CrossRef]
- Akinboye, E.S.; Rosen, M.D.; Denmeade, S.R.; Kwabi-Addo, B.; Bakare, O. Design, Synthesis, and Evaluation of PH-Dependent Hydrolyzable Emetine Analogues as Treatment for Prostate Cancer. J. Med. Chem. 2012, 55, 7450–7459. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Niu, J.; Wang, C.; Huang, B.; Wang, W.; Zhu, N.; Deng, Y.; Wang, H.; Ye, F.; Cen, S.; et al. High-Throughput Screening and Identification of Potent Broad-Spectrum Inhibitors of Coronaviruses. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [Green Version]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; da Silva Costa, J.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2018, 23, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, E.L.; Santos, C.F.; Braga, F.S.; Costa, J.S.; Silva, R.C.; Favacho, H.A.S.; Hage-Melim, L.I.S.; Carvalho, J.C.T.; da Silva, C.H.T.P.; Santos, C.B.R. Computational Investigation of Antifungal Compounds Using Molecular Modeling and Prediction of ADME/Tox Properties. J. Comput. Theor. Nanosci. 2015, 12, 3682–3691. [Google Scholar] [CrossRef]
- Stenberg, P.; Norinder, U.; Luthman, K.; Artursson, P. Experimental and Computational Screening Models for the Prediction of Intestinal Drug Absorption. J. Med. Chem. 2001, 44, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Förster, C. Advances and Applications in Bioinformatics and Chemistry Dovepress in Silico Predictive Model to Determine Vector-Mediated Transport Properties for the Blood-Brain Barrier Choline Transporter. Adv. Appl. Bioinforma. Chem. 2014, 7, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical Course and Outcomes of Critically Ill Patients with SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Platt, M.P.; Bolding, K.A.; Wayne, C.R.; Chaudhry, S.; Cutforth, T.; Franks, K.M.; Agalliu, D. Th17 Lymphocytes Drive Vascular and Neuronal Deficits in a Mouse Model of Postinfectious Autoimmune Encephalitis. Proc. Natl. Acad. Sci. USA 2020, 117, 6708–6716. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.; Li, J.; Liu, X.; Zhu, C. Prominent Changes in Blood Coagulation of Patients with SARS-CoV-2 Infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannan Baig, A.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host−Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Tovar-Moll, F.; Moll, J.; Munoz, D.P.; Ferreira, S.T. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Central Nervous System. Trends Neurosci. 2020, 43, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Rojas, H.; Ritter, C.; Pizzol, F.D. Mechanisms of Dysfunction of the Blood-Brain Barrier in Critically Ill Patients: Emphasis on the Role of Matrix Metalloproteinases. Rev. Bras. Ter. Intensiva 2011, 23, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hou, T. Drug and Drug Candidate Building Block Analysis. J. Chem. Inf. Model. 2010, 50, 55–67. [Google Scholar] [CrossRef]
- Ren, X.; Glende, J.; Al-Falah, M.; de Vries, V.; Schwegmann-Wessels, C.; Qu, X.; Tan, L.; Tschernig, T.; Deng, H.; Naim, H.Y.; et al. Analysis of ACE2 in Polarized Epithelial Cells: Surface Expression and Function as Receptor for Severe Acute Respiratory Syndrome-Associated Coronavirus. J. Gen. Virol. 2006, 87, 1691–1695. [Google Scholar] [CrossRef]
- Uemura, K.; Sasaki, M.; Sanaki, T.; Toba, S.; Takahashi, Y.; Orba, Y.; Hall, W.W.; Maenaka, K.; Sawa, H.; Sato, A. MRC5 Cells Engineered to Express ACE2 Serve as a Model System for the Discovery of Antivirals Targeting SARS-CoV-2. Sci. Rep. 2021, 11, 5376. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic Efficacy of the Small Molecule GS-5734 against Ebola Virus in Rhesus Monkeys. Nature 2016, 531, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, B.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Keminer, O.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; et al. A SARS-CoV-2 Cytopathicity Dataset Generated by High-Content Screening of a Large Drug Repurposing Collection. Sci. Data 2021, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Fujita, T. P -σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Box, K.; Comer, J. Using Measured PKa, LogP and Solubility to Investigate Supersaturation and Predict BCS Class. Curr. Drug Metab. 2008, 9, 869–878. [Google Scholar] [CrossRef]
- Khalifa, I.; Zhu, W.; Hamed, H.; Mohammed, H.; Dutta, K.; Chunmei, L.; Li, C. Tannins Inhibit SARS-CoV-2 through Binding with Catalytic Dyad Residues of 3CL pro: An in Silico Approach with 19 Structural Different Hydrolysable Tannins Practical Applications. J. Food Biochem. 2020, 44, 13432. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural Basis of SARS-CoV-2 3CLpro and Anti-COVID-19 Drug Discovery from Medicinal Plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 491. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and Molecular Dynamics Simulation Studies Reveal Potential Covalent and FDA-Approved Inhibitors of SARS-CoV-2 Main Protease 3CL Pro. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zheng, W.; Huang, R. High-throughput Screening Assays for SARS-CoV-2 Drug Development: Current Status and Future Directions. Drug Discov. Today 2021, 26, 2439–2444. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and Electrophilic Fragment Screening of the SARS-CoV-2 Main Protease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Borrel, A.; Regad, L.; Xhaard, H.; Petitjean, M.; Camproux, A.-C. PockDrug: A Model for Predicting Pocket Druggability That Overcomes Pocket Estimation Uncertainties. J. Chem. Inf. Model. 2015, 55, 882–895. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Ngan, C.H.; Bohnuud, T.; Mottarella, S.E.; Beglov, D.; Villar, E.A.; Hall, D.R.; Kozakov, D.; Vajda, S. FTMAP: Extended Protein Mapping with User-Selected Probe Molecules. Nucleic Acids Res. 2012, 40, W271–W275. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.-Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-Based Identification of Druggable ‘Hot Spots’ of Proteins Using Fourier Domain Correlation Techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Chuang, G.-Y.; Cencic, R.; Brenke, R.; Grove, L.E.; Beglov, D.; Pelletier, J.; Whitty, A.; Vajda, S. Structural Conservation of Druggable Hot Spots in Protein-Protein Interfaces. Proc. Natl. Acad. Sci. USA 2011, 108, 13528–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schärfer, C.; Schulz-Gasch, T.; Ehrlich, H.-C.; Guba, W.; Rarey, M.; Stahl, M. Torsion Angle Preferences in Druglike Chemical Space: A Comprehensive Guide. J. Med. Chem. 2013, 56, 2016–2028. [Google Scholar] [CrossRef]
- Brethon, A.; Chantalat, L.; Christin, O.; Clary, L.; Fournier, J.-F.; Gastreich, M.; Harris, C.S.; Isabet, T.; Pascau, J.; Thoreau, E.; et al. New Caspase-1 Inhibitor by Scaffold Hopping into Bio-Inspired 3D-Fragment Space. Bioorg. Med. Chem. Lett. 2017, 27, 5373–5377. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Macêdo, W.J.C.; Silva, R.C.; Taft, C.A.; Silva, C.H.T.P.; Rodriguez, A.F.R.; Campos, J.M.; dos Santos, C.B.R. High-Throughput-Based Virtual Screening via Molecular Docking for Oxidative Stress Mediated by ROS Enzyme. In Functional Properties of Advanced Engineering Materials and Biomolecules; Engineering Materials; La Porta, F.A., Taft, C.A., Eds.; Springer: Cham, Switzerland; Hillerød, Denmark, 2021; pp. 489–513. [Google Scholar] [CrossRef]
- Schrödinger QikProp: Rapid ADME Predictions of Drug Candidates. Available online: https://www.schrodinger.com/products/qikprop (accessed on 18 October 2021).
- Hou, T.; Wang, J. Structure—ADME Relationship: Still a Long Way to Go? Expert Opin. Drug Metab. Toxicol. 2008, 4, 759–770. [Google Scholar] [CrossRef]
- Greene, N.; Judson, P.N.; Langowski, J.J.; Marchant, C.A. Knowledge-Based Expert Systems for Toxicity and Metabolism Prediction: DEREK, StAR and METEOR. SAR QSAR Environ. Res. 1999, 10, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and Validation of the Automated Topology Builder (ATB) Version 2.0: Prediction of Hydration Free Enthalpies. J. Comput. Aided. Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Tayar, N.E.; Tsai, R.-S.; Testa, B.; Carrupt, P.-A.; Leo, A. Partitioning of Solutes in Different Solvent Systems: The Contribution of Hydrogen-Bonding Capacity and Polarity. J. Pharm. Sci. 1991, 80, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Seiler, P. Interconversion of Lipophilicities from Hydrocarbon/Water Systems into the Octanol/Water System. Eur. J. Med. Chem. 1974, 9, 473–479. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Compounds | Chemical Structure | Hydrogen Bond Donors (HBD) 1 | Hydrogen Bond Acceptors (HBA) | Lipinki’s Rule 2 Violation | Molecular Surface Area (Å2) 3 | Solvent Accessible Surface (Å3) 4 | Estimated Affinity Range 5 (µM) | Inter Clash Type 6 |
|---|---|---|---|---|---|---|---|---|---|
| b01 | VE0DIA0AF |  | 1 | 2 | 1 | 134.92 | 478.2 | 3.29–326.4 | + |
| b02 | VE0PPA0AF |  | 5 | 5 | 0 | 150.25 | 481.3 | 4.42–439.6 | 0 |
| b03 | VE0ISA0AF | 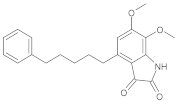 | 1 | 5 | 0 | 152.87 | 645.3 | 7.24–719.2 | + |
| b04 | VE0FKA0AF |  | 2 | 5 | 0 | 150.34 | 576.2 | 17.25–1714.3 | + |
| b05 | VE0FEA0SF |  | 4 | 6 | 0 | 150.17 | 519.5 | 18.18–1806.5 | 0 |
| b06 | VE0JDA0SF | 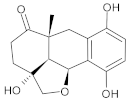 | 3 | 5 | 0 | 121.72 | 355.9 | 20.82–2068.7 | 0 |
| b07 | VE0ZDA0AF | 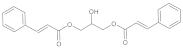 | 1 | 5 | 0 | 151.74 | 652.2 | 41.3–4103.0 | + |
| b08 | VE0NCA0SF | 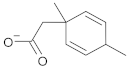 | 1 | 2 | 0 | 72.59 | 309.2 | 51.97–5164.2 | + |
| b09 | VE0KJA0SI | 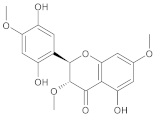 | 3 | 8 | 0 | 148.67 | 599.8 | 52.40–5206.5 | 0 |
| b10 | VE0NHA0SF |  | 3 | 6 | 0 | 122.04 | 543.6 | 53.85–5350.2 | + |
| 255Control(RZS) | - | 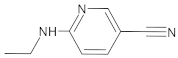 | 1 | 3 | 0 | 65.69 | 350.4 | 504.29–50,103.8 | + |
| 562Positive control(E-64) | - | 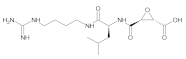 | 7 | 10 | 1 | 145.91 | 638.3 | 96,164.969, 455,201.9 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, R.C.; Freitas, H.F.; Campos, J.M.; Kimani, N.M.; Silva, C.H.T.P.; Borges, R.S.; Pita, S.S.R.; Santos, C.B.R. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021, 22, 11739. https://doi.org/10.3390/ijms222111739
Silva RC, Freitas HF, Campos JM, Kimani NM, Silva CHTP, Borges RS, Pita SSR, Santos CBR. Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. International Journal of Molecular Sciences. 2021; 22(21):11739. https://doi.org/10.3390/ijms222111739
Chicago/Turabian StyleSilva, Rai C., Humberto F. Freitas, Joaquín M. Campos, Njogu M. Kimani, Carlos H. T. P. Silva, Rosivaldo S. Borges, Samuel S. R. Pita, and Cleydson B. R. Santos. 2021. "Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro" International Journal of Molecular Sciences 22, no. 21: 11739. https://doi.org/10.3390/ijms222111739
APA StyleSilva, R. C., Freitas, H. F., Campos, J. M., Kimani, N. M., Silva, C. H. T. P., Borges, R. S., Pita, S. S. R., & Santos, C. B. R. (2021). Natural Products-Based Drug Design against SARS-CoV-2 Mpro 3CLpro. International Journal of Molecular Sciences, 22(21), 11739. https://doi.org/10.3390/ijms222111739