
PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway


 , , , , ,
, , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Result
2.1. Aberrant PDK1 Expression in Human OSCC Positively Correlates with Disease Progression
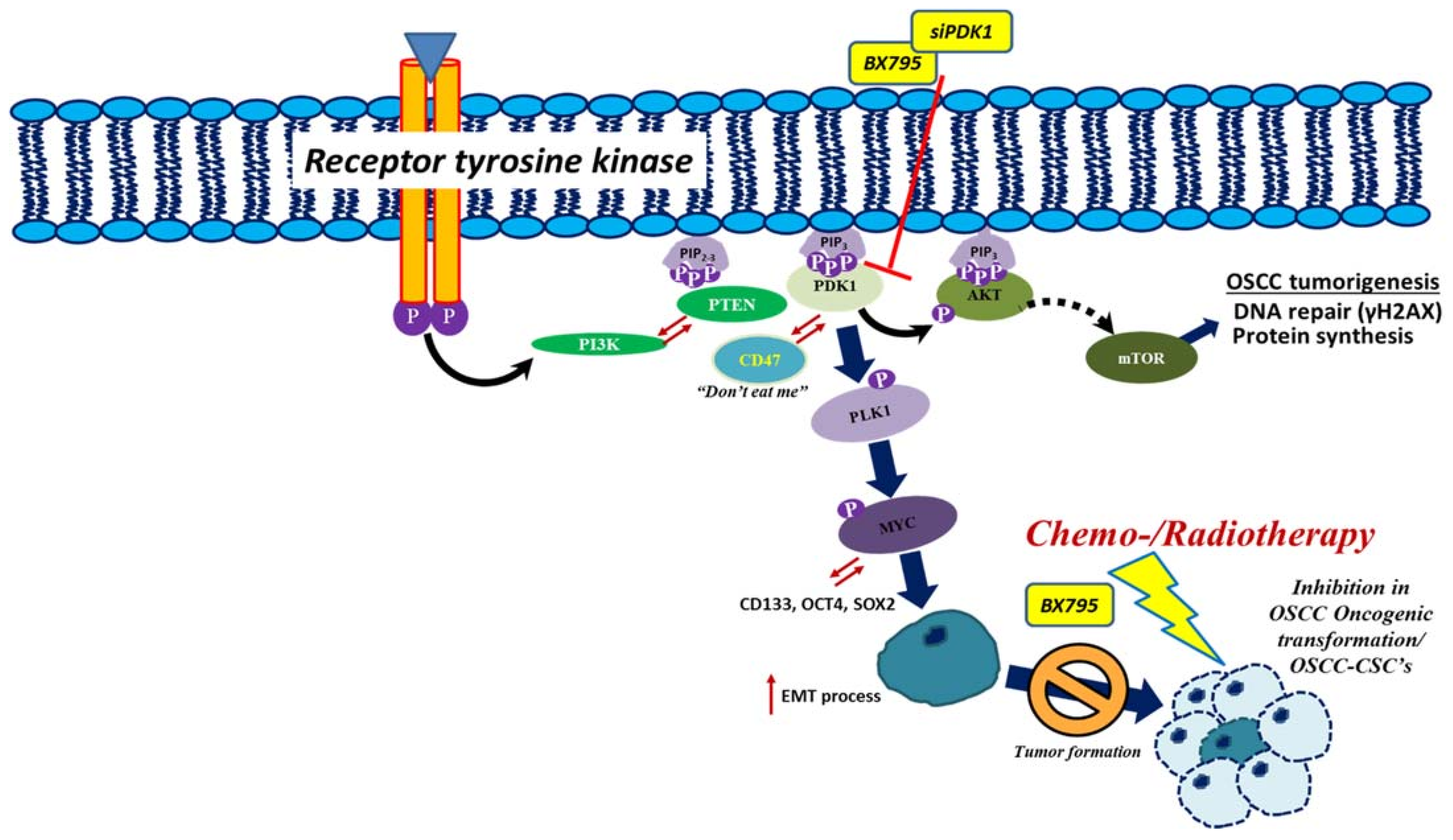
2.2. PDK1 Expression Associated with CD47 Modulation through Akt/Glycolysis Signaling Axis Activation
2.3. PDK1 Silencing Attenuates CSC’s Phenotypes Maintenance in OSCC
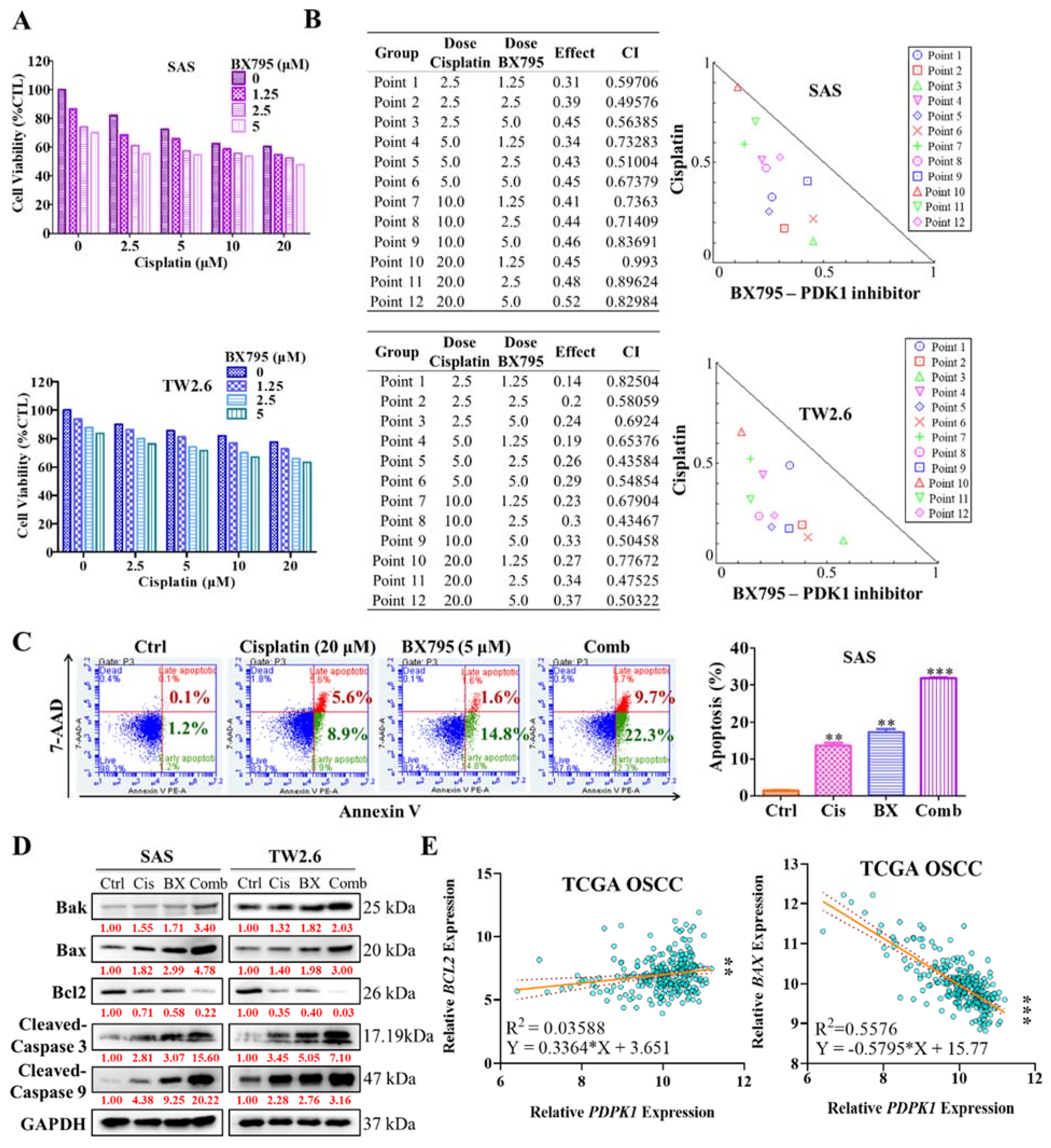
2.4. Pharmacologic Inhibition of PDK1 Acts Synergistically with Cisplatin in the Human OSCC Cell Line
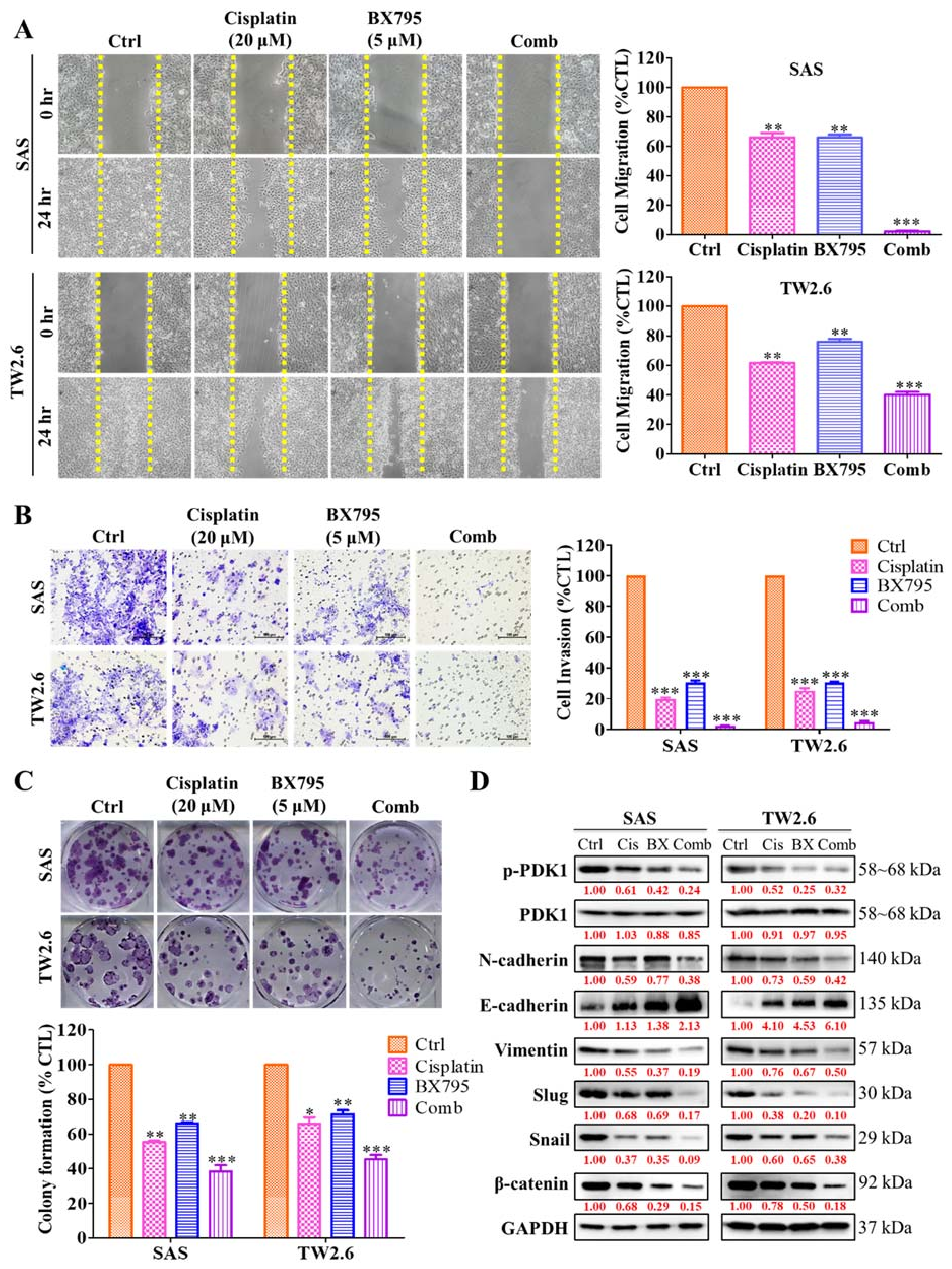
2.5. BX795 and Cisplatin Combination Therapy Effectively Diminishes Human OSCC Cell Line Epithelial–Mesenchymal Transition Properties
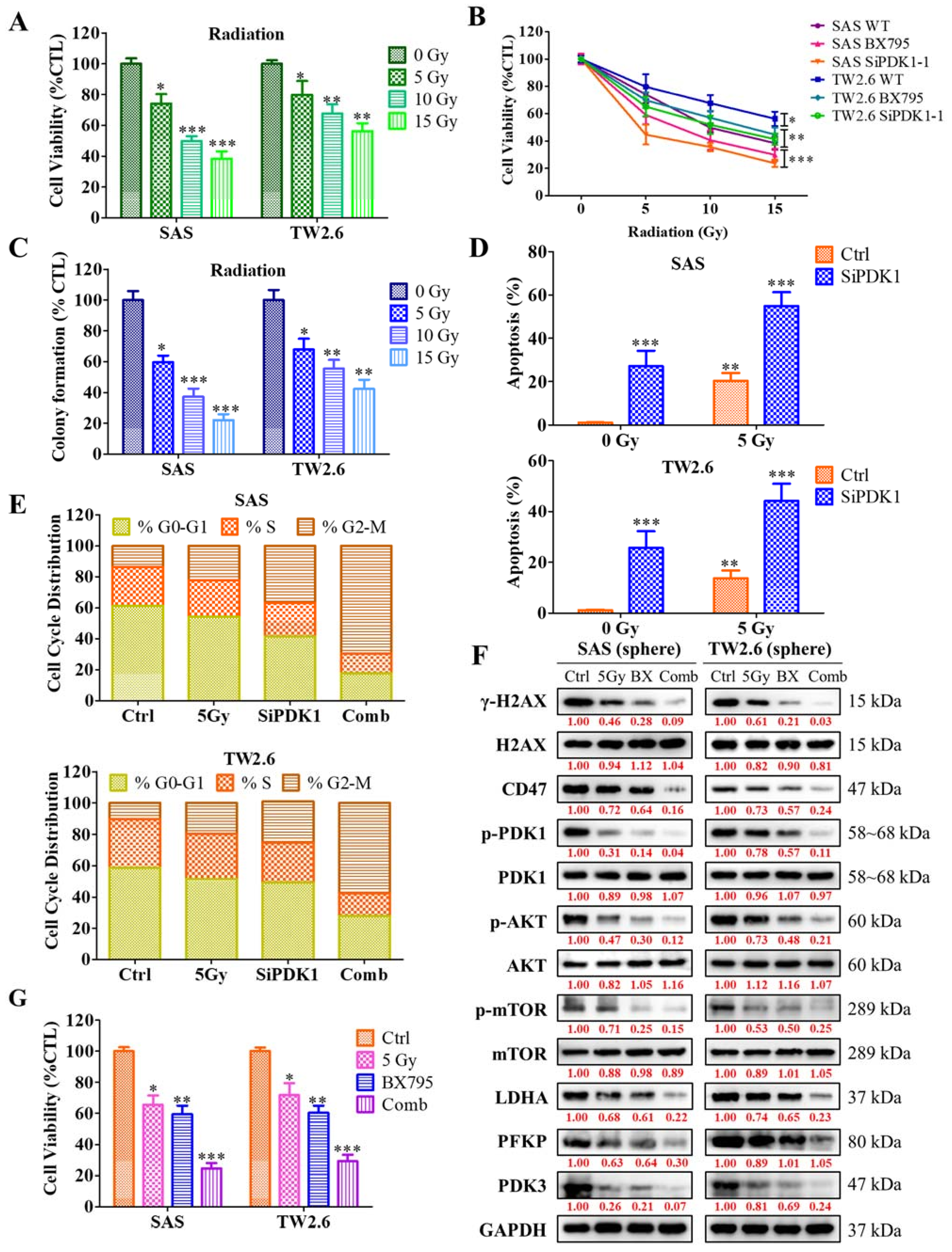
2.6. Inhibition of PDK1 Expression Enhances the Sensitivity of OSCC Cells toward Radiotherapy
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Immunohistochemical Staining
4.3. Cell Lines and Culture
4.4. siRNA Transfection of OSCC Cell Lines
4.5. Orosphere Formation and Self-Renewal Assay
4.6. Cell Cycle and Apoptosis Analysis
4.7. Sulforhodamine B Cytotoxicity Assay
4.8. Western Blot Analysis
4.9. Wound Healing Migration Assay
4.10. Matrigel Invasion and Migration Assay
4.11. Immunofluorescence Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shield, K.D.; Ferlay, J.; Jemal, A.; Sankaranarayanan, R.; Chaturvedi, A.K.; Bray, F.; Soerjomataram, I. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin. 2017, 67, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Tangthongkum, M.; Kirtsreesakul, V.; Supanimitjaroenporn, P.; Leelasawatsuk, P. Treatment outcome of advance staged oral cavity cancer: Concurrent chemoradiotherapy compared with primary surgery. Eur. Arch. Otorhinolaryngol. 2017, 274, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidi, A.; Falasca, M. Targeting pdk1 for chemosensitization of cancer cells. Cancers 2017, 9, 140. [Google Scholar] [CrossRef]
- Bamodu, O.A.; Chang, H.L.; Ong, J.R.; Lee, W.H.; Yeh, C.T.; Tsai, J.T. Elevated pdk1 expression drives pi3k/akt/mtor signaling promotes radiation-resistant and dedifferentiated phenotype of hepatocellular carcinoma. Cells 2020, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Muranen, T.A.; Greco, D.; Fagerholm, R.; Kilpivaara, O.; Kampjarvi, K.; Aittomaki, K.; Blomqvist, C.; Heikkila, P.; Borg, A.; Nevanlinna, H. Breast tumors from chek2 1100delc-mutation carriers: Genomic landscape and clinical implications. Breast Cancer Res. 2011, 13, R90. [Google Scholar] [CrossRef] [Green Version]
- Choucair, K.A.; Guerard, K.P.; Ejdelman, J.; Chevalier, S.; Yoshimoto, M.; Scarlata, E.; Fazli, L.; Sircar, K.; Squire, J.A.; Brimo, F.; et al. The 16p13.3 (PDPK1) genomic gain in prostate cancer: A potential role in disease progression. Transl. Oncol. 2012, 5, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y.; et al. 3-phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R. Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth medal lecture. Biochem. Soc. Trans. 2001, 29, 1–14. [Google Scholar] [CrossRef]
- Di Blasio, L.; Gagliardi, P.A.; Puliafito, A.; Primo, L. Serine/threonine kinase 3-phosphoinositide-dependent protein kinase-1 (PDK1) as a key regulator of cell migration and cancer dissemination. Cancers 2017, 9, 25. [Google Scholar] [CrossRef]
- Pinner, S.; Sahai, E. Pdk1 regulates cancer cell motility by antagonising inhibition of rock1 by rhoe. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.A.; Kowalski, J.; Smith, B.D.; Matsui, W. Concise review: Emerging concepts in clinical targeting of cancer stem cells. Stem Cells 2011, 29, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Shukla, G.; Khera, H.K.; Srivastava, A.K.; Khare, P.; Patidar, R.; Saxena, R. Therapeutic potential, challenges and future perspective of cancer stem cells in translational oncology: A critical review. Curr. Stem Cell Res. Ther. 2017, 12, 207–224. [Google Scholar] [CrossRef]
- Wu, C.X.; Wang, X.Q.; Chok, S.H.; Man, K.; Tsang, S.H.Y.; Chan, A.C.Y.; Ma, K.W.; Xia, W.; Cheung, T.T. Blocking CDK1/PDK1/beta-catenin signaling by CDK1 inhibitor ro3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018, 8, 3737–3750. [Google Scholar] [CrossRef]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.; Wee, Z.N.; et al. PDK1 signaling toward plk1-myc activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mtor-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [Green Version]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; De Majo, M.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CDK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (cscs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Sur, S.; Nakanishi, H.; Flaveny, C.; Ippolito, J.E.; McHowat, J.; Ford, D.A.; Ray, R.B. Inhibition of the key metabolic pathways, glycolysis and lipogenesis, of oral cancer by bitter melon extract. Cell Commun. Signal. 2019, 17, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. ΓH2ax: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, N.; Meunier, A.; Lynch, T.H.; Hollywood, D.; Marignol, L. Isogenic radiation resistant cell lines: Development and validation strategies. Int. J. Radiat. Biol. 2014, 90, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Jiang-Hua, M.; Ge-Liang, H.; Qing, C.; Ya-Ming, L. Mir-34a inhibits spinal cord injury and blocks spinal cord neuron apoptosis by activating phatidylinositol 3-kinase (pi3k)/akt pathway through targeting CD47. Curr. Neurovasc. Res. 2019, 16, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, X.; Wang, Y.; Li, Y.; Chen, X.; Yang, W.; Jiang, L. CD47 promotes human glioblastoma invasion through activation of the pi3k/akt pathway. Oncol. Res. 2019, 27, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Kong, X.; Li, E.; Liu, Y.; Du, X.; Kang, Z.; Tang, Y.; Kuang, Y.; Yang, Z.; et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci. Rep. 2016, 6, 29719. [Google Scholar] [CrossRef]
- Pai, S.; Bamodu, O.A.; Lin, Y.K.; Lin, C.S.; Chu, P.Y.; Chien, M.H.; Wang, L.S.; Hsiao, M.; Yeh, C.T.; Tsai, J.T. CD47-sirpalpha signaling induces epithelial-mesenchymal transition and cancer stemness and links to a poor prognosis in patients with oral squamous cell carcinoma. Cells 2019, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Elkahloun, A.G.; Singh, S.P.; Chen, Q.-R.; Meerzaman, D.M.; Song, T.; Manu, N.; Wu, W.; Mannan, P.; Garfield, S.H.; et al. A function-blocking CD47 antibody suppresses stem cell and egf signaling in triple-negative breast cancer. Oncotarget 2016, 7, 10133–10152. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chang, Y.; He, X.; Cai, Y.; Jiang, H.; Jia, R.; Leng, J. CD47 enhances cell viability and migration ability but inhibits apoptosis in endometrial carcinoma cells via the pi3k/akt/mtor signaling pathway. Front. Oncol. 2020, 10, 1525. [Google Scholar] [CrossRef]
- Chen, C.; Méndez, E.; Houck, J.; Fan, W.; Lohavanichbutr, P.; Doody, D.; Yueh, B.; Futran, N.D.; Upton, M.; Farwell, D.G.; et al. Gene expression profiling identifies genes predictive of oral squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2152–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. Ncbi geo: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-H.; O’Sullivan, B. Oral cancer: Current role of radiotherapy and chemotherapy. Med. Oral. Patol. Oral. Cir. Bucal. 2013, 18, e233–e240. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The pi3k-akt network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2018, 48, 27–35. [Google Scholar] [CrossRef]
- Casamayor, A.; Morrice, N.A.; Alessi, D.R. Phosphorylation of ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem. J. 1999, 342 Pt 2, 287–292. [Google Scholar] [CrossRef]
- Arencibia, J.M.; Pastor-Flores, D.; Bauer, A.F.; Schulze, J.O.; Biondi, R.M. Agc protein kinases: From structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim. Biophys. Acta 2013, 1834, 1302–1321. [Google Scholar] [CrossRef]
- Bramhecha, Y.M.; Guerard, K.P.; Rouzbeh, S.; Scarlata, E.; Brimo, F.; Chevalier, S.; Hamel, L.; Dragomir, A.; Aprikian, A.G.; Lapointe, J. Genomic gain of 16p13.3 in prostate cancer predicts poor clinical outcome after surgical intervention. Mol. Cancer Res. 2018, 16, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wu, Z.; Liu, T.; Han, L.; Wang, C.; Yang, B.; Zheng, F. Upregulation of PDK1 associates with poor prognosis in esophageal squamous cell carcinoma with facilitating tumorigenicity in vitro. Med. Oncol. 2014, 31, 337. [Google Scholar] [CrossRef]
- Scortegagna, M.; Ruller, C.; Feng, Y.; Lazova, R.; Kluger, H.; Li, J.L.; De, S.K.; Rickert, R.; Pellecchia, M.; Bosenberg, M.; et al. Genetic inactivation or pharmacological inhibition of PDK1 delays development and inhibits metastasis of braf(v600e): Pten(-/-) melanoma. Oncogene 2014, 33, 4330–4339. [Google Scholar] [CrossRef] [Green Version]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Lo, C.M.; Chen, L.; Ngan, E.S.; Xu, A.; Poon, R.Y. CDK1-PDK1-pi3k/akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ. 2017, 24, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Sestito, S.; Pietrobono, D.; Giacomelli, C.; Chiellini, G.; Di Maio, D.; Marinelli, L.; Novellino, E.; Martini, C.; Rapposelli, S. Dual inhibition of PDK1 and aurora kinase A: An effective strategy to induce differentiation and apoptosis of human glioblastoma multiforme stem cells. ACS Chem. Neurosci. 2017, 8, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase a by erbb2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- An, J.; He, H.; Yao, W.; Shang, Y.; Jiang, Y.; Yu, Z. Pi3k/akt/foxo pathway mediates glycolytic metabolism in hepg2 cells exposed to triclosan (tcs). Environ. Int. 2020, 136, 105428. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.L.; Fan, S.; Zhang, S.Y.; Chen, W.X.; Li, Q.X.; Pan, G.K.; Zhang, H.Q.; Wang, W.W.; Weng, B.; Zhang, Z.; et al. Sox8 regulates cancer stem-like properties and cisplatin-induced emt in tongue squamous cell carcinoma by acting on the wnt/beta-catenin pathway. Int. J. Cancer 2018, 142, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Xu, X.; Li, J.; Chen, C.; Chen, W.; Wang, F.; Xie, Y.; Li, F. The PDK1/cjun pathway activated by tgfbeta induces emt and promotes proliferation and invasion in human glioblastoma. Int. J. Oncol. 2018, 53, 2067–2080. [Google Scholar]
- Lian, S.; Shao, Y.; Liu, H.; He, J.; Lu, W.; Zhang, Y.; Jiang, Y.; Zhu, J. PDK1 induces junb, emt, cell migration and invasion in human gallbladder cancer. Oncotarget 2015, 6, 29076–29086. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.S.; Zhang, X.H.; Yang, S.N.; Liu, D.; Diao, C.R.; Wang, H.; Zheng, F.P. Cyanidin inhibits emt induced by oxaliplatin via targeting the PDK1-pi3k/akt signaling pathway. Food Funct. 2019, 10, 592–601. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. Pi3k-akt-mtor inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Dahl, K.N.; Westhoff, C.M.; Discher, D.E. Fractional attachment of CD47 (iap) to the erythrocyte cytoskeleton and visual colocalization with rh protein complexes. Blood 2003, 101, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, J. Functions of immune checkpoint molecules beyond immune evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bras, M.; Yuste, V.J.; Roue, G.; Barbier, S.; Sancho, P.; Virely, C.; Rubio, M.; Baudet, S.; Esquerda, J.E.; Merle-Beral, H.; et al. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol. Cell. Biol. 2007, 27, 7073–7088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, V.; Brown, E.J.; Biron, G.; Rubio, M.; Fischer, A.; Deist, F.L.; Sarfati, M. Mechanisms of CD47-induced caspase-independent cell death in normal and leukemic cells: Link between phosphatidylserine exposure and cytoskeleton organization. Blood 2002, 100, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Cheung, V.C.; Lu, P.; Lau, E.Y.; Ma, S.; Tang, K.H.; Tong, M.; Lo, J.; Ng, I.O. Blockade of CD47-mediated cathepsin s/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 2014, 60, 179–191. [Google Scholar] [CrossRef]
- Tan, W.; Tang, H.; Jiang, X.; Ye, F.; Huang, L.; Shi, D.; Li, L.; Huang, X.; Li, L.; Xie, X.; et al. Metformin mediates induction of mir-708 to inhibit self-renewal and chemoresistance of breast cancer stem cells through targeting CD47. J. Cell. Mol. Med. 2019, 23, 5994–6004. [Google Scholar] [CrossRef] [PubMed]
- Sick, E.; Boukhari, A.; Deramaudt, T.; Ronde, P.; Bucher, B.; Andre, P.; Gies, J.P.; Takeda, K. Activation of CD47 receptors causes proliferation of human astrocytoma but not normal astrocytes via an akt-dependent pathway. Glia 2011, 59, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Variables | No. (n = 62) | PDK1 | x2 | p-Value | |
|---|---|---|---|---|---|
| High Expression | Low Expression | ||||
| Median Age | 0.104 | 0.747 | |||
| ≤50 | 33 | 15 (45) | 18 (55) | ||
| >50 | 29 | 11 (41) | 17 (59) | ||
| Gender | 0.112 | 0.737 | |||
| Male | 56 | 24 (43) | 32 (57) | ||
| Female | 6 | 3 (50) | 3 (50) | ||
| AJCC stage | 8.308 | 0.040 | |||
| I | 8 | 2 (25) | 6 (75) | ||
| II | 16 | 3 (19) | 13 (81) | ||
| III | 14 | 8 (57) | 6 (43) | ||
| IV | 24 | 14 (58) | 10 (42) | ||
| T stage | 2.410 | 0.492 | |||
| 1 | 12 | 3 (25) | 9 (75) | ||
| 2 | 28 | 13 (46) | 15 (54) | ||
| 3 | 5 | 3 (60) | 2 (40) | ||
| 4 | 17 | 8 (47) | 9 (53) | ||
| LN metastasis | 0.230 | 0.632 | |||
| No | 32 | 13 (41) | 19 (59) | ||
| Yes | 30 | 14 (47) | 16 (53) | ||
| M, Metastasis | 5.018 | 0.025 | |||
| No | 53 | 20 (38) | 33 (62) | ||
| Yes | 9 | 7 (78) | 2 (22) | ||
| Primary site | 4.525 | 0.718 | |||
| Buccal | 26 | 13 (50) | 13 (50) | ||
| Palata | 1 | 0 (0) | 1 (100) | ||
| Tongue | 27 | 10 (37) | 17 (63) | ||
| Gingiva | 6 | 2 (33) | 4 (67) | ||
| Mouth Floor | 0 | 0 (0) | 0 (0) | ||
| Maxilla | 0 | 0 (0) | 0 (0) | ||
| Lip | 1 | 1 (100) | 0 (0) | ||
| Tonsil | 1 | 1 (100) | 0 (0) | ||
| Univariate | Multivariate | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Covariate | Coefficient | SE | Wald | p-Value | HR | 95%CI | Coefficient | SE | Wald | p-Value | HR | 95%CI | ||
| Age (≤50 vs. >50) | −1.32 | 0.717 | 3.385 | 0.066 | 0.267 | 0.0655 | 1.09 | |||||||
| Gender (Male vs. Female) | −1.243 | 0.902 | 1.9 | 0.168 | 0.288 | 0.0492 | 1.69 | |||||||
| AJCC stage (I/II vs. III/IV) | −1.676 | 0.871 | 3.705 | 0.054 | 0.187 | 0.034 | 1.031 | −2.031 | 0.604 | 11.305 | <0.001 | 0.131 | 0.0402 | 0.429 |
| T stage (T1 vs. ≥T1) | −1.007 | 0.91 | 1.226 | 0.268 | 0.365 | 0.0614 | 2.172 | |||||||
| N stage (0 vs. ≥1) | 0.164 | 0.607 | 0.0731 | 0.787 | 1.178 | 0.359 | 3.869 | |||||||
| M stage (0 vs. 1) | 1.15 | 0.491 | 5.482 | 0.019 | 3.157 | 1.206 | 8.263 | 1.139 | 0.462 | 6.076 | 0.014 | 3.122 | 1.263 | 7.721 |
| PDK1 expression (high vs. low) | −1.529 | 0.53 | 8.33 | 0.004 | 0.217 | 0.0767 | 0.612 | −1.282 | 0.449 | 8.16 | 0.004 | 0.278 | 0.115 | 0.669 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pai, S.; Yadav, V.K.; Kuo, K.-T.; Pikatan, N.W.; Lin, C.-S.; Chien, M.-H.; Lee, W.-H.; Hsiao, M.; Chiu, S.-C.; Yeh, C.-T.; et al. PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 11492. https://doi.org/10.3390/ijms222111492
Pai S, Yadav VK, Kuo K-T, Pikatan NW, Lin C-S, Chien M-H, Lee W-H, Hsiao M, Chiu S-C, Yeh C-T, et al. PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(21):11492. https://doi.org/10.3390/ijms222111492
Chicago/Turabian StylePai, Shin, Vijesh Kumar Yadav, Kuang-Tai Kuo, Narpati Wesa Pikatan, Chun-Shu Lin, Ming-Hsien Chien, Wei-Hwa Lee, Michael Hsiao, Shao-Chih Chiu, Chi-Tai Yeh, and et al. 2021. "PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway" International Journal of Molecular Sciences 22, no. 21: 11492. https://doi.org/10.3390/ijms222111492
APA StylePai, S., Yadav, V. K., Kuo, K.-T., Pikatan, N. W., Lin, C.-S., Chien, M.-H., Lee, W.-H., Hsiao, M., Chiu, S.-C., Yeh, C.-T., & Tsai, J.-T. (2021). PDK1 Inhibitor BX795 Improves Cisplatin and Radio-Efficacy in Oral Squamous Cell Carcinoma by Downregulating the PDK1/CD47/Akt-Mediated Glycolysis Signaling Pathway. International Journal of Molecular Sciences, 22(21), 11492. https://doi.org/10.3390/ijms222111492