
Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
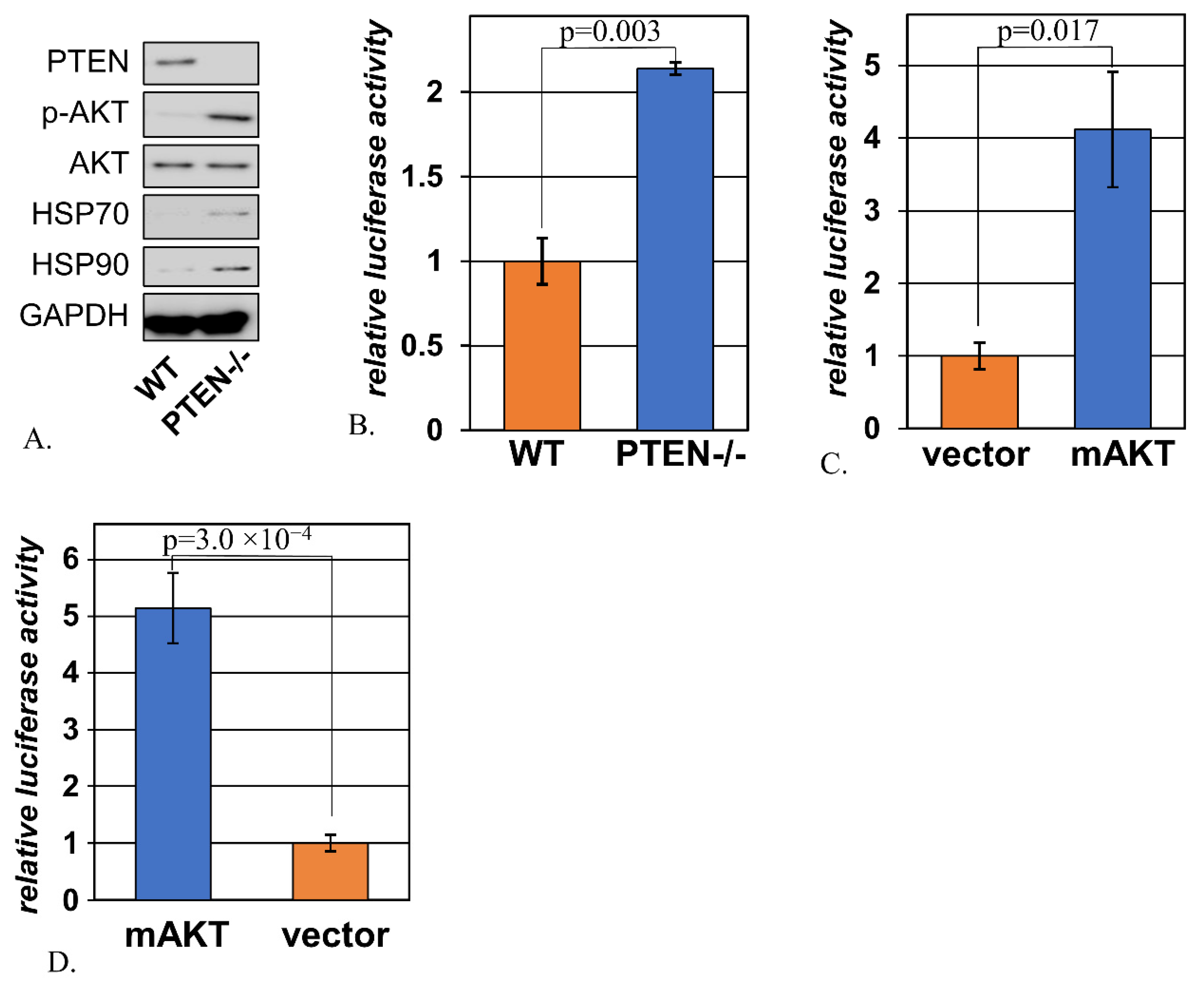
2.1. Signs of Activated Heat Shock Response Accompany Hyperactivation of AKT
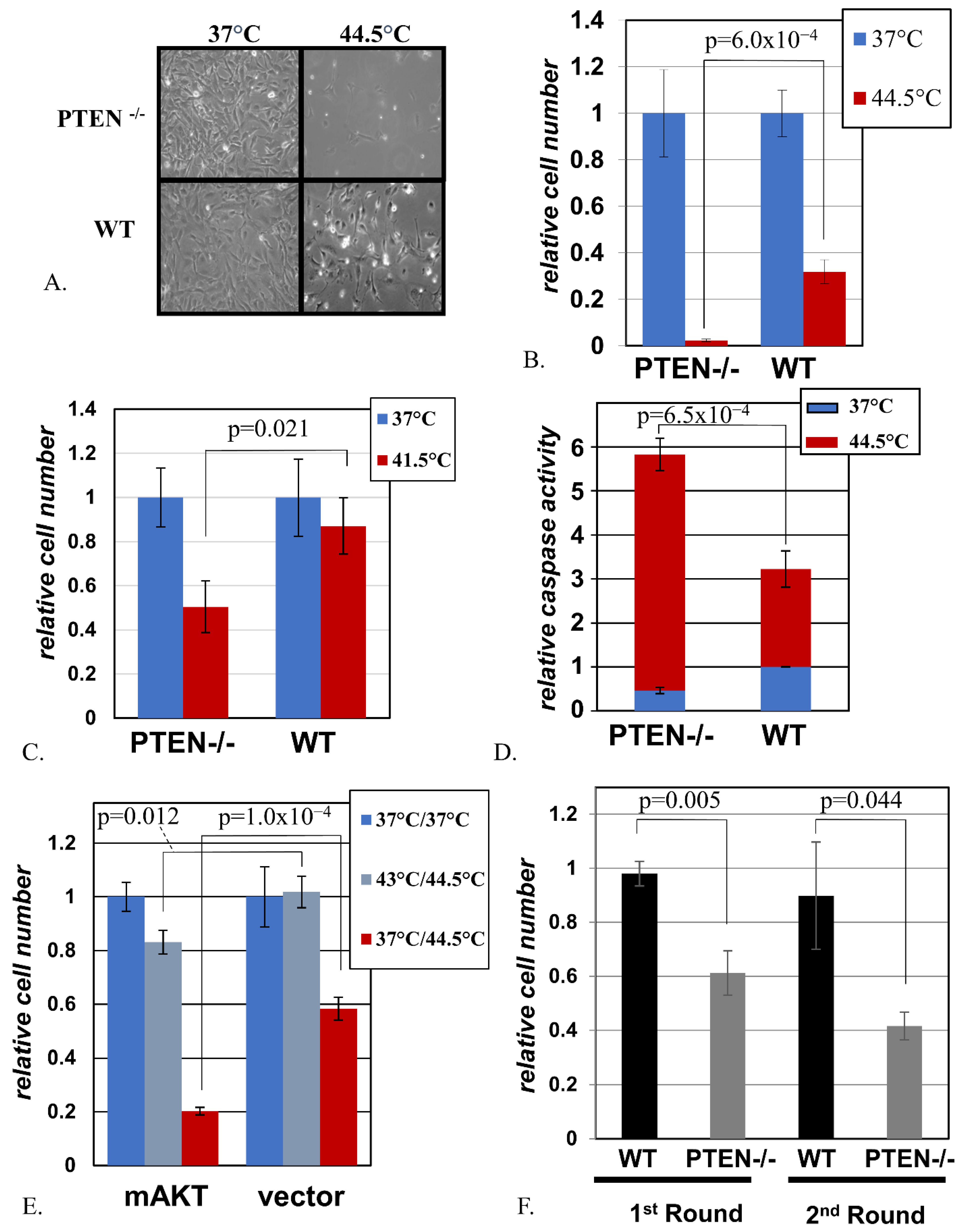
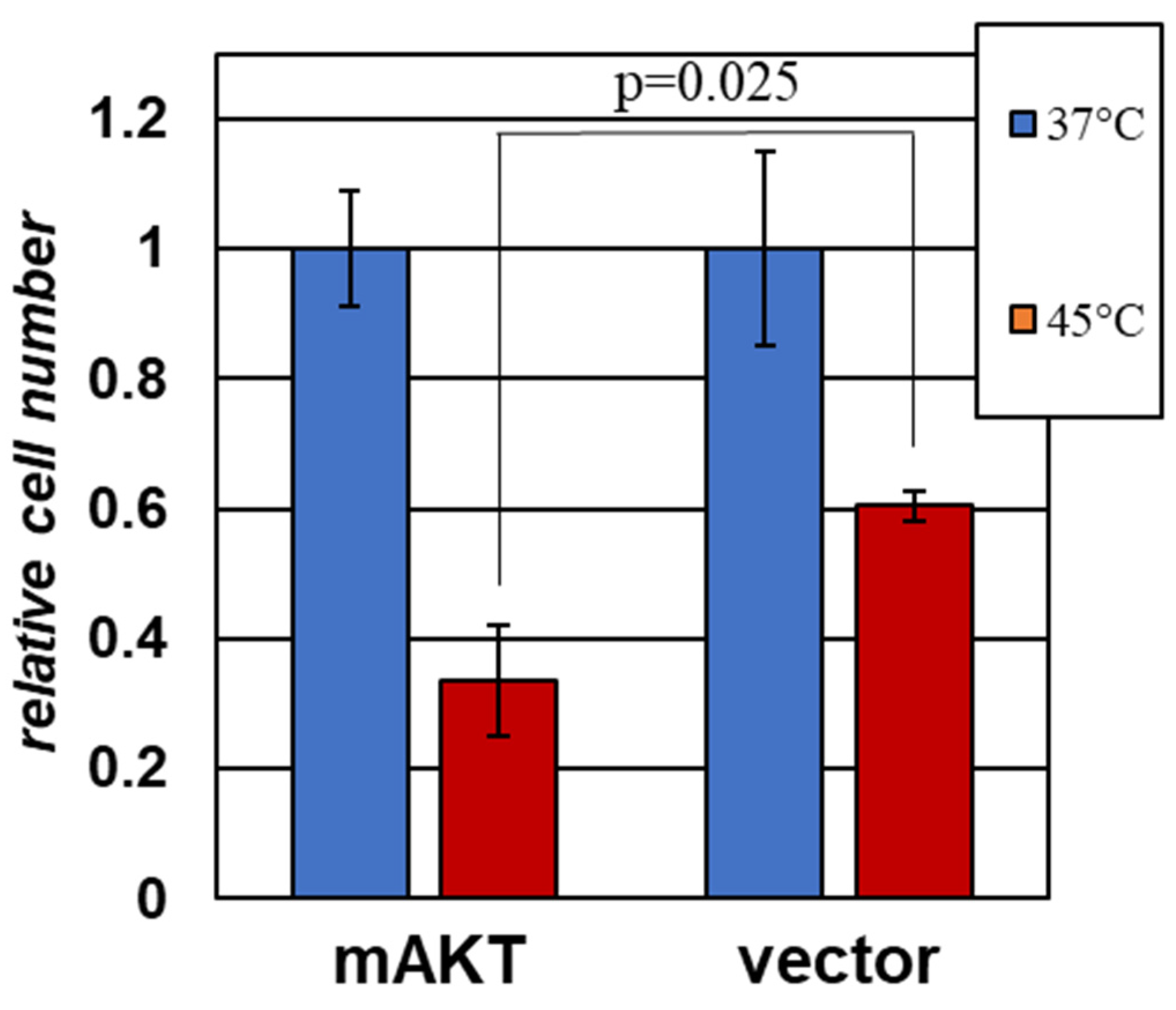
2.2. Cells with Activated AKT Are Hypersensitive to Hyperthermia
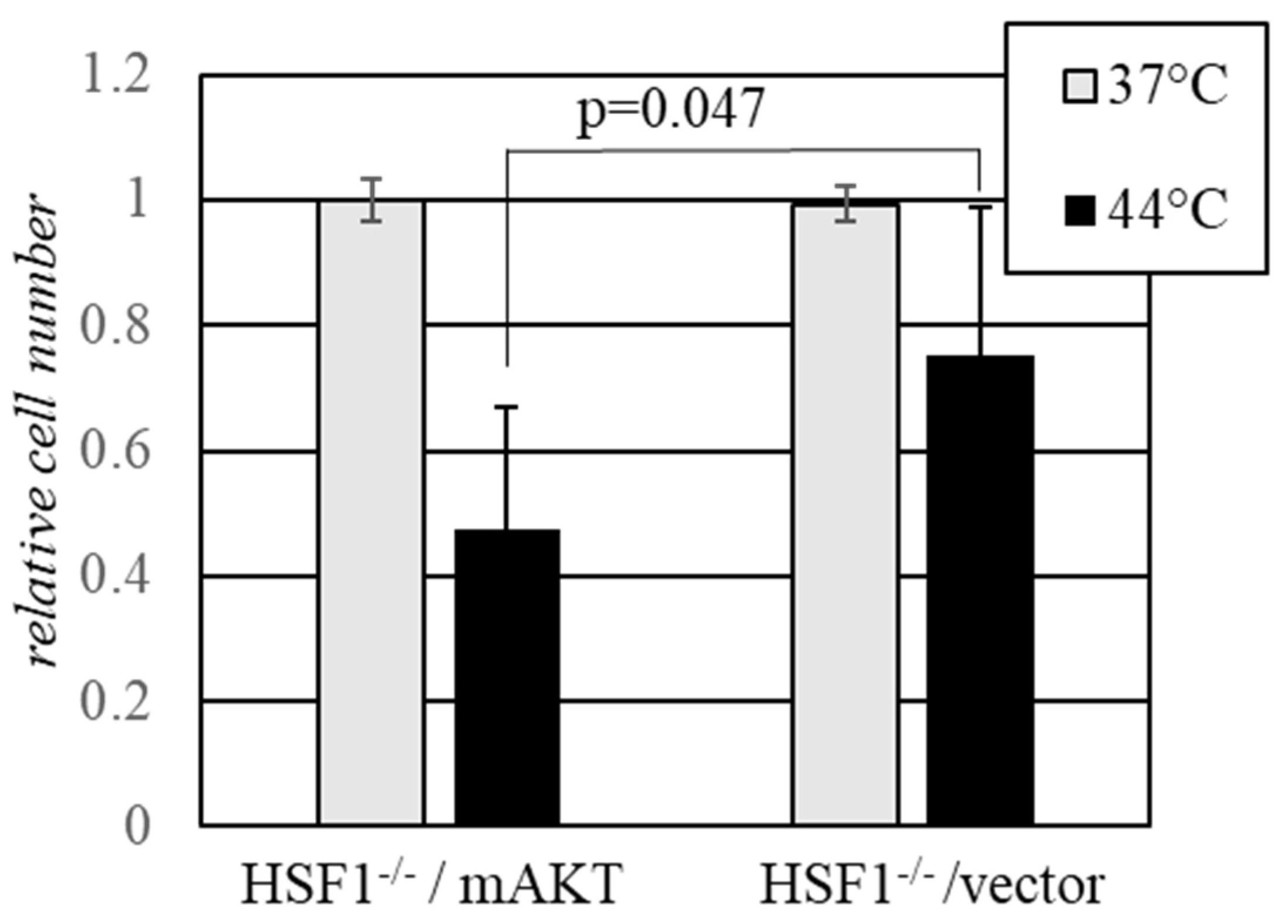
2.3. Activated AKT Sensitizes Cells to Hyperthermia in the Absence of HSF1
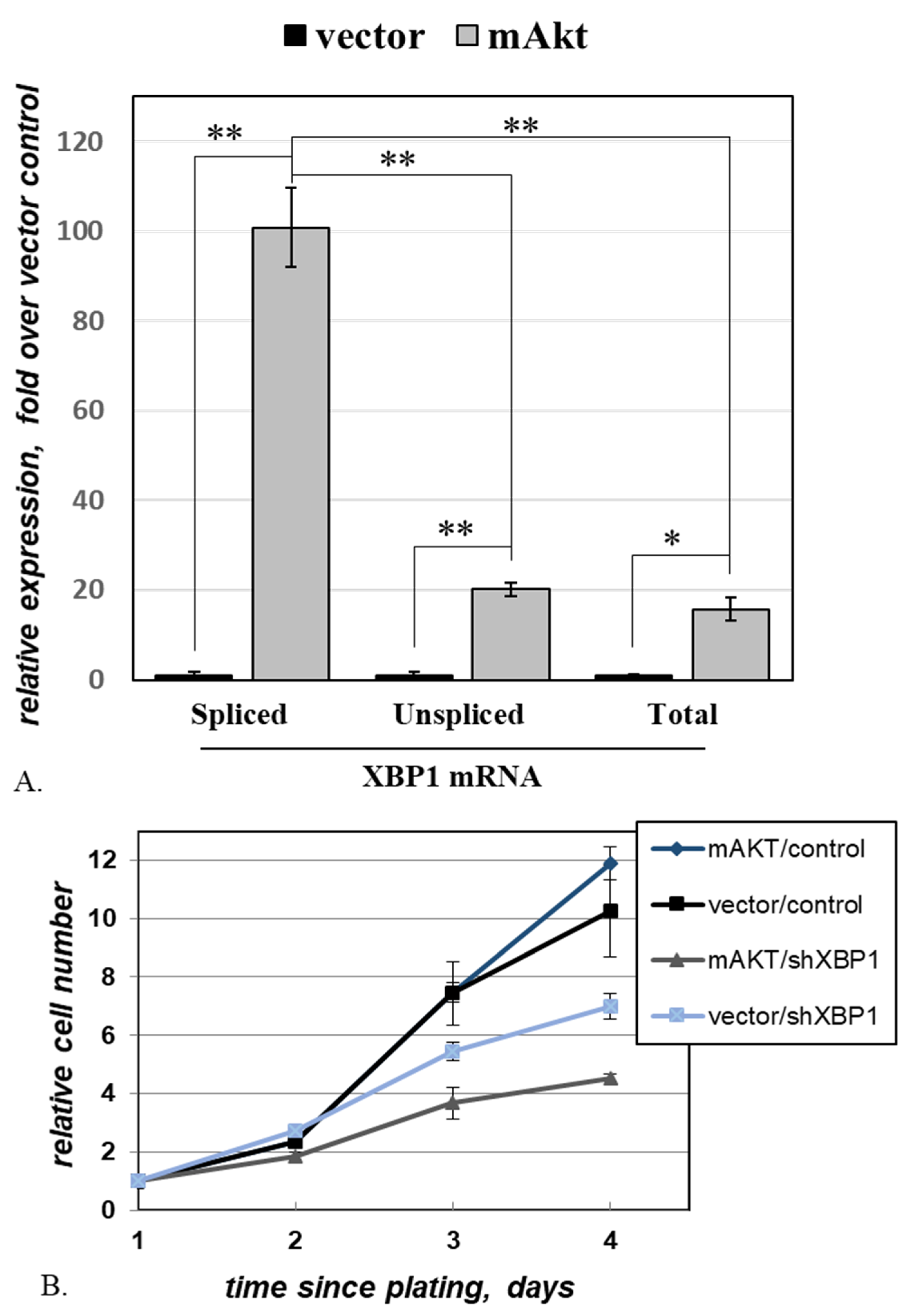
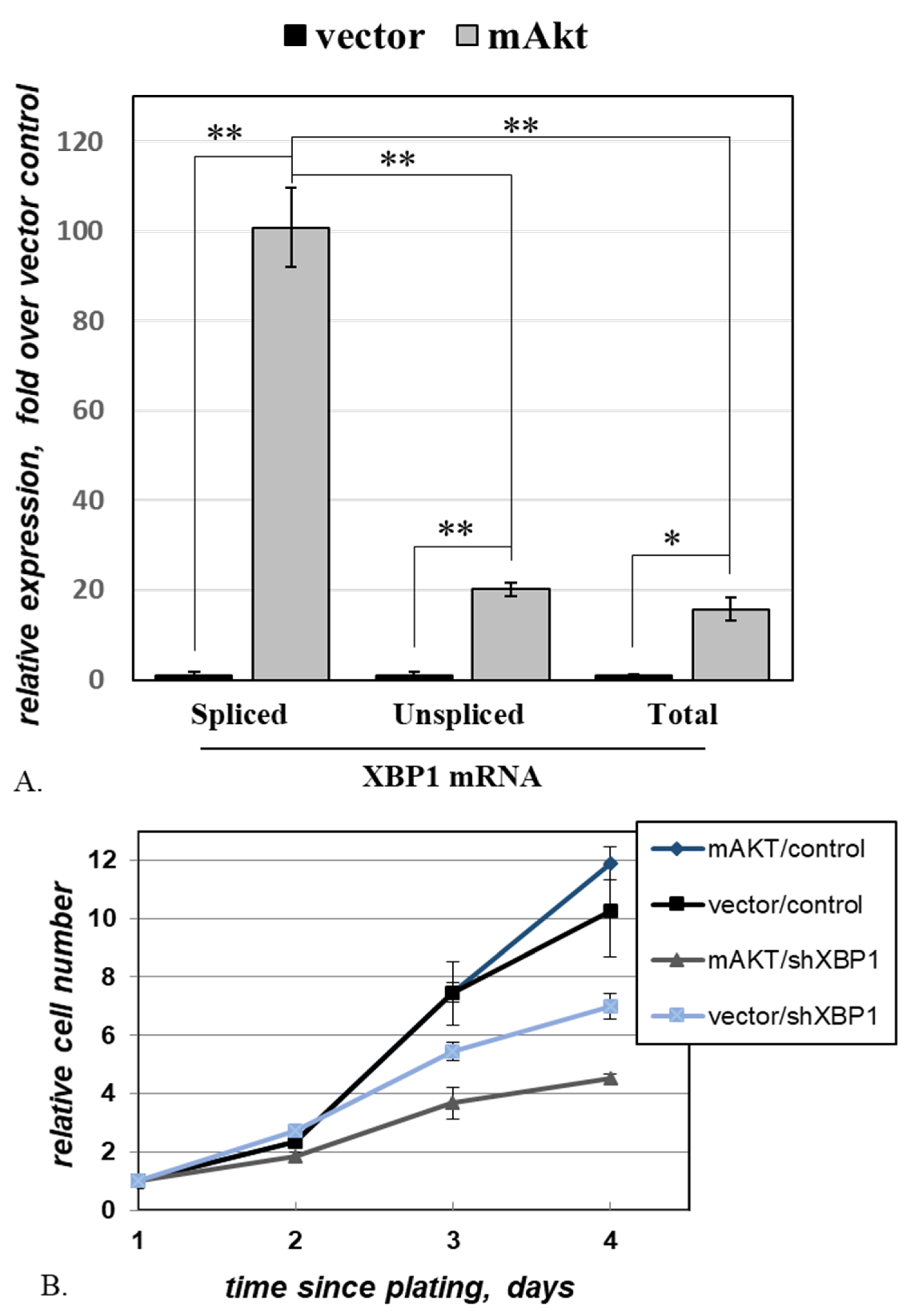
2.4. Activated AKT Increases Cell Dependence on XBP1
2.5. Activated AKT Sensitizes a Human Melanoma Cell Line to Hyperthermia
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Virus Production
4.3. Heat Shock
4.4. Comparison of Cell Numbers
4.5. Apoptosis Assays
4.6. Quantitative RT-PCR
4.7. Immunoblotting
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kandel, E.S.; Hay, N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Harari, P.M.; Wheeler, D.L.; Toulany, M. Targeting AKT/PKB to improve treatment outcomes for solid tumors. Mutat. Res. 2020, 819–820, 111690. [Google Scholar] [CrossRef]
- Alvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It's all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Somanath, P.R.; Vijai, J.; Kichina, J.V.; Byzova, T.; Kandel, E.S. The role of PAK-1 in activation of MAP kinase cascade and oncogenic transformation by Akt. Oncogene 2009, 28, 2365–2369. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Truitt, M.L.; Ruggero, D. Oncogenic AKTivation of translation as a therapeutic target. Br. J. Cancer 2011, 105, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”? Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.Y.; Walter, R.B.; Faderl, S.H.; Jabbour, E.J.; Zeng, Z.; Borthakur, G.; Huang, X.; Kadia, T.M.; Ruvolo, P.P.; Feliu, J.B.; et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin. Cancer Res. 2014, 20, 2226–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Speranza, G.; Bishop, R.; Khin, S.; Rubinstein, L.; Kinders, R.J.; Datiles, M.; Eugeni, M.; Lam, M.H.; Doyle, L.A.; et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investig. New Drugs 2015, 33, 720–728. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Kennecke, H.F.; Iqbal, S.; Baranda, J.C.; Seery, T.E.; Lim, H.J.; Hezel, A.F.; Vaccaro, G.M.; Blanke, C.D. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: A SWOG cooperative group trial (S1005). Cancer 2015, 121, 2193–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Alarcon, E.; Armstrong, A.C.; Philco, M.; Lopez Chuken, Y.A.; Sablin, M.P.; Tamura, K.; Gomez Villanueva, A.; Perez-Fidalgo, J.A.; Cheung, S.Y.A.; et al. BEECH: A dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann. Oncol. 2019, 30, 774–780. [Google Scholar]
- Bang, Y.J.; Kang, Y.K.; Ng, M.; Chung, H.C.; Wainberg, Z.A.; Gendreau, S.; Chan, W.Y.; Xu, N.; Maslyar, D.; Meng, R.; et al. A phase II, randomised study of mFOLFOX6 with or without the Akt inhibitor ipatasertib in patients with locally advanced or metastatic gastric or gastroesophageal junction cancer. Eur. J. Cancer 2019, 108, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Xie, Y.; Wang, X.; Khaleque, M.A.; Chou, S.D.; Murshid, A.; Prince, T.; Zhang, Y. Signal Transduction Pathways Leading to Heat Shock Transcription. Sign Transduct. Insights 2010, 2, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. [Google Scholar] [CrossRef] [PubMed]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep. 2018, 25, 212–223.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate cancer, PI3K, PTEN and prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef]
- Lesche, R.; Groszer, M.; Gao, J.; Wang, Y.; Messing, A.; Sun, H.; Liu, X.; Wu, H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 2002, 32, 148–149. [Google Scholar] [CrossRef]
- Zuehlke, A.D.; Beebe, K.; Neckers, L.; Prince, T. Regulation and function of the human HSP90AA1 gene. Gene 2015, 570, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sistonen, L.; Sarge, K.D.; Morimoto, R.I. Human heat shock factors 1 and 2 are differentially activated and can synergistically induce hsp70 gene transcription. Mol. Cell Biol. 1994, 14, 2087–2099. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Aparicio, C.; Renner, O.; Leal, J.F.; Carnero, A. PTEN, more than the AKT pathway. Carcinogenesis 2007, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettaieb, A.; Averill-Bates, D.A. Thermotolerance induced at a mild temperature of 40 degrees C alleviates heat shock-induced ER stress and apoptosis in HeLa cells. Biochim. Biophys. Acta 2015, 1853, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Paw, I.; Dewhirst, M.W.; Lo, H.W. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 2015, 34, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Babagana, M.; Johnson, S.; Slabodkin, H.; Bshara, W.; Morrison, C.; Kandel, E.S. P21-activated kinase 1 regulates resistance to BRAF inhibition in human cancer cells. Mol. Carcinog. 2017, 56, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Andrulis, M.; Stuhmer, T.; Muller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Frezzato, F.; Raggi, F.; Martini, V.; Severin, F.; Trimarco, V.; Visentin, A.; Scomazzon, E.; Accordi, B.; Bresolin, S.; Piazza, F.; et al. HSP70/HSF1 axis, regulated via a PI3K/AKT pathway, is a druggable target in chronic lymphocytic leukemia. Int. J. Cancer 2019, 145, 3089–3100. [Google Scholar] [CrossRef]
- Kennedy, S.G.; Wagner, A.J.; Conzen, S.D.; Jordan, J.; Bellacosa, A.; Tsichlis, P.N.; Hay, N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997, 11, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [Green Version]
- Brunelle, J.K.; Santore, M.T.; Budinger, G.R.; Tang, Y.; Barrett, T.A.; Zong, W.X.; Kandel, E.; Keith, B.; Simon, M.C.; Thompson, C.B.; et al. c-Myc sensitization to oxygen deprivation-induced cell death is dependent on Bax/Bak, but is independent of p53 and hypoxia-inducible factor-1. J. Biol. Chem. 2004, 279, 4305–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zynda, E.R.; Maloy, M.H.; Kandel, E.S. The role of PAK1 in the sensitivity of kidney epithelial cells to ischemia-like conditions. Cell Cycle 2019, 18, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.K.; Feliciano, C.S.; Najmabadi, F.; Haegebarth, A.; Kandel, E.S.; Tyner, A.L.; Gartel, A.L. Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S phase of the cell cycle. Oncogene 2004, 23, 4173–4176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, H.; Minamino, T.; Tateno, K.; Kunieda, T.; Toko, H.; Komuro, I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. Embo J. 2004, 23, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Driessens, N.; Costa, M.; De Deken, X.; Detours, V.; Corvilain, B.; Maenhaut, C.; Miot, F.; Van Sande, J.; Many, M.C.; et al. Roles of hydrogen peroxide in thyroid physiology and disease. J. Clin. Endocrinol. Metab. 2007, 92, 3764–3773. [Google Scholar] [CrossRef]
- Balakrishnan, P.B.; Sweeney, E.E.; Ramanujam, A.S.; Fernandes, R. Photothermal therapies to improve immune checkpoint blockade for cancer. Int. J. Hyperthermia 2020, 37, 34–49. [Google Scholar] [CrossRef]
- Park, H.J.; Carr, J.R.; Wang, Z.; Nogueira, V.; Hay, N.; Tyner, A.L.; Lau, L.F.; Costa, R.H.; Raychaudhuri, P. FoxM1, a critical regulator of oxidative stress during oncogenesis. Embo J. 2009, 28, 2908–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewska, S.; Kroliczewski, J.; Crossman, D.K.; Pogorzelska, A.; Baginski, M.; Collawn, J.F.; Bartoszewski, R. Triazoloacridone C-1305 impairs XBP1 splicing by acting as a potential IRE1alpha endoribonuclease inhibitor. Cell Mol. Biol. Lett. 2021, 26, 11. [Google Scholar] [CrossRef]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J.; et al. Acridine Derivatives as Inhibitors of the IRE1alpha-XBP1 Pathway Are Cytotoxic to Human Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef] [Green Version]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schurer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Jana, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernandez, E.; Sassano, M.L.; et al. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Yi, P.; Dong, W.; Nalin, A.P.; Zhang, J.; Zhu, Z.; Chen, L.; Benson, D.M.; Mundy-Bosse, B.L.; et al. The IL-15-AKT-XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nature Immunol. 2019, 20, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, J.M.D.; Wiseman, R.L. Small molecule strategies to harness the unfolded protein response: Where do we go from here? J. Biol. Chem. 2020, 295, 15692–15711. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Andrews, K.L.; Beckmann, H.; Bellon, S.F.; Beltran, P.J.; Booker, S.; Chen, H.; Chung, Y.A.; D’Angelo, N.D.; Dao, J.; et al. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2015, 58, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [Green Version]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife 2016, 5. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; You, Q.D.; Xu, X.L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef]
- Doi, T.; Kurokawa, Y.; Sawaki, A.; Komatsu, Y.; Ozaka, M.; Takahashi, T.; Naito, Y.; Ohkubo, S.; Nishida, T. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: A phase II, single-arm trial. Eur. J. Cancer 2019, 121, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, A.; Yamamoto, N.; Kondo, S.; Fujiwara, Y.; Suzuki, S.; Yanagitani, N.; Horiike, A.; Kitazono, S.; Ohyanagi, F.; Doi, T.; et al. First-in-Human Phase I Study of an Oral HSP90 Inhibitor, TAS-116, in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2019, 18, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef]
- Durkin, M.E.; Qian, X.; Popescu, N.C.; Lowy, D.R. Isolation of Mouse Embryo Fibroblasts. Bio Protoc. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossovskaya, V.S.; Mazo, I.A.; Chernov, M.V.; Chernova, O.B.; Strezoska, Z.; Kondratov, R.; Stark, G.R.; Chumakov, P.M.; Gudkov, A.V. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc. Natl. Acad. Sci. USA 1996, 93, 10309–10314. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [Green Version]
- Kandel, E.S.; Lu, T.; Wan, Y.; Agarwal, M.K.; Jackson, M.W.; Stark, G.R. Mutagenesis by reversible promoter insertion to study the activation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2005, 102, 6425–6430. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.; Deng, X.; Chenchik, A.A.; Kandel, E.S. Long-distance effects of insertional mutagenesis. PLoS ONE 2011, 6, e15832. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Costanzi, E.; Haas, M.; Verma, I.M. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 1996, 70, 5701–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamovsky, I.; Ivannikov, M.; Kandel, E.S.; Gershon, D.; Nudler, E. RNA-mediated response to heat shock in mammalian cells. Nature 2006, 440, 556–560. [Google Scholar] [CrossRef]
- Zynda, E.R.; Schott, B.; Babagana, M.; Gruener, S.; Wernher, E.; Nguyen, G.D.; Ebeling, M.; Kandel, E.S. An RNA interference screen identifies new avenues for nephroprotection. Cell Death Differ 2016, 23, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babagana, M.; Brown, L.R.; Slabodkin, H.Z.; Kichina, J.V.; Kandel, E.S. Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT. Int. J. Mol. Sci. 2021, 22, 11376. https://doi.org/10.3390/ijms222111376
Babagana M, Brown LR, Slabodkin HZ, Kichina JV, Kandel ES. Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT. International Journal of Molecular Sciences. 2021; 22(21):11376. https://doi.org/10.3390/ijms222111376
Chicago/Turabian StyleBabagana, Mahamat, Lorin R. Brown, Hannah Z. Slabodkin, Julia V. Kichina, and Eugene S. Kandel. 2021. "Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT" International Journal of Molecular Sciences 22, no. 21: 11376. https://doi.org/10.3390/ijms222111376
APA StyleBabagana, M., Brown, L. R., Slabodkin, H. Z., Kichina, J. V., & Kandel, E. S. (2021). Proteotoxic Stress as an Exploitable Vulnerability in Cells with Hyperactive AKT. International Journal of Molecular Sciences, 22(21), 11376. https://doi.org/10.3390/ijms222111376