
Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease


Abstract
:1. Introduction
2. The Role of Microglia in the Brain
3. Microglial Phenotype Polarization during Neuroinflammation/Neurodegeneration and the Role in Intercellular Communication
4. The Role of Aβ Peptides and Other Proteins with Altered Conformations in the Neuron–Microglia Dialogue in AD and Neuroinflammation
5. Glutamatergic Signaling in Microglia–Neuron Communication in AD
6. GABAergic Signaling in Microglial Dialogue with Neurons in AD and Neuroinflammation
7. Therapeutic Compounds Affecting Glutamate/GABA Neurotransmission in Microglial–Neuron Communication
8. Concluding Remarks and Future Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Adams, J.D. Probable Causes of Alzheimer’s Disease. Science 2021, 3, 16. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Genomic mechanisms in Alzheimer’s disease. Brain Pathol. 2020, 30, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Moyse, E.; Haddad, M.; Benlabiod, C.; Ramassamy, C.; Krantic, S. Common Pathological Mechanisms and Risk Factors for Alzheimer’s Disease and Type-2 Diabetes: Focus on Inflammation. Curr. Alzheimer Res. 2019, 16, 986–1006. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Sini, P.; Dang, T.B.C.; Fais, M.; Galioto, M.; Padedda, B.M.; Lugliè, A.; Iaccarino, C.; Crosio, C. Cyanobacteria, Cyanotoxins, and Neurodegenerative Diseases: Dangerous Liaisons. Int. J. Mol. Sci. 2021, 22, 8726. [Google Scholar] [CrossRef]
- Ambrad Giovannetti, E.; Fuhrmann, M. Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease. Brain Res. 2019, 1707, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M. Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhäuser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef] [PubMed]
- Jesudasan, S.J.B.; Gupta, S.J.; Churchward, M.A.; Todd, K.G.; Winship, I.R. Inflammatory Cytokine Profile and Plasticity of Brain and Spinal Microglia in Response to ATP and Glutamate. Front. Cell. Neurosci. 2021, 15, 634020. [Google Scholar] [CrossRef] [PubMed]
- Noda, M. Dysfunction of Glutamate Receptors in Microglia May Cause Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 381–386. [Google Scholar] [CrossRef]
- Stolero, N.; Frenkel, D. The dialog between neurons and microglia in Alzheimer’s disease: The neurotransmitters view. J. Neurochem. 2020, 158, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Younger, D.; Murugan, M.; Rama Rao, K.V.; Wu, L.J.; Chandra, N. Microglia Receptors in Animal Models of Traumatic Brain Injury. Mol. Neurobiol. 2019, 56, 5202–5228. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, T.L.; Carrier, M.; Tremblay, M. Physiology of Microglia. Adv. Exp. Med. Biol. 2019, 1175, 129–148. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Adle-Biassette, H.; Delezoide, A.L.; Evrard, P.; Gressens, P.; Verney, C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J. Neuropathol. Exp. Neurol. 2007, 66, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Nikodemova, M.; Kimyon, R.S.; De, I.; Small, A.L.; Collier, L.S.; Watters, J.J. Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J. Neuroimmunol. 2015, 278, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Udeochu, J.C.; Shea, J.M.; Villeda, S.A. Microglia communication: Parallels between aging and Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2016, 7, 114–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Strekalova, T.; Ponomarev, E.D. The Role of Neuronal Factors in the Epigenetic Reprogramming of Microglia in the Normal and Diseased Central Nervous System. Front. Cell. Neurosci. 2019, 13, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahaya, M.A.F.; Zolkiffly, S.Z.I.; Moklas, M.A.M.; Hamid, H.A.; Stanslas, J.; Zainol, M.; Mehat, M.Z. Possible Epigenetic Role of Vitexin in Regulating Neuroinflammation in Alzheimer’s Disease. J. Immunol. Res. 2020, 2020, 9469210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, M.; Liang, J.; Wang, C.; Deng, Y.; Chen, Z. Epigenetic Regulation of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4956. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ventura, J.; Amo-Aparicio, J.; Navarro, X.; Penas, C. BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J. Neuroinflamm. 2019, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, B.; Mota, M.; Pizzi, M. Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim. Biophys. Acta 2016, 1862, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Kwon, S.; Iba, M.; Kim, C.; Masliah, E. Immunotherapies for Aging-Related Neurodegenerative Diseases-Emerging Perspectives and New Targets. Neurotherapeutics 2020, 17, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, C.; Kindred, N.; Curry, J.; Williams, C.; Taylor, J.P.; Atkinson, P.; Randall, F.; Erskine, D.; Morris, C.M.; Reeve, A.K.; et al. Hippocampal network hyperexcitability in young transgenic mice expressing human mutant alpha-synuclein. Neurobiol. Dis. 2021, 149, 105226. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.J.; Yang, C.H.; Fang, Y.H.; Cho, K.H.; Chien, W.L.; Wang, W.T.; Wu, T.W.; Lin, C.P.; Fu, W.M.; Shen, C.K. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 2010, 207, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Hebron, M.; Chen, W.; Miessau, M.J.; Lonskaya, I.; Moussa, C.E. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J. Neurochem. 2014, 129, 350–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammisetty, S.S.; Pedragosa, J.; Weng, Y.C.; Calon, F.; Planas, A.; Kriz, J. Age-related deregulation of TDP-43 after stroke enhances NF-κB-mediated inflammation and neuronal damage. J. Neuroinflamm. 2018, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, D. Amyloid-β Impairs Synaptic Inhibition via GABA(A) Receptor Endocytosis. J. Neurosci. 2015, 35, 9205–9210. [Google Scholar] [CrossRef] [Green Version]
- Hascup, K.N.; Hascup, E.R. Soluble Amyloid-β42 Stimulates Glutamate Release through Activation of the α7 Nicotinic Acetylcholine Receptor. J. Alzheimers Dis. 2016, 53, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Borutaite, V. Extracellular tau-induced microglia-mediated neuronal death. In Proceedings of the Book of Abstracts of Virtual FENS Regional Meeting, Kraków, Poland, 25–27 August 2021; p. 31. [Google Scholar]
- Kazmierczak, A.; Strosznajder, J.B.; Adamczyk, A. alpha-Synuclein enhances secretion and toxicity of amyloid beta peptides in PC12 cells. Neurochem. Int. 2008, 53, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Strosznajder, J.B.; Adamczyk, A. Toxicity of extracellular secreted alpha-synuclein: Its role in nitrosative stress and neurodegeneration. Neurochem. Int. 2013, 62, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Jęśko, H.; Cieślik, M.; Gromadzka, G.; Adamczyk, A. Dysfunctional proteins in neuropsychiatric disorders: From neurodegeneration to autism spectrum disorders. Neurochem. Int. 2020, 141, 104853. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.B.; Nowrangi, M.A.; Lyketsos, C.G. Neuropsychiatric symptoms in Alzheimer’s disease: What might be associated brain circuits? Mol. Aspects Med. 2015, 43-44, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, M.; Shao, H.; Zandi, P.; Lyketsos, C.G.; Welsh-Bohmer, K.A.; Norton, M.C.; Breitner, J.C.; Steffens, D.C.; Tschanz, J.T. Point and 5-year period prevalence of neuropsychiatric symptoms in dementia: The Cache County Study. Int. J. Geriatr. Psychiatry 2008, 23, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. Psych.-Gerichtl. Med. 1907, 64, 146–148. [Google Scholar]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Zhao, J.W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276-1290.e1217. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Walker, K.A.; Gottesman, R.F.; Wu, A.; Knopman, D.S.; Gross, A.L.; Mosley, T.H., Jr.; Selvin, E.; Windham, B.G. Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology 2019, 92, e1256–e1267. [Google Scholar] [CrossRef]
- Marsland, A.L.; Gianaros, P.J.; Kuan, D.C.; Sheu, L.K.; Krajina, K.; Manuck, S.B. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain Behav. Immun. 2015, 48, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puris, E.; Auriola, S.; Korhonen, P.; Loppi, S.; Kanninen, K.M.; Malm, T.; Koistinaho, J.; Gynther, M. Systemic inflammation induced changes in protein expression of ABC transporters and ionotropic glutamate receptor subunit 1 in the cerebral cortex of familial Alzheimer‘s disease mouse model. J. Pharm. Sci. 2021. S0022-3549(21)00414-7. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Zurek, A.A.; Lecker, I.; Yu, J.; Abramian, A.M.; Avramescu, S.; Davies, P.A.; Moss, S.J.; Lu, W.Y.; Orser, B.A. Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2012, 2, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoi, Y.; Saito, S. An alteration in the gamma-aminobutyric acid receptor system in experimentally induced septic shock in rats. Crit. Care Med. 1996, 24, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharm. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, D.; Zhang, B.; Zhu, J.; Zhou, Z.; Cui, L. Regulation of microglia by glutamate and its signal pathway in neurodegenerative diseases. Drug Discov. Today 2020, 25, 1074–1085. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Bu, X.L.; Yao, X.Q.; Jiao, S.S.; Zeng, F.; Liu, Y.H.; Xiang, Y.; Liang, C.R.; Wang, Q.H.; Wang, X.; Cao, H.Y.; et al. A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 2015, 22, 1519–1525. [Google Scholar] [CrossRef]
- Kanagasingam, S.; Chukkapalli, S.S.; Welbury, R.; Singhrao, S.K. Porphyromonas gingivalis is a Strong Risk Factor for Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2020, 4, 501–511. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vico Varela, E.; Etter, G.; Williams, S. Excitatory-inhibitory imbalance in Alzheimer’s disease and therapeutic significance. Neurobiol. Dis. 2019, 127, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Styr, B.; Slutsky, I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 2018, 21, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Vélez-Fort, M.; Audinat, E.; Angulo, M.C. Central role of GABA in neuron-glia interactions. Neuroscientist 2012, 18, 237–250. [Google Scholar] [CrossRef]
- Hodo, T.W.; de Aquino, M.T.P.; Shimamoto, A.; Shanker, A. Critical Neurotransmitters in the Neuroimmune Network. Front. Immunol. 2020, 11, 1869. [Google Scholar] [CrossRef]
- Danysz, W.; Zajaczkowski, W.; Parsons, C.G. Modulation of learning processes by ionotropic glutamate receptor ligands. Behav. Pharmacol. 1995, 6, 455–474. [Google Scholar] [CrossRef]
- Lazarewicz, J.W.; Salinska, E.; Wroblewski, J.T. NMDA receptor-mediated arachidonic acid release in neurons: Role in signal transduction and pathological aspects. Adv. Exp. Med. Biol. 1992, 318, 73–89. [Google Scholar] [CrossRef]
- Jędrzejewska-Szmek, J. Molecular control of NMDAR-dependent gabaergic plasticity. In Proceedings of the Book of Abstracts of Virtual FENS Regional Meeting, Kraków, Poland, 25–27 August 2021; p. 56. [Google Scholar]
- Robinson, M.B. The family of sodium-dependent glutamate transporters: A focus on the GLT-1/EAAT2 subtype. Neurochem. Int. 1998, 33, 479–491. [Google Scholar] [CrossRef]
- Li, S.; Mallory, M.; Alford, M.; Tanaka, S.; Masliah, E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 1997, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, N.C.; Coleman, P.D.; Cribbs, D.H.; Rogers, J.; Gillen, D.L.; Cotman, C.W. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuba, G.; Vernay, A.; Kraftsik, R.; Tardif, E.; Riederer, B.M.; Savioz, A. Pathological reorganization of NMDA receptors subunits and postsynaptic protein PSD-95 distribution in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Palma, E.; Bustos, B.I.; Villamán, C.F.; Alarcón, M.A.; Avila, M.E.; Ugarte, G.D.; Reyes, A.E.; Opazo, C.; De Ferrari, G.V. Overrepresentation of glutamate signaling in Alzheimer’s disease: Network-based pathway enrichment using meta-analysis of genome-wide association studies. PLoS ONE 2014, 9, e95413. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.J.; Dimayuga, E.; Morganti, J.M.; Van Eldik, L.J. Microglial-associated responses to comorbid amyloid pathology and hyperhomocysteinemia in an aged knock-in mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 274. [Google Scholar] [CrossRef]
- Sebastian Monasor, L.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 2020, 9, e54083. [Google Scholar] [CrossRef]
- Rienecker, K.D.A.; Paladini, M.S.; Grue, K.; Krukowski, K.; Rosi, S. Microglia: Ally and Enemy in Deep Space. Neurosci. Biobehav. Rev. 2021, 126, 509–514. [Google Scholar] [CrossRef]
- Parsons, C.G.; Stöffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system--too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723. [Google Scholar] [CrossRef]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Melgar, A.; Albasanz, J.L.; Palomera-Ávalos, V.; Pallàs, M.; Martín, M. Resveratrol Modulates and Reverses the Age-Related Effect on Adenosine-Mediated Signalling in SAMP8 Mice. Mol. Neurobiol. 2019, 56, 2881–2895. [Google Scholar] [CrossRef] [PubMed]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Czapski, G.A.; Adamczyk, A. Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: Facts and hypotheses. J. Neurochem. 2016, 136, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Achour, S.; Pascual, O. Glia: The many ways to modulate synaptic plasticity. Neurochem. Int. 2010, 57, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Basilico, B.; Pagani, F.; Grimaldi, A.; Cortese, B.; Di Angelantonio, S.; Weinhard, L.; Gross, C.; Limatola, C.; Maggi, L.; Ragozzino, D. Microglia shape presynaptic properties at developing glutamatergic synapses. Glia 2019, 67, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Basilico, B.; Ferrucci, L.; Ratano, P.; Golia, M.T.; Grimaldi, A.; Rosito, M.; Ferretti, V.; Reverte, I.; Marrone, M.C.; Giubettini, M.; et al. Microglia control glutamatergic synapses in the adult mouse hippocampus. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.R.; Blanco-Luquin, I.; Mendioroz, M. The Participation of Microglia in Neurogenesis: A Review. Brain Sci. 2021, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Allam, S.L.; Bouteiller, J.M.; Hu, E.Y.; Ambert, N.; Greget, R.; Bischoff, S.; Baudry, M.; Berger, T.W. Synaptic Efficacy as a Function of Ionotropic Receptor Distribution: A Computational Study. PLoS ONE 2015, 10, e0140333. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Walker, A.K. Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J. Neural Transm. 2014, 121, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zmarowski, A.; Wu, H.Q.; Brooks, J.M.; Potter, M.C.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur. J. Neurosci. 2009, 29, 529–538. [Google Scholar] [CrossRef]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R. Kynurenines and Glutamate: Multiple Links and Therapeutic Implications. Adv. Pharmacol. 2016, 76, 13–37. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J. Quinolinic acid: Neurotoxicity. FEBS J. 2012, 279, 1355. [Google Scholar] [CrossRef]
- Ferreira, F.S.; Schmitz, F.; Marques, E.P.; Siebert, C.; Wyse, A.T.S. Intrastriatal Quinolinic Acid Administration Impairs Redox Homeostasis and Induces Inflammatory Changes: Prevention by Kynurenic Acid. Neurotox. Res. 2020, 38, 50–58. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ß production. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Perry, V.H.; O’Connor, V. The role of microglia in synaptic stripping and synaptic degeneration: A revised perspective. ASN Neuro. 2010, 2, e00047. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Neves, A.C.; Carecho, R.; Correia, S.C.; Carvalho, C.; Campos, E.J.; Baptista, F.I.; Moreira, P.I.; Ambrósio, A.F. Retina and Brain Display Early and Differential Molecular and Cellular Changes in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3043–3060. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, S.; Wencel, P.L.; Wieczorek, I.; Strosznajder, J.B.; Strosznajder, R.P. Early alterations in transcription of genes coding NAD dependent- enzymes and mitochondria proteins in Alzheimer’s dis-ease animal model. Novel target(s) for neuroprotection. In Proceedings of the Book of Abstracts of Virtual FENS Regional Meeting, Kraków, Poland, 25–27 August 2021; p. 306. [Google Scholar]
- Govindpani, K.; Calvo-Flores Guzmán, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a Better Understanding of GABAergic Remodeling in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrer, T.E.; Palpagama, T.H.; Waldvogel, H.J.; Synek, B.J.L.; Turner, C.; Faull, R.L.; Kwakowsky, A. Impaired expression of GABA transporters in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. Neuroscience 2017, 351, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Ikonomovic, M.D.; Grayson, D.R.; Sheffield, R.; Armstrong, D.M. Immunohistochemical study of GABAA receptor alpha1 subunit in the hippocampal formation of aged brains with Alzheimer-related neuropathologic changes. Brain Res. 1998, 799, 148–155. [Google Scholar] [CrossRef]
- Rissman, R.A.; Mishizen-Eberz, A.J.; Carter, T.L.; Wolfe, B.B.; De Blas, A.L.; Miralles, C.P.; Ikonomovic, M.D.; Armstrong, D.M. Biochemical analysis of GABA(A) receptor subunits alpha 1, alpha 5, beta 1, beta 2 in the hippocampus of patients with Alzheimer’s disease neuropathology. Neuroscience 2003, 120, 695–704. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, M.; Mizukami, K.; Ikonomovic, M.D.; Ishikawa, M.; Hidaka, S.; Abrahamson, E.E.; DeKosky, S.T.; Asada, T. Changes in hippocampal GABABR1 subunit expression in Alzheimer’s patients: Association with Braak staging. Acta Neuropathol. 2005, 109, 467–474. [Google Scholar] [CrossRef]
- Chu, D.C.; Penney, J.B., Jr.; Young, A.B. Cortical GABAB and GABAA receptors in Alzheimer’s disease: A quantitative autoradiographic study. Neurology 1987, 37, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Cowburn, R.; Barton, A.; Reynolds, G.; Dodd, P.; Wester, P.; O’Carroll, A.M.; Lofdahl, E.; Winblad, B. A disorder of cortical GABAergic innervation in Alzheimer’s disease. Neurosci. Lett. 1987, 73, 192–196. [Google Scholar] [CrossRef]
- Simpson, M.D.; Cross, A.J.; Slater, P.; Deakin, J.F. Loss of cortical GABA uptake sites in Alzheimer’s disease. J. Neural Transm. 1988, 71, 219–226. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Calvo-Flores Guzmán, B.; Pandya, M.; Turner, C.; Waldvogel, H.J.; Faull, R.L. GABA(A) receptor subunit expression changes in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J. Neurochem. 2018, 145, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Charles, K.J.; Deuchars, J.; Davies, C.H.; Pangalos, M.N. GABA B receptor subunit expression in glia. Mol. Cell. Neurosci. 2003, 24, 214–223. [Google Scholar] [CrossRef]
- Kuhn, S.A.; van Landeghem, F.K.; Zacharias, R.; Färber, K.; Rappert, A.; Pavlovic, S.; Hoffmann, A.; Nolte, C.; Kettenmann, H. Microglia express GABA(B) receptors to modulate interleukin release. Mol. Cell. Neurosci. 2004, 25, 312–322. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; McGeer, P.L. Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011, 59, 152–165. [Google Scholar] [CrossRef]
- Schmidt, M. GABA(C) receptors in retina and brain. Results Probl. Cell Differ. 2008, 44, 49–67. [Google Scholar] [CrossRef]
- Fontainhas, A.M.; Wang, M.; Liang, K.J.; Chen, S.; Mettu, P.; Damani, M.; Fariss, R.N.; Li, W.; Wong, W.T. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS ONE 2011, 6, e15973. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Zhuo, M. Resting microglial motility is independent of synaptic plasticity in mammalian brain. J. Neurophysiol. 2008, 99, 2026–2032. [Google Scholar] [CrossRef]
- Wong, W.T.; Wang, M.; Li, W. Regulation of microglia by ionotropic glutamatergic and GABAergic neurotransmission. Neuron Glia Biol. 2011, 7, 41–46. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, L.; Xu, B.; Yuan, J.; Li, S.; Lian, S.; Chen, Y.; Guo, J.; Yang, H. GABA-mediated activated microglia induce neuroinflammation in the hippocampus of mice following cold exposure through the NLRP3 inflammasome and NF-κB signaling pathways. Int. Immunopharmacol. 2020, 89, 106908. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. FDA Approval of Aduhelm Paves a New Path for Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2714–2715. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef]
- Armstrong, R.A. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol. 2014, 52, 211–225. [Google Scholar] [CrossRef]
- Mullard, A. FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat. Rev. Drug Discov. 2021, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- in t’ Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Côté, S.; Carmichael, P.H.; Verreault, R.; Lindsay, J.; Lefebvre, J.; Laurin, D. Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 219–226. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169. [Google Scholar] [CrossRef]
- Small, G.W.; Siddarth, P.; Silverman, D.H.; Ercoli, L.M.; Miller, K.J.; Lavretsky, H.; Bookheimer, S.Y.; Huang, S.C.; Barrio, J.R.; Phelps, M.E. Cognitive and cerebral metabolic effects of celecoxib versus placebo in people with age-related memory loss: Randomized controlled study. Am. J. Geriatr. Psychiatry 2008, 16, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, M.P.; Carriba, P.; Masgrau, R.; Pujol, A.; Galea, E. Staging anti-inflammatory therapy in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 142. [Google Scholar] [CrossRef] [Green Version]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; Green, R.C.; et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Leoutsakos, J.M.; Muthen, B.O.; Breitner, J.C.; Lyketsos, C.G. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: Findings from the randomized controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial. Int. J. Geriatr. Psychiatry 2012, 27, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Glinz, D.; Gloy, V.L.; Monsch, A.U.; Kressig, R.W.; Patel, C.; McCord, K.A.; Ademi, Z.; Tomonaga, Y.; Schwenkglenks, M.; Bucher, H.C.; et al. Acetylcholinesterase inhibitors combined with memantine for moderate to severe Alzheimer’s disease: A meta-analysis. Swiss Med. Wkly. 2019, 149, w20093. [Google Scholar] [CrossRef] [Green Version]
- Rosi, S.; Vazdarjanova, A.; Ramirez-Amaya, V.; Worley, P.F.; Barnes, C.A.; Wenk, G.L. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience 2006, 142, 1303–1315. [Google Scholar] [CrossRef]
- Arif, M.; Chikuma, T.; Ahmed, M.M.; Nakazato, M.; Smith, M.A.; Kato, T. Effects of memantine on soluble Alphabeta(25-35)-induced changes in peptidergic and glial cells in Alzheimer’s disease model rat brain regions. Neuroscience 2009, 164, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Chang, H.F.; Wu, S.N. The inhibition of inwardly rectifying K+ channels by memantine in macrophages and microglial cells. Cell. Physiol. Biochem. 2013, 31, 938–951. [Google Scholar] [CrossRef]
- Rosi, S.; Ramirez-Amaya, V.; Vazdarjanova, A.; Esparza, E.E.; Larkin, P.B.; Fike, J.R.; Wenk, G.L.; Barnes, C.A. Accuracy of hippocampal network activity is disrupted by neuroinflammation: Rescue by memantine. Brain 2009, 132, 2464–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakawa-Hirachi, T.; Mizoguchi, Y.; Ohgidani, M.; Haraguchi, Y.; Monji, A. Effect of memantine, an anti-Alzheimer’s drug, on rodent microglial cells in vitro. Sci. Rep. 2021, 11, 6151. [Google Scholar] [CrossRef]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Tzeng, N.S.; Qian, L.; Wei, S.J.; Hu, X.; Chen, S.H.; Rawls, S.M.; Flood, P.; Hong, J.S.; Lu, R.B. Novel neuroprotective mechanisms of memantine: Increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 2009, 34, 2344–2357. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Mao, X.; Dowd, K.; Tsakanikas, D.; Jiang, C.S.; Meuser, C.; Andrews, R.D.; Lukic, A.S.; Lee, J.; Hampilos, N.; et al. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain 2021. [Google Scholar] [CrossRef] [PubMed]
- Hunsberger, H.C.; Weitzner, D.S.; Rudy, C.C.; Hickman, J.E.; Libell, E.M.; Speer, R.R.; Gerhardt, G.A.; Reed, M.N. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015, 135, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Gray, J.D.; Larson, C.S.; Kazim, S.F.; Soya, H.; McEwen, B.S.; Pereira, A.C. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef]
- Cao, Y.J.; Dreixler, J.C.; Couey, J.J.; Houamed, K.M. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur. J. Pharmacol. 2002, 449, 47–54. [Google Scholar] [CrossRef]
- Haas, L.T.; Salazar, S.V.; Smith, L.M.; Zhao, H.R.; Cox, T.O.; Herber, C.S.; Degnan, A.P.; Balakrishnan, A.; Macor, J.E.; Albright, C.F.; et al. Silent Allosteric Modulation of mGluR5 Maintains Glutamate Signaling while Rescuing Alzheimer’s Mouse Phenotypes. Cell Rep. 2017, 20, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Lanctôt, K.L.; Herrmann, N.; Mazzotta, P.; Khan, L.R.; Ingber, N. GABAergic function in Alzheimer’s disease: Evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can. J. Psychiatry 2004, 49, 439–453. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, J.M.; Irwin, R.W.; Yao, J.; Liu, L.; Brinton, R.D. Allopregnanolone promotes regeneration and reduces β-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS ONE 2011, 6, e24293. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Liu, L.; Wang, J.M.; Irwin, R.W.; Yao, J.; Chen, S.; Henry, S.; Thompson, R.F.; Brinton, R.D. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol. Aging 2012, 33, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, S.K.; Johansson, M.; Backstrom, T.; Nitsch, R.M.; Wang, M. Brief but chronic increase in allopregnanolone cause accelerated AD pathology differently in two mouse models. Curr. Alzheimer Res. 2013, 10, 38–47. [Google Scholar] [CrossRef]
- Bengtsson, S.K.; Johansson, M.; Bäckström, T. Long-term continuous allopregnanolone elevation causes memory decline and hippocampus shrinkage, in female wild-type B6 mice. Horm. Behav. 2016, 78, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Jolivel, V.; Brun, S.; Binamé, F.; Benyounes, J.; Taleb, O.; Bagnard, D.; De Sèze, J.; Patte-Mensah, C.; Mensah-Nyagan, A.G. Microglial Cell Morphology and Phagocytic Activity Are Critically Regulated by the Neurosteroid Allopregnanolone: A Possible Role in Neuroprotection. Cells 2021, 10, 698. [Google Scholar] [CrossRef]
- Ghai, R.; Nagarajan, K.; Arora, M.; Grover, P.; Ali, N.; Kapoor, G. Current Strategies and Novel Drug Approaches for Alzheimer Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.R.; Islam, T.; Zaman, T.; Shahjaman, M.; Karim, M.R.; Huq, F.; Quinn, J.M.W.; Holsinger, R.M.D.; Gov, E.; Moni, M.A. Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer’s disease: Insights from a systems biomedicine perspective. Genomics 2020, 112, 1290–1299. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement. 2021, 7, e12179. [Google Scholar] [CrossRef]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.; Jeitner, T.M. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—2: Beyond amyloid—Re-defining AD and its causality to discover effective therapeutics. Biochem. Pharmacol. 2018, 158, 376–401. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s disease. Front. Neurol. 2017, 8, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Raichle, M.E. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Ríos, J.A.; Cisternas, P.; Arrese, M.; Barja, S.; Inestrosa, N.C. Is Alzheimer’s disease related to metabolic syndrome? A Wnt signaling conundrum. Prog. Neurobiol. 2014, 121, 125–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Barua, S.; Jeong, Y.J.; Lee, J.E. Adiponectin: The Potential Regulator and Therapeutic Target of Obesity and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6419. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Li, D.; Chen, H.S.; Huang, J.G.; Xu, J.F.; Zhu, W.W.; Chen, J.G.; Wang, F. Metformin produces anxiolytic-like effects in rats by facilitating GABA(A) receptor trafficking to membrane. Br. J. Pharmacol. 2019, 176, 297–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhou, T.; Guo, A.M.; Chen, W.B.; Lin, D.; Liu, Z.Y.; Fei, E.K. Metformin Ameliorates Lipopolysaccharide-Induced Depressive-Like Behaviors and Abnormal Glutamatergic Transmission. Biology 2020, 9, 359. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Preclinical Models of Alzheimer’s disease: Relevance and Translational Validity. Curr. Protoc. Pharmacol. 2019, 84, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straathof, M.; Meerwaldt, A.E.; De Feyter, H.M.; de Graaf, R.A.; Dijkhuizen, R.M. Deuterium Metabolic Imaging of the Healthy and Diseased Brain. Neuroscience 2021, 474, 94–99. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, R.S.; Mecca, A.P.; Chen, M.K.; Naganawa, M.; Toyonaga, T.; Lu, Y.; Godek, T.A.; Harris, J.E.; Bartlett, H.H.; Banks, E.R.; et al. Association of Aβ deposition and regional synaptic density in early Alzheimer’s disease: A PET imaging study with [(11)C]UCB-J. Alzheimers Res. Ther. 2021, 13, 11. [Google Scholar] [CrossRef] [PubMed]
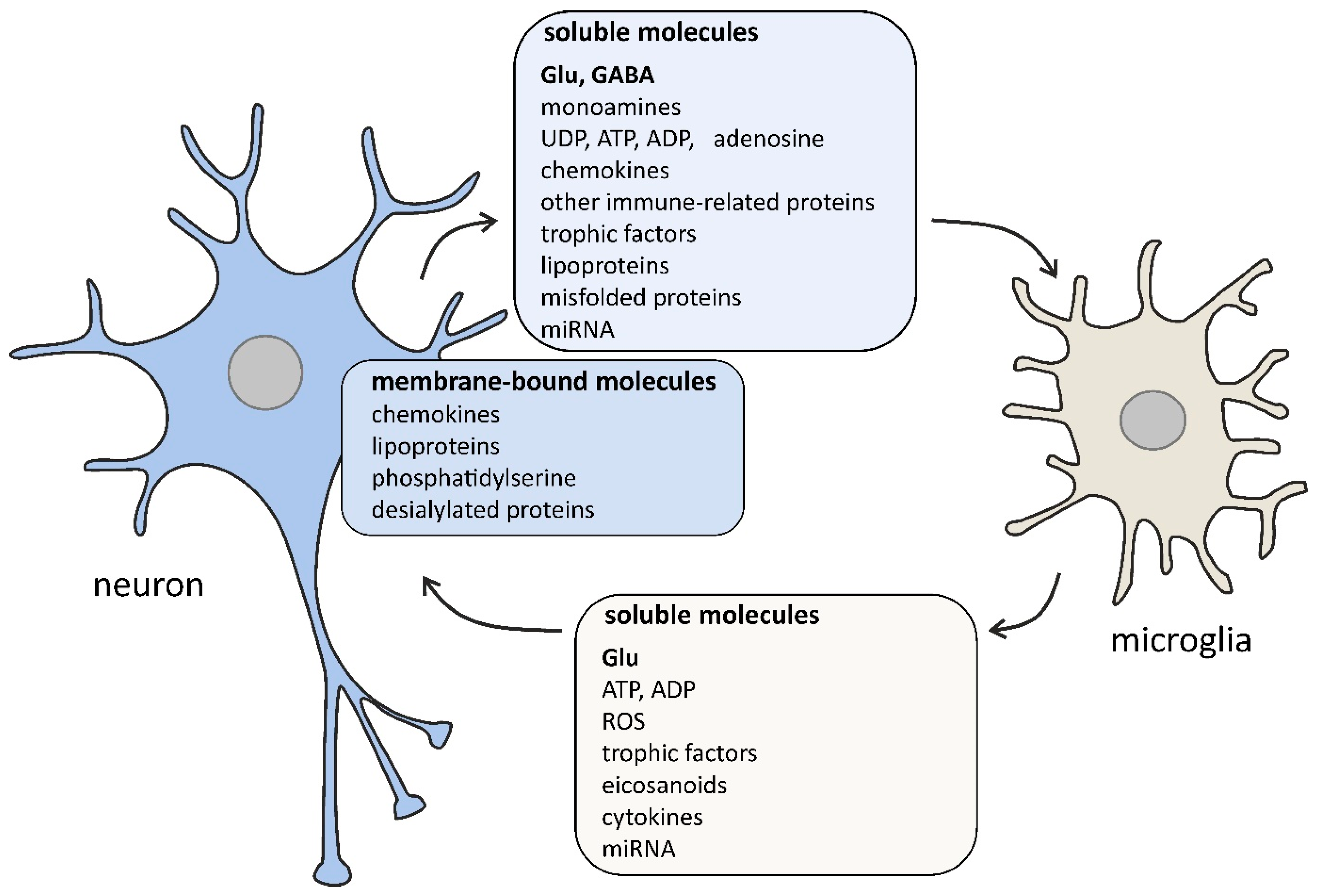
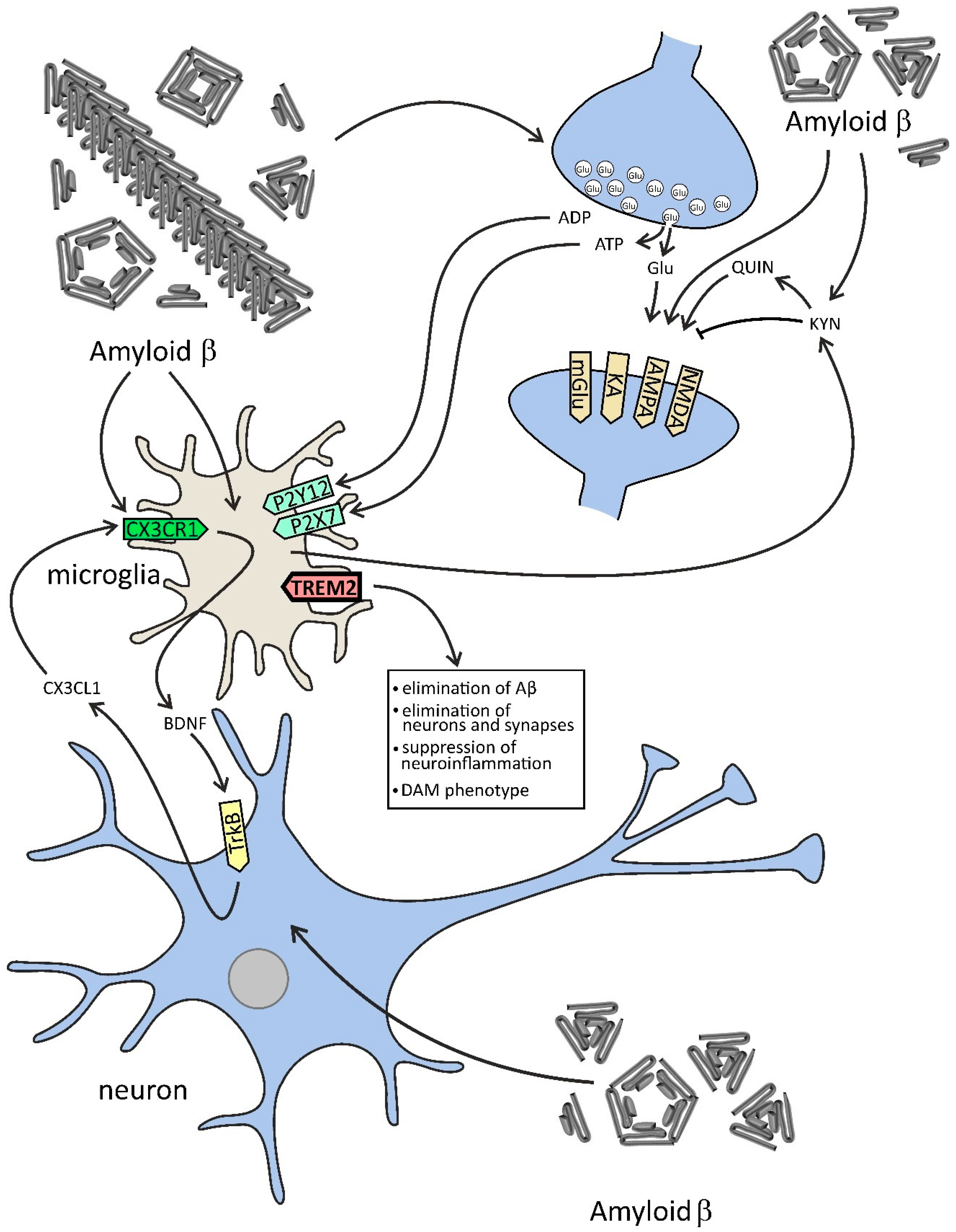
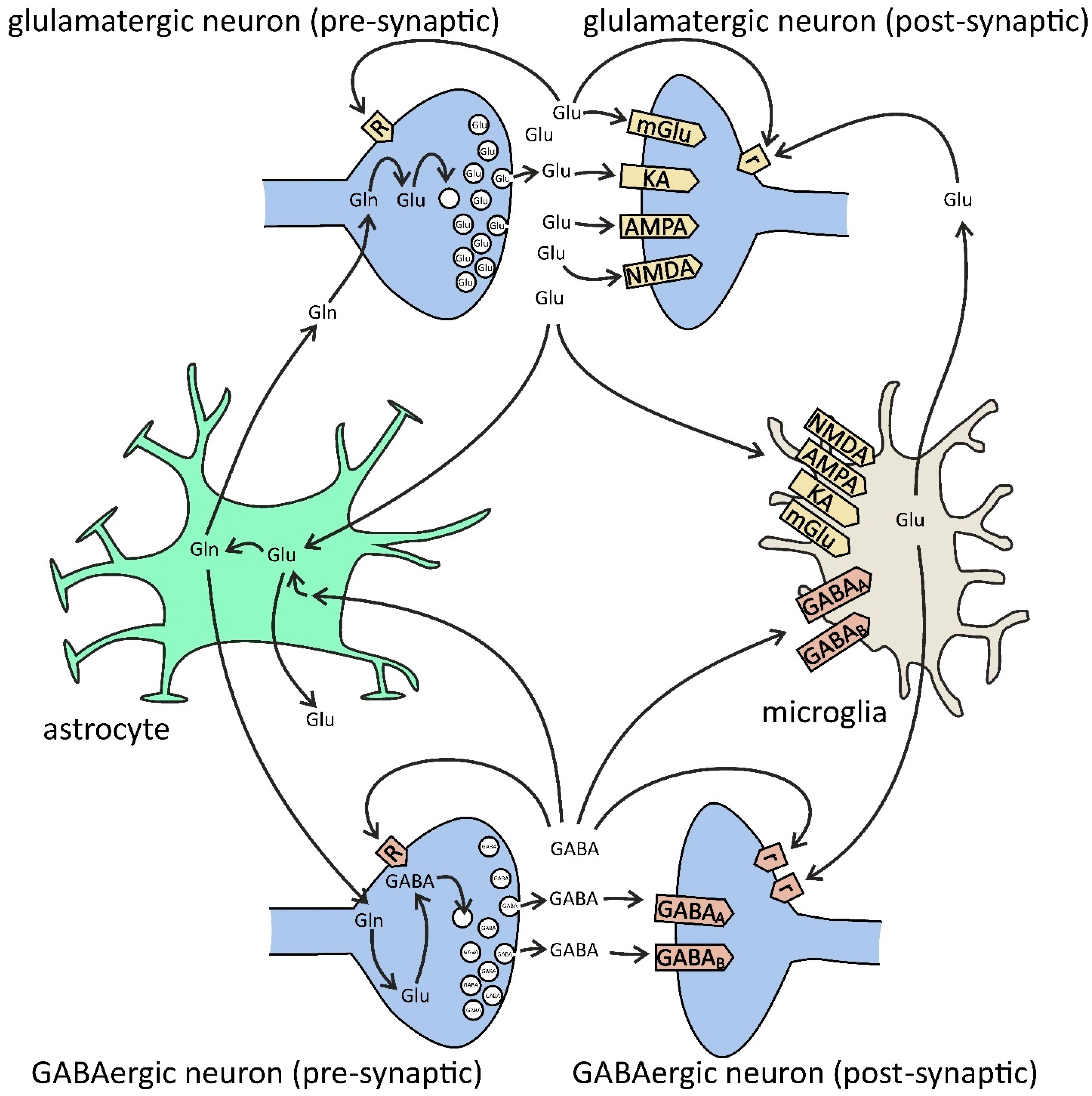

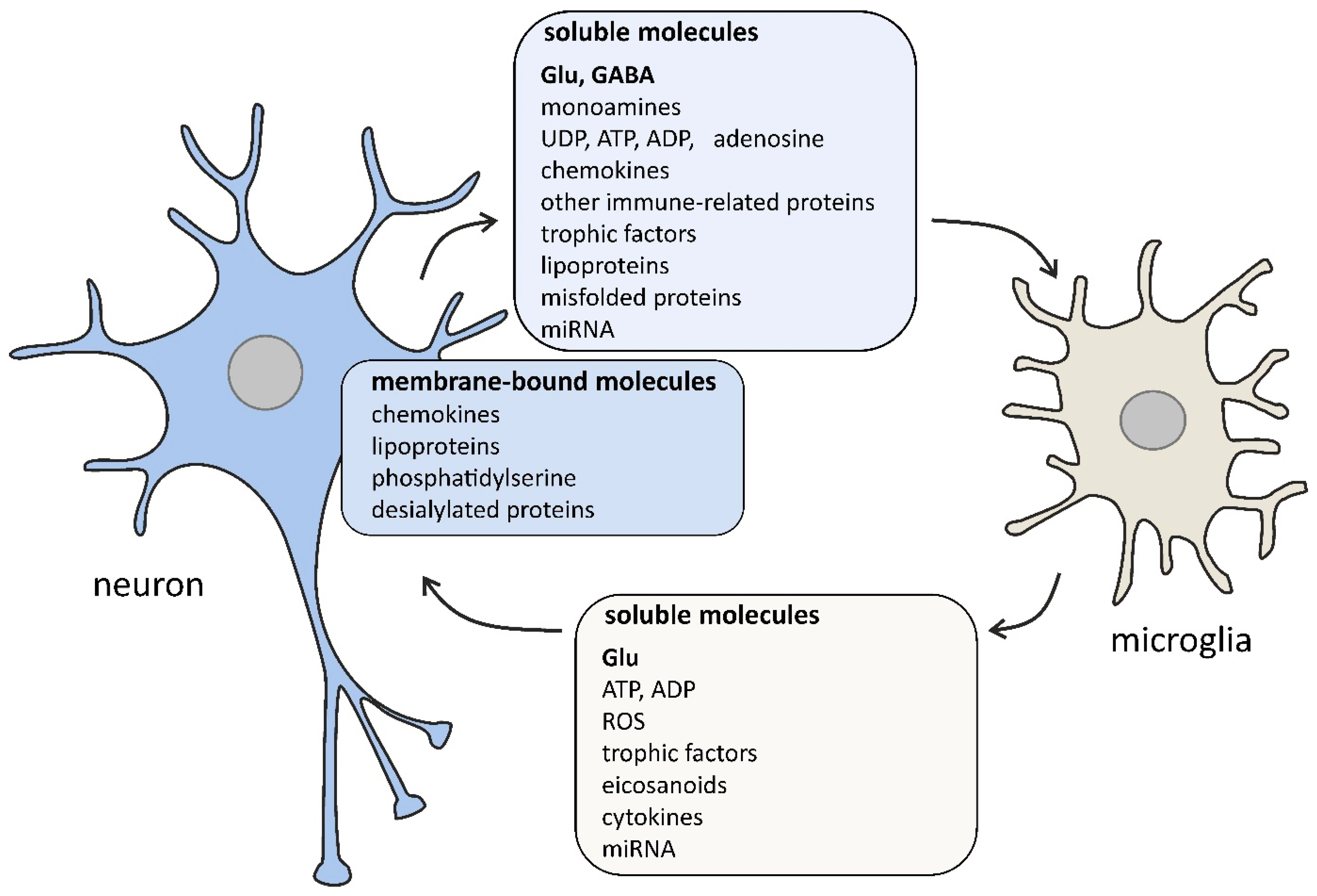{kind=link}
{kind=link}
{kind=link}
| Glutamate Components | Localization | Expression | References |
|---|---|---|---|
| Transporters | |||
| EAAT2 | Presynaptic | ↓ | [93] |
| EAAT3 | Postsynaptic | ↓ | [94] |
| Ionotropic receptors | |||
| NMDA | Post- and extrasynaptic | ↑ ↓ | [94,95] |
| AMPA | Pre-, post-, and extrasynaptic | ↑ ↓ | [94,96] |
| KA | Pre-, post-, and extrasynaptic | ↓ | [94] |
| Metabotropic receptors | |||
| mGluR3 (group II) | Pre- and postsynaptic | ↑ | [96] |
| GABA Components | Localization | Expression | References |
|---|---|---|---|
| Transporters | |||
| GAT1 | Pre-, post-, and extrasynaptic | ↓ | [136] |
| GAT3 | Pre- and extrasynaptic | ↓ | [135,136] |
| BGT1 | Extrasynaptic | ↑ | [136] |
| Ionotropic receptors | |||
| GABAA | Pre-, post-, and extrasynaptic | ↓ | [137,138,139] |
| Metabotropic receptors | |||
| GABAB | Pre-, post-, and extrasynaptic | ↓ ↑ | [140,141] |
| Compound | Effect on Glutamatergic/GABAergic Signaling | Non-Canonical Effects |
|---|---|---|
| Memantine | NMDA receptor antagonist | Inhibition of LPS-induced production of ROS and proinflammatory factors |
| Inhibition of microglia activation and neuroinflammation | ||
| Inhibition of Aβ-evoked iNOS activation and expression of glial marker proteins | ||
| The effect on inwardly rectifying K+ Kir2.1 channels in microglial cells | ||
| Inhibition of Aβ-evoked phosphorylation of Tau protein | ||
| Inhibition of the activity of cellular histone deacetylase leading to histone hyperacetylation and consequently to increased production of GDNF | ||
| Riluzole | An inhibitor of both glutamate release and postsynaptic glutamate receptor signaling | Modulation of the activity of small conductance, Ca2+-activated K+ channels (SK channels) |
| Decrease in Aβ levels (oligomers and plaques) | ||
| Decrease in Tau pathology (total level and phosphorylation) | ||
| Modulation of the gene expression patterns (including immune-related pathways implicated in AD) | ||
| BMS-984923 | The allosteric modulator of mGluR5 receptor | Inhibition of Aβ oligomer-dependent impairment of intracellular signaling |
| Decrease in Tau pathology (total level and phosphorylation) | ||
| Allopregnanolone (APα) | The positive GABAA receptor modulating steroid | In a single dose: decrease in Aβ generation |
| In a single dose: neuroregeneration | ||
| In a single dose: improvement of learning and memory | ||
| In a chronic treatment: memory decline and accelerated dementia | ||
| Modulation of microglial morphology (protrusions extension) | ||
| Decrease in migratory capacity of microglia | ||
| Decrease in phagocyting activity of microglia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11677. https://doi.org/10.3390/ijms222111677
Czapski GA, Strosznajder JB. Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(21):11677. https://doi.org/10.3390/ijms222111677
Chicago/Turabian StyleCzapski, Grzegorz A., and Joanna B. Strosznajder. 2021. "Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 21: 11677. https://doi.org/10.3390/ijms222111677
APA StyleCzapski, G. A., & Strosznajder, J. B. (2021). Glutamate and GABA in Microglia-Neuron Cross-Talk in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(21), 11677. https://doi.org/10.3390/ijms222111677