
The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases

Abstract
:1. The Interferon Pathway and Its Role in Autoimmunity: Systemic Lupus Erythematosus and Beyond
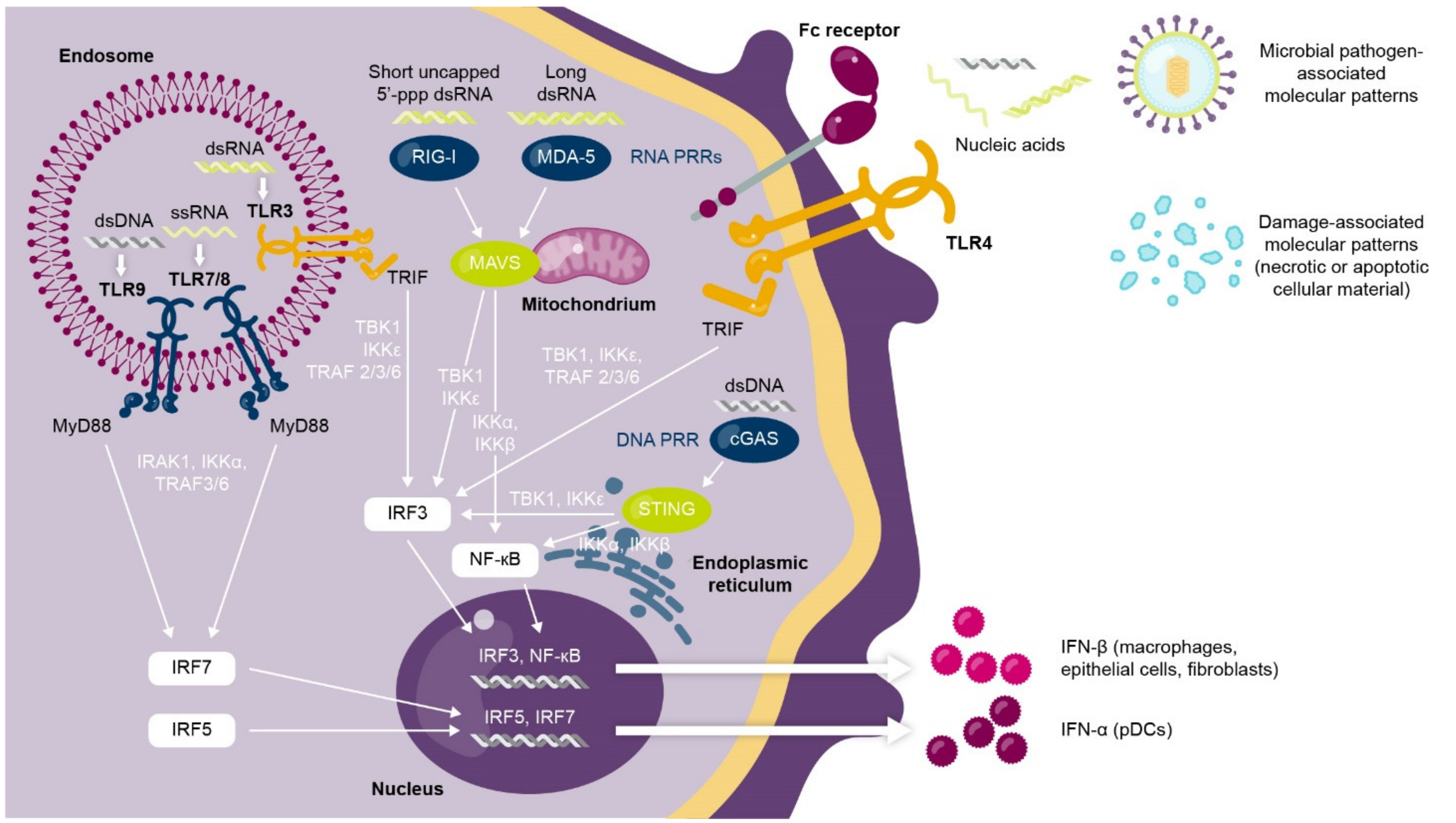
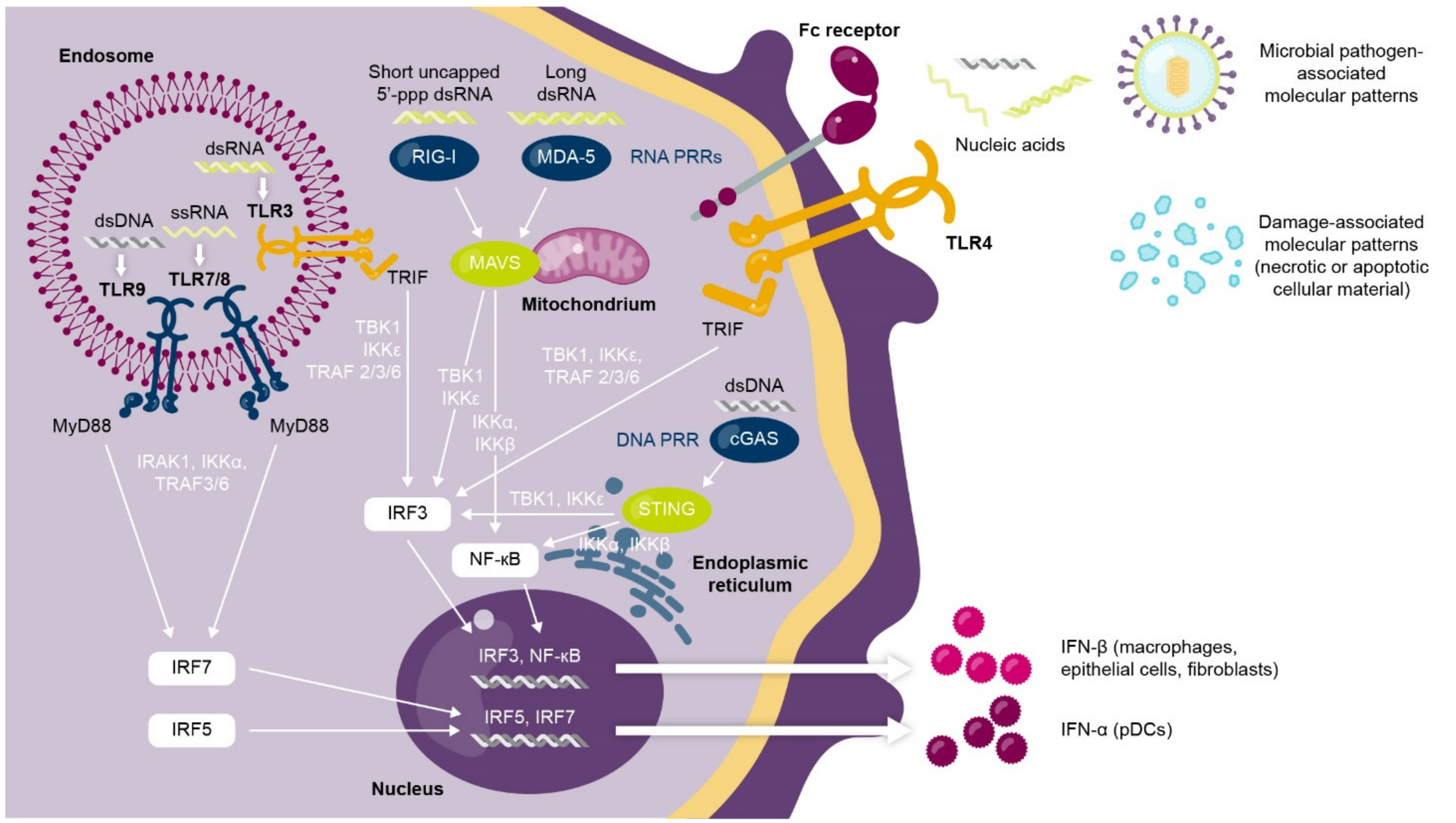
1.1. Interferons Are a Complex Family of Cytokines with Antiviral and Immunomodulatory Functions
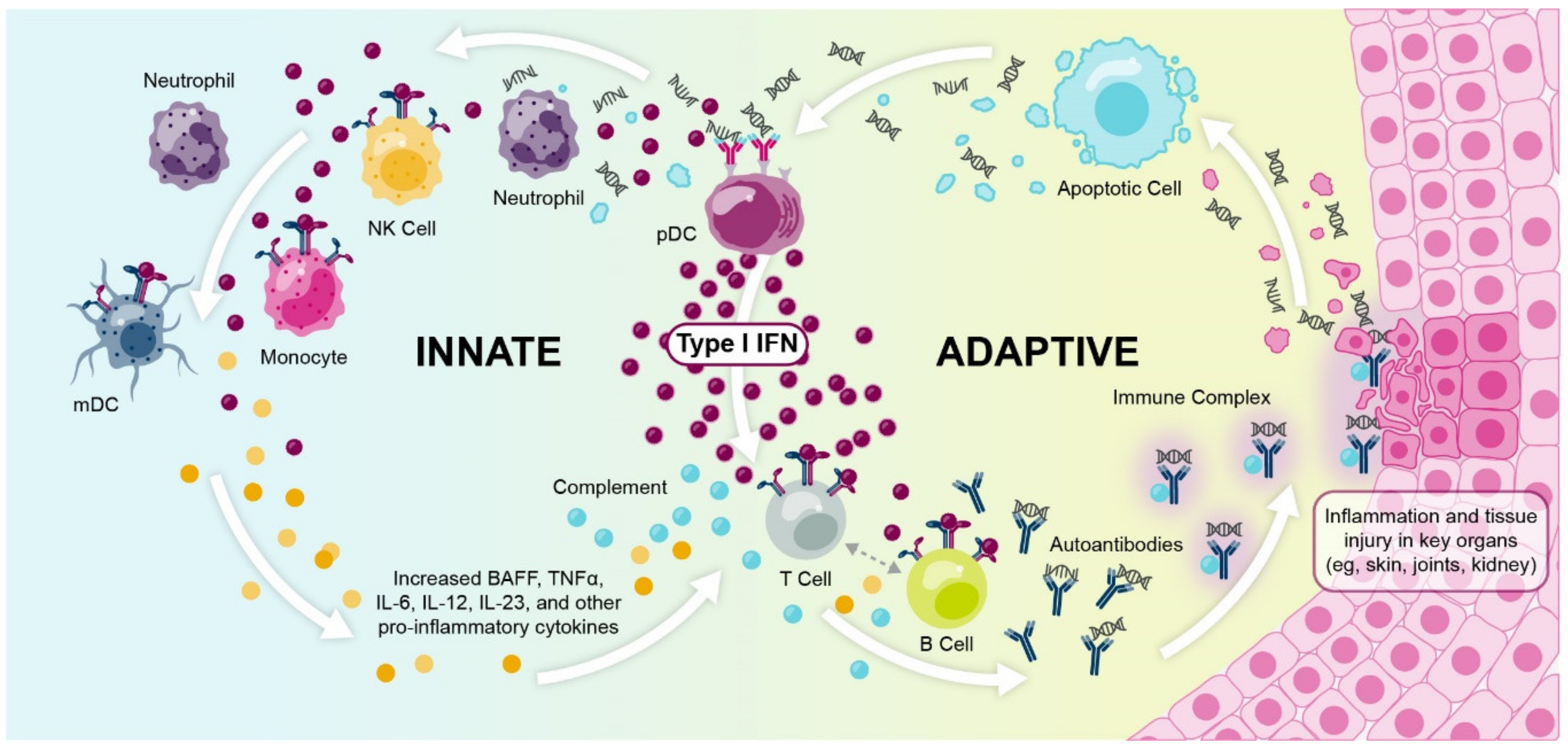
1.2. Dysregulated Type I Interferon Signaling Is Associated with Loss of Immune Tolerance and Autoimmunity
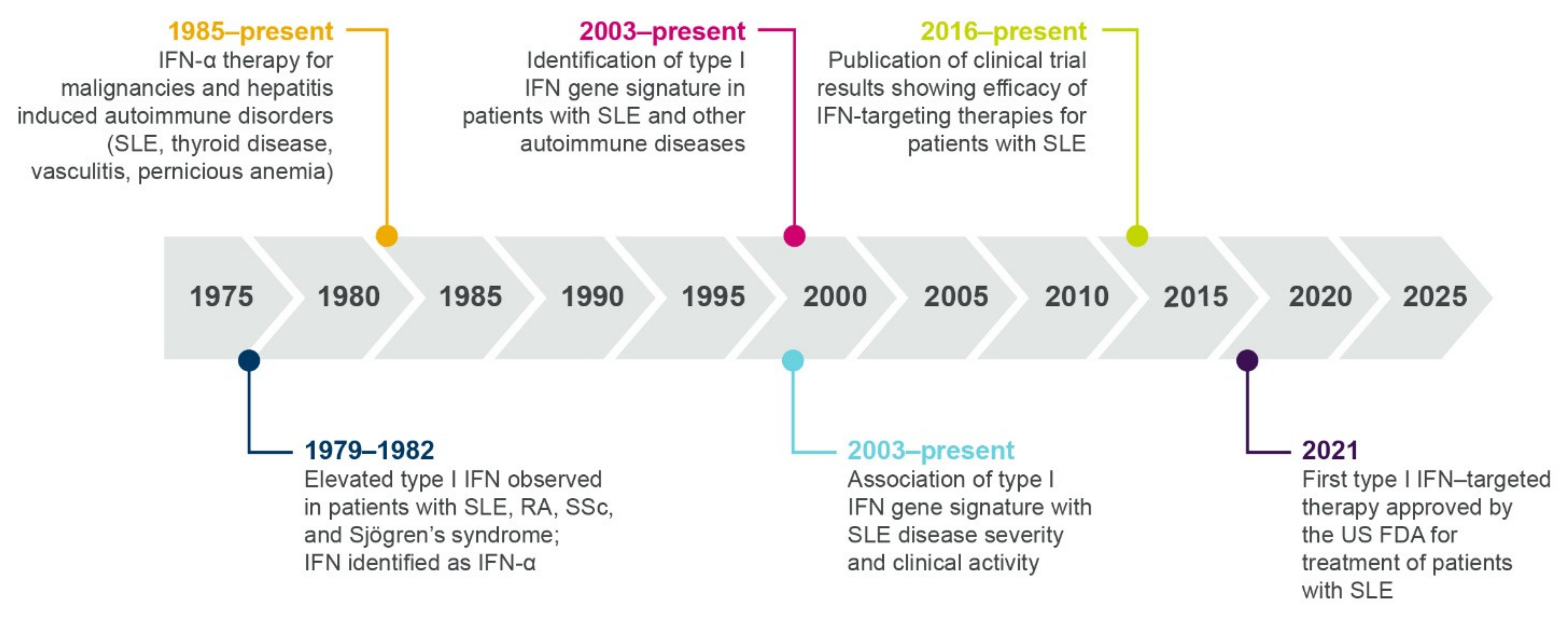
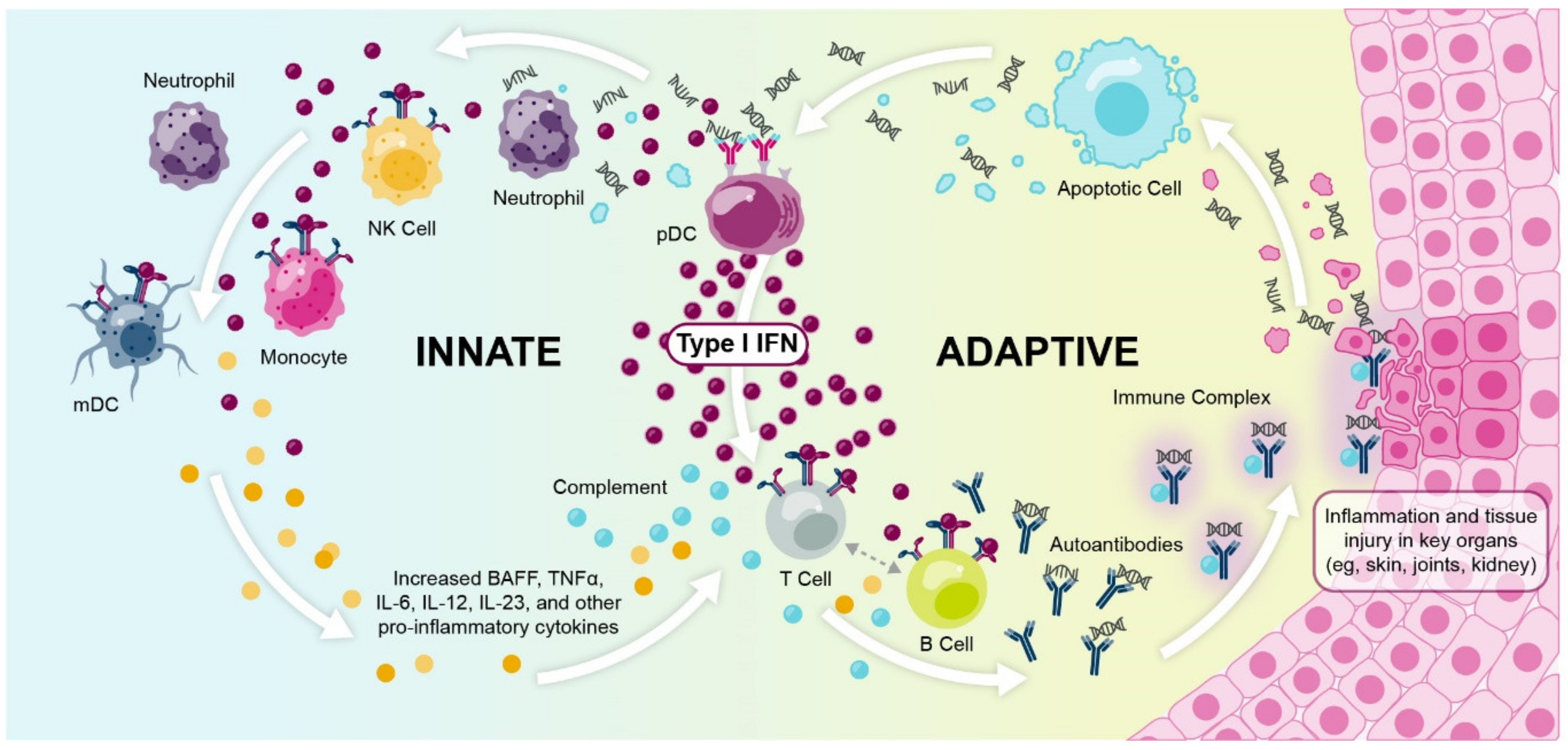
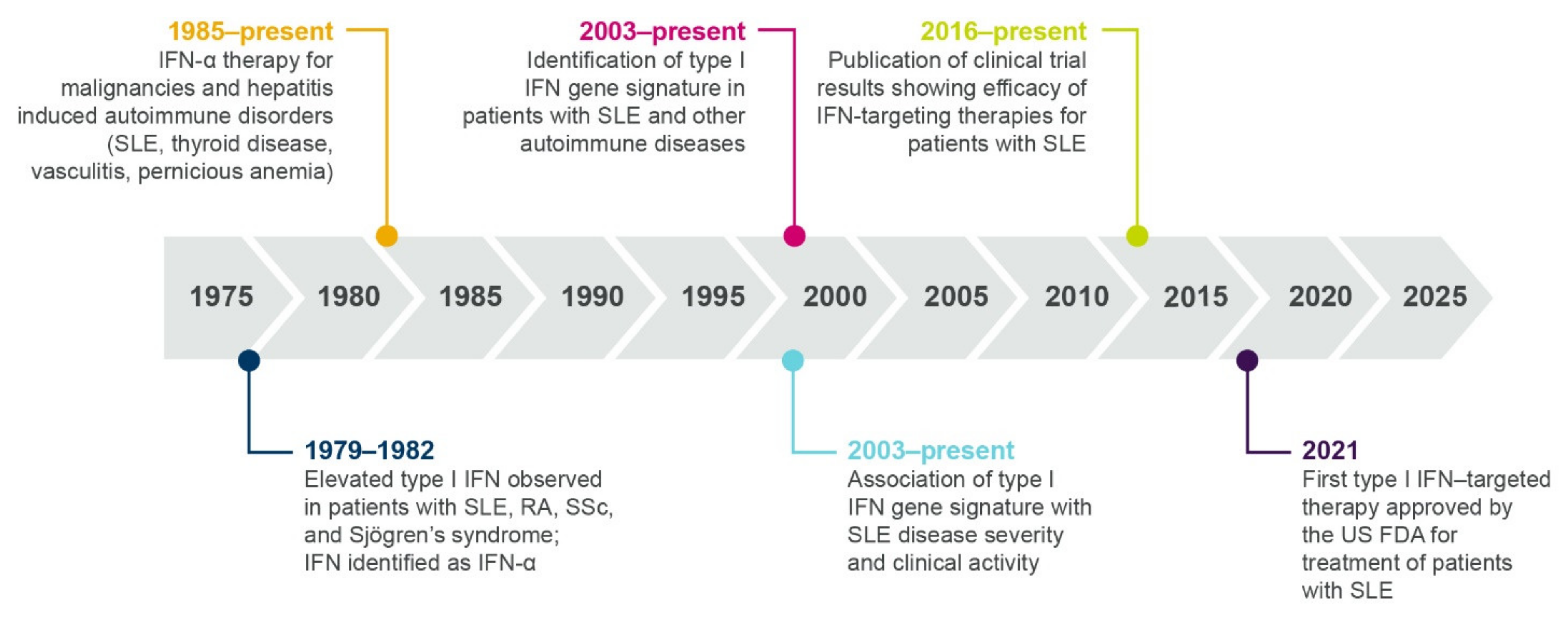
1.3. Multiple Lines of Evidence Support the Role of Interferons in SLE and Autoimmunity
1.3.1. Elevated Interferon Protein Is Detected in Patients with Autoimmune Diseases
1.3.2. Therapeutic Interferon Can Induce/Exacerbate Autoimmunity
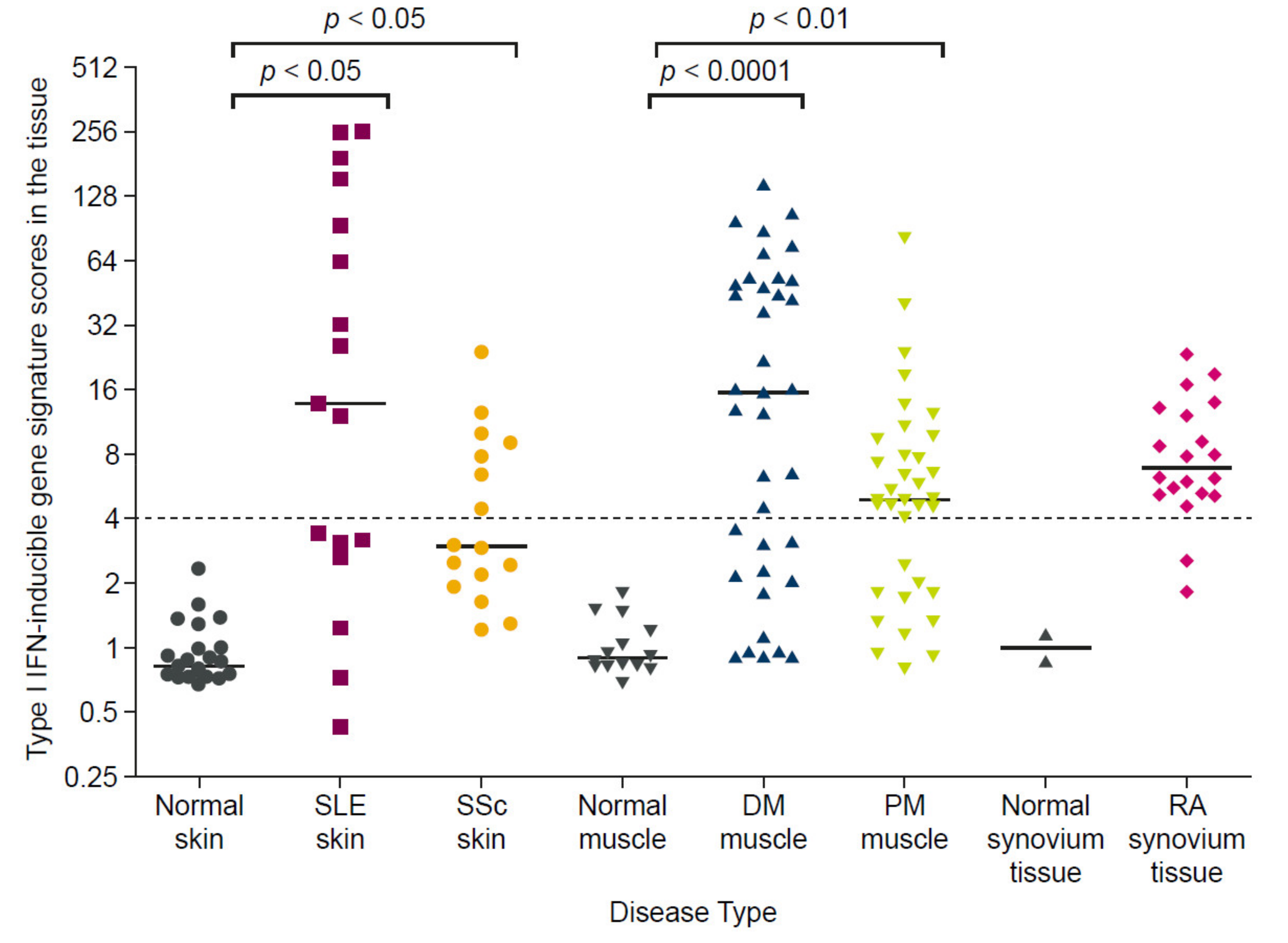
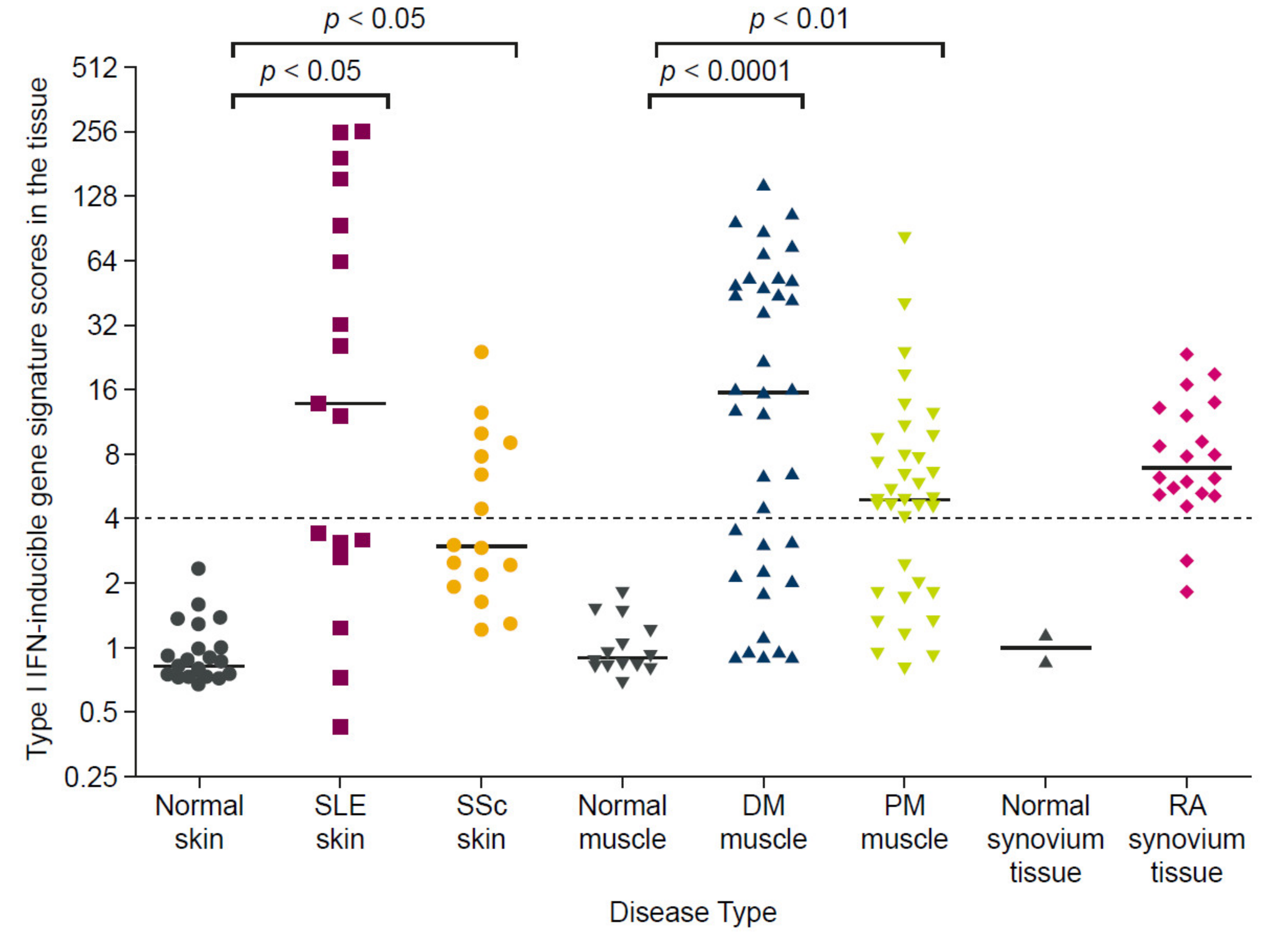
1.3.3. Interferon Gene Signatures Are Upregulated in Patients with Autoimmune Diseases
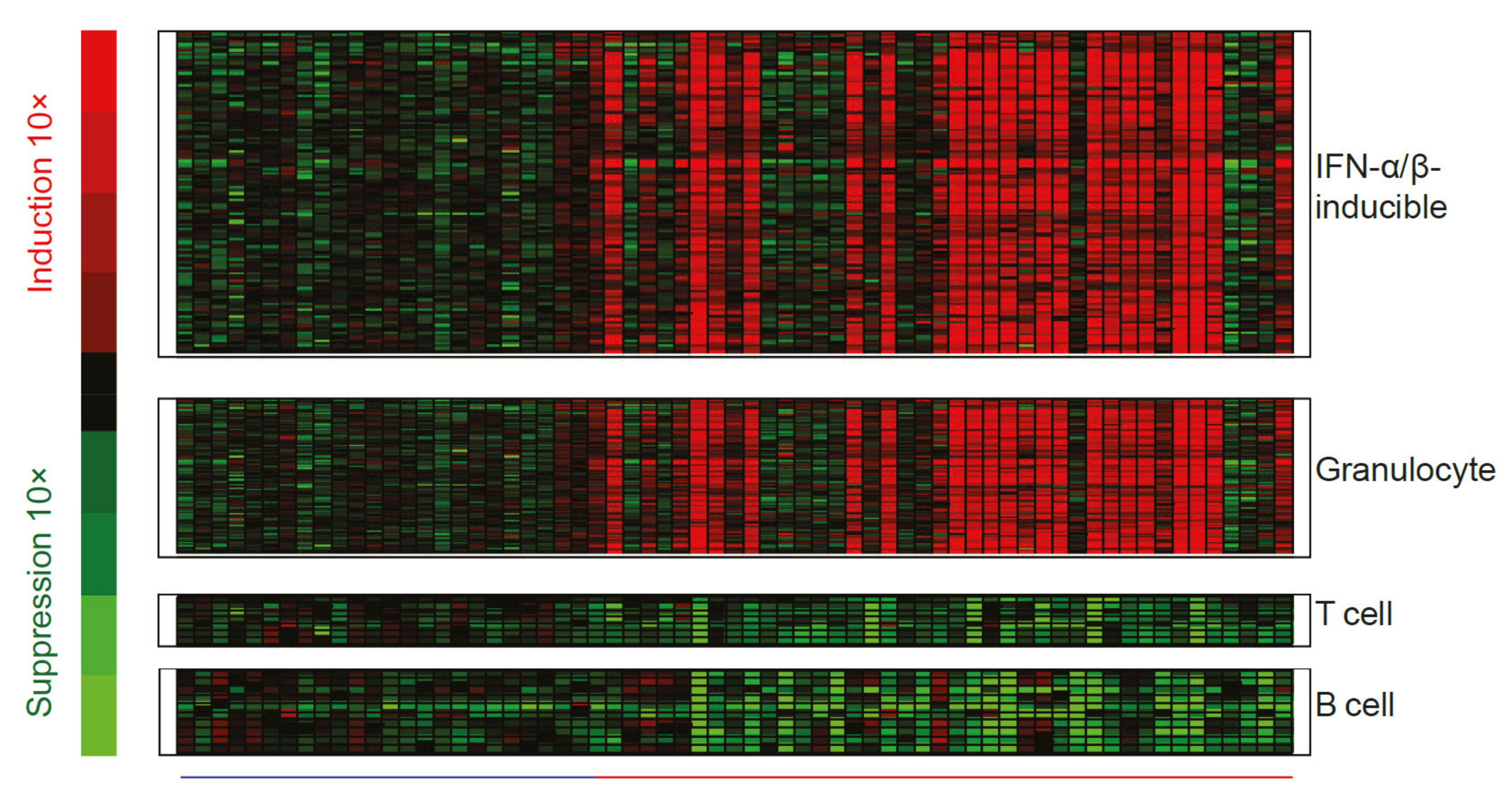
1.3.4. Many Immune Cell Types Express Interferon Gene Signatures in Autoimmune Disease
1.3.5. Polymorphisms in Interferon-Related Genes Associate with Increased Risk of Autoimmune Disease
1.3.6. Type I Interferon Gene Signature Expression Occurs within a Broader Context of Immune Dysregulation
1.4. Interferon Dysregulation Associates with Clinical Disease Activity and Response to Standard Therapy in Patients with SLE
2. Therapeutic Highlights: Past, Present, and Future
2.1. Current Standard Therapies, Approved Biologics, and Impact on Interferon Signaling
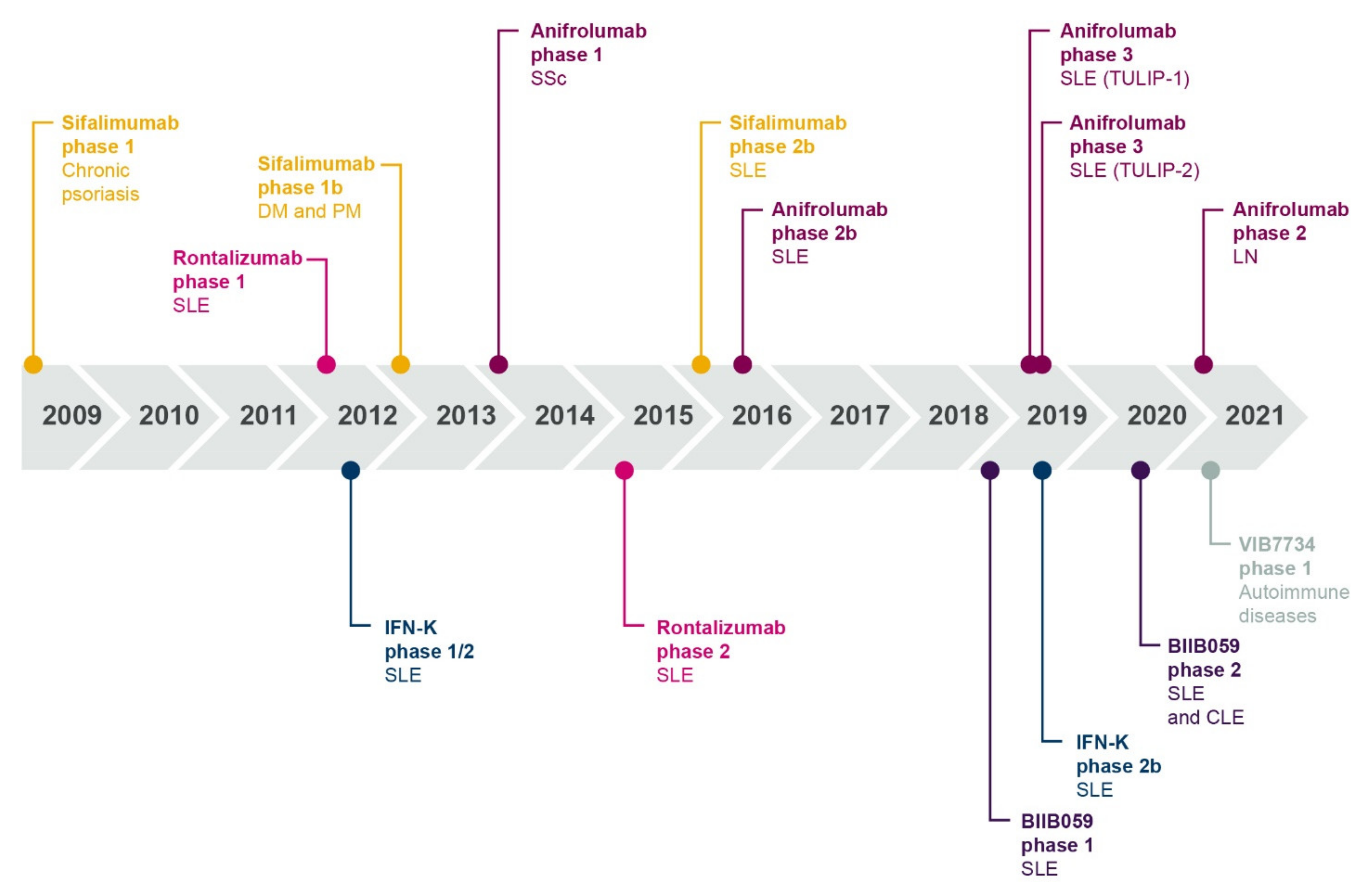
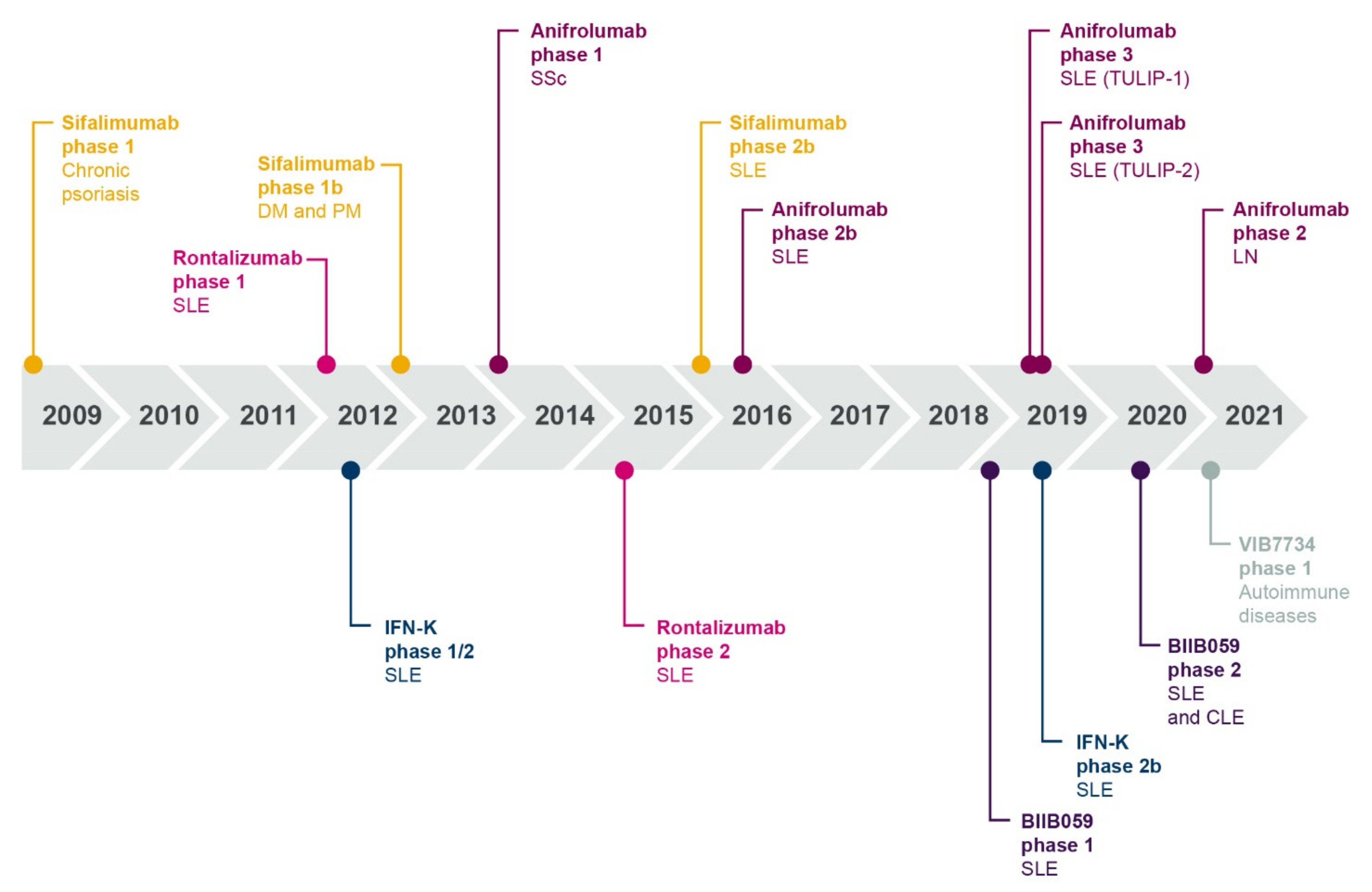
2.2. An Interferon-Targeting Therapeutic Is Now Approved for the Treatment of SLE and Others Are in Clinical Development
2.3. The Future of Interferon Blockade and Lupus Therapies
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.; Leonard, D. Interferon Pathway in SLE: One Key to Unlocking the Mystery of the Disease. Lupus Sci. Med. 2019, 6, e000270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglia, M.; Merlotti, G.; De Andrea, M.; Borgogna, C.; Cantaluppi, V. Viral Infections and Systemic Lupus Erythematosus: New Players in an Old Story. Viruses 2021, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.E.; Roman, R.M.; Rudenga, B.J.; Holers, V.M.; Craft, J.E. Epstein-Barr Virus Promotes Interferon-Alpha Production by Plasmacytoid Dendritic Cells. Arthritis Rheum. 2010, 62, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Liao, A.P.; Salajegheh, M.; Morehouse, C.; Nazareno, R.; Jubin, R.G.; Jallal, B.; Yao, Y.; Greenberg, S.A. Human Plasmacytoid Dendritic Cell Accumulation Amplifies Their Type 1 Interferon Production. Clin. Immunol. 2010, 136, 130–138. [Google Scholar] [CrossRef] [Green Version]
- De Weerd, N.A.; Nguyen, T. The Interferons and Their Receptors—Distribution and Regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef]
- Greth, W.; Robbie, G.J.; Brohawn, P.; Hultquist, M.; Yao, B. Targeting the Interferon Pathway with Sifalimumab for the Treatment of Systemic Lupus Erythematosus. Immunotherapy 2017, 9, 57–70. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I Interferons in Host Defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Goër de Herve, M.G.; Durali, D.; Dembele, B.; Giuliani, M.; Tran, T.A.; Azzarone, B.; Eid, P.; Tardieu, M.; Delfraissy, J.F.; Taoufik, Y. Interferon-Alpha Triggers B Cell Effector 1 (Be1) Commitment. PLoS ONE 2011, 6, e19366. [Google Scholar] [CrossRef]
- Crow, M.K. Type I Interferon in the Pathogenesis of Lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef]
- Niewold, T.B. Interferon Alpha as a Primary Pathogenic Factor in Human Lupus. J. Interferon Cytokine Res. 2011, 31, 887–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B. Type I Interferon Modulation of Cellular Responses to Cytokines and Infectious Pathogens: Potential Role in SLE Pathogenesis. Autoimmunity 2003, 36, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting Neutrophils are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of Plasmacytoid Dendritic Cells by Apoptotic Particles—Mechanism for the Loss of Immunological Tolerance in Sjögren’s Syndrome. Clin. Exp. Immunol. 2018, 191, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Schiller, M.; Parcina, M.; Heyder, P.; Foermer, S.; Ostrop, J.; Leo, A.; Heeg, K.; Herrmann, M.; Lorenz, H.M.; Bekeredjian-Ding, I. Induction of Type I IFN is a Physiological Immune Reaction to Apoptotic Cell-Derived Membrane Microparticles. J. Immunol. 2012, 189, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eloranta, M.L.; Rönnblom, L. Cause and Consequences of the Activated Type I Interferon System in SLE. J. Mol. Med. 2016, 94, 1103–1110. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of Autoantibodies Before the Clinical Onset of Systemic Lupus Erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Rönnblom, L.; Alm, G.V. A Pivotal Role for the Natural Interferon Alpha-Producing Cells (Plasmacytoid Dendritic Cells) in the Pathogenesis of Lupus. J. Exp. Med. 2001, 194, F59–F63. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Tsokos, G.C.; Tsuboi, N. Mechanisms of Immune Complex-Mediated Neutrophil Recruitment and Tissue Injury. Circulation 2009, 120, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Uva, L.; Miguel, D.; Pinheiro, C.; Freitas, J.P.; Marques Gomes, M.; Filipe, P. Cutaneous Manifestations of Systemic Lupus Erythematosus. Autoimmune Dis. 2012, 2012, 834291. [Google Scholar] [CrossRef] [PubMed]
- McCauliffe, D.P. Cutaneous Lupus Erythematosus. Semin. Cutan. Med. Surg. 2001, 20, 14–26. [Google Scholar] [CrossRef]
- Zayat, A.S.; Md Yusof, M.Y.; Wakefield, R.J.; Conaghan, P.G.; Emery, P.; Vital, E.M. The Role of Ultrasound in Assessing Musculoskeletal Symptoms of Systemic Lupus Erythematosus: A Systematic Literature Review. Rheumatology 2016, 55, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Jawad, A.S. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology 2017, 56, i67–i77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacre, K.; Criswell, L.A.; McCune, J.M. Hydroxychloroquine is Associated With Impaired Interferon-Alpha and Tumor Necrosis Factor-Alpha Production by Plasmacytoid Dendritic Cells in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2012, 14, R155. [Google Scholar] [CrossRef] [Green Version]
- Psarras, A.; Alase, A.; Antanaviciute, A.; Carr, I.M.; Md Yusof, M.Y.; Wittmann, M.; Emery, P.; Tsokos, G.C.; Vital, E.M. Functionally Impaired Plasmacytoid Dendritic Cells and Non-Haematopoietic Sources of Type I Interferon Characterize Human Autoimmunity. Nat. Commun. 2020, 11, 6149. [Google Scholar] [CrossRef]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune Interferon in the Circulation of Patients With Autoimmune Disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Schnitzer, T.J. Serum Interferon Levels in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 1982, 25, 401–406. [Google Scholar] [CrossRef]
- Smith, M.A.; Henault, J.; Karnell, J.L.; Parker, M.L.; Riggs, J.M.; Sinibaldi, D.; Taylor, D.K.; Ettinger, R.; Grant, E.P.; Sanjuan, M.A.; et al. SLE Plasma Profiling Identifies Unique Signatures of Lupus Nephritis and Discoid Lupus. Sci. Rep. 2019, 9, 14433. [Google Scholar] [CrossRef] [Green Version]
- Higgs, B.W.; Liu, Z.; White, B.; Zhu, W.; White, W.I.; Morehouse, C.; Brohawn, P.; Kiener, P.A.; Richman, L.; Fiorentino, D.; et al. Patients With Systemic Lupus Erythematosus, Myositis, Rheumatoid Arthritis and Scleroderma Share Activation of a Common Type I Interferon Pathway. Ann. Rheum. Dis. 2011, 70, 2029–2036. [Google Scholar] [CrossRef]
- Smith, M.A.; Chiang, C.-C.; Zerrouki, K.; Rahman, S.; White, W.I.; Streicher, K.; Rees, W.A.; Schiffenbauer, A.; Rider, L.G.; Miller, F.W.; et al. Using the Circulating Proteome to Assess Type I Interferon Activity in Systemic Lupus Erythematosus. Sci. Rep. 2020, 10, 4462. [Google Scholar] [CrossRef]
- Skaug, B.; Assassi, S. Type I Interferon Dysregulation in Systemic Sclerosis. Cytokine 2020, 132, 154635. [Google Scholar] [CrossRef]
- Brkic, Z.; van Bon, L.; Cossu, M.; van Helden-Meeuwsen, C.G.; Vonk, M.C.; Knaapen, H.; van den Berg, W.; Dalm, V.A.; Van Daele, P.L.; Severino, A.; et al. The Interferon Type I Signature is Present in Systemic Sclerosis Before Overt Fibrosis and Might Contribute to its Pathogenesis Through High BAFF Gene Expression and High Collagen Synthesis. Ann. Rheum. Dis. 2016, 75, 1567–1573. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, Z.; Jallal, B.; Shen, N.; Rönnblom, L. Type I Interferons In Sjögren’s Syndrome. Autoimmun. Rev. 2013, 12, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Hjorton, K.; Hagberg, N.; Pucholt, P.; Eloranta, M.L.; Rönnblom, L. The Regulation and Pharmacological Modulation of Immune Complex Induced Type III IFN Production by Plasmacytoid Dendritic Cells. Arthritis Res. Ther. 2020, 22, 130. [Google Scholar] [CrossRef]
- Weckerle, C.E.; Franek, B.S.; Kelly, J.A.; Kumabe, M.; Mikolaitis, R.A.; Green, S.L.; Utset, T.O.; Jolly, M.; James, J.A.; Harley, J.B.; et al. Network Analysis of Associations Between Serum Interferon-A Activity, Autoantibodies, and Clinical Features in Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 1044–1053. [Google Scholar] [CrossRef]
- Ko, K.; Koldobskaya, Y.; Rosenzweig, E.; Niewold, T.B. Activation of the Interferon Pathway is Dependent Upon Autoantibodies in African-American SLE Patients, but not in European-American SLE Patients. Front. Immunol. 2013, 4, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimley, P.M.; Davis, G.L.; Kang, Y.H.; Dooley, J.S.; Strohmaier, J.; Hoofnagle, J.H. Tubuloreticular Inclusions in Peripheral Blood Mononuclear Cells Related to Systemic Therapy With Alpha-Interferon. Lab Invest 1985, 52, 638–649. [Google Scholar] [PubMed]
- Rönnblom, L.E.; Alm, G.V.; Oberg, K.E. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann. Intern. Med. 1991, 115, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Kojima, A.; Hirokawa, T.; Oyama, T.; Naganuma, A.; Maruta, S.; Okada, K.; Ban, S.; Yoshida, K.; Takagi, H.; et al. Systemic Sclerosis After Interferon Alphacon-1 Therapy for Hepatitis C. Intern. Med. 2007, 46, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnblom, L.E.; Alm, G.V.; Oberg, K.E. Possible Induction of Systemic Lupus Erythematosus by Interferon-Alpha Treatment in a Patient With a Malignant Carcinoid Tumour. J. Intern. Med. 1990, 227, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Black, C.M.; Silman, A.J.; Herrick, A.I.; Denton, C.P.; Wilson, H.; Newman, J.; Pompon, L.; Shi-Wen, X. Interferon-Alpha Does not Improve Outcome at One Year in Patients With Diffuse Cutaneous Scleroderma: Results of a Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheum. 1999, 42, 299–305. [Google Scholar] [CrossRef]
- Warabi, Y.; Matsubara, S.; Mizutani, T.; Hayashi, H. Inclusion Body Myositis After Interferon-Alpha Treatment in a Patient with HCV and HTLV-1 Infection. Rinsho Shinkeigaku 2004, 44, 609–614. [Google Scholar] [PubMed]
- Greenberg, S.A. Dermatomyositis and Type 1 Interferons. Curr. Rheumatol. Rep. 2010, 12, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-Inducible Gene Expression Signature in Peripheral Blood Cells of Patients With Severe Lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, M.; Singh, S.; Tesfasyone, H.; Dedrick, R.; Fry, K.; Lal, P.; Williams, G.; Bauer, J.; Gregersen, P.; Behrens, T.; et al. Longitudinal Expression of Type I Interferon Responsive Genes in Systemic Lupus Erythematosus. Lupus 2009, 18, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, L.; Asaduzzaman, A.; Noamani, B.; Fortin, P.R.; Gladman, D.D.; Touma, Z.; Urowitz, M.B.; Wither, J. The Baseline Interferon Signature Predicts Disease Severity Over the Subsequent 5 Years in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2021, 23, 29. [Google Scholar] [CrossRef]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular Transcriptional Repertoire Analyses of Adults With Systemic Lupus Erythematosus Reveal Distinct Type I and Type II Interferon Signatures. Arthritis Rheum. 2014, 66, 1583–1595. [Google Scholar] [CrossRef]
- Merrill, J.T.; Immermann, F.; Whitley, M.; Zhou, T.; Hill, A.; O’Toole, M.; Reddy, P.; Honczarenko, M.; Thanou, A.; Rawdon, J.; et al. The Biomarkers of Lupus Disease Study: A Bold Approach May Mitigate Interference of Background Immunosuppressants in Clinical Trials. Arthritis Rheum. 2017, 69, 1257–1266. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef]
- Furie, R.; Morand, E.; Bruce, I.N.; Manzi, S.; Kalunian, K.; Vital, E.M.; Lawrence Ford, T.; Gupta, R.; Hiepe, F.; Santiago, M.; et al. Type I Interferon Inhibitor Anifrolumab in Active Systemic Lupus Erythematosus (TULIP-1): A Randomised, Controlled, Phase 3 Trial. Lancet Rheumatol. 2019, 1, e208–e219. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S. Anifrolumab, an Anti-Interferon-A Receptor Monoclonal Antibody, in Moderate-To-Severe Systemic Lupus Erythematosus. Arthritis Rheum. 2017, 69, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W. Sifalimumab, an Anti-Interferon-A Monoclonal Antibody, in Moderate to Severe Systemic Lupus Erythematosus: A Randomised, Double-Blind, Placebo-Controlled Study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Higgs, B.W.; Morehouse, C.; de Los Reyes, M.; Trigona, W.; Brohawn, P.; White, W.; Zhang, J.; White, B.; Coyle, A.J.; et al. Development of Potential Pharmacodynamic and Diagnostic Markers for Anti-IFN-α Monoclonal Antibody Trials in Systemic Lupus Erythematosus. Hum. Genomics Proteom. 2009, 2009, 374312. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Gao, C.; Zhang, C.; Di, X.; Liang, W.; Sun, W.; Wang, Q.; Zheng, Z. Identification of Molecular Markers Associated with the Pathophysiology and Treatment of Lupus Nephritis Based on Integrated Transcriptome Analysis. Front. Genet. 2020, 11, 583629. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriens, C.; Raja, Q.; Husain, S.; George, B.; Abedi, M.; Jacobs, A.; Guyon, T.; Wijesuriya, H.; Aberle, T.; Thanou, A.; et al. Increased Risk of Progression to Lupus Nephritis for Lupus Patients with Elevated Interferon Signature. Arthritis Rheum. 2019, 71 (Suppl. 10). [Google Scholar]
- Abedi, M.; Borisov, L.; Doyle, A.; Flores, F.; Fujimoto, J.; Jacobs, A.; Naranatt, P.; Pan, L.; Ricketts, W.; Spangler, J.; et al. Type 1 Interferon Levels Correlates With Age of Diagnosis and Ethnicity in Systemic Lupus Erythematous. Arthritis Rheum. 2018, 70 (Suppl. 10). [Google Scholar] [CrossRef] [Green Version]
- Szymczak, F.; Colli, M.L.; Mamula, M.J.; Evans-Molina, C.; Eizirik, D.L. Gene Expression Signatures of Target Tissues in Type 1 Diabetes, Lupus Erythematosus, Multiple Sclerosis, and Rheumatoid Arthritis. Sci. Adv. 2021, 7, eabd7600. [Google Scholar] [CrossRef] [PubMed]
- Lambers, W.M.; de Leeuw, K.; Doornbos-van der Meer, B.; Diercks, G.F.H.; Bootsma, H.; Westra, J. Interferon score is increased in incomplete systemic lupus erythematosus and correlates with myxovirus-resistance protein A in blood and skin. Arthritis Res. Ther. 2019, 21, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Md Yusof, M.Y.; Psarras, A.; El-Sherbiny, Y.M.; Hensor, E.M.A.; Dutton, K.; Ul-Hassan, S.; Zayat, A.S.; Shalbaf, M.; Alase, A.; Wittmann, M.; et al. Prediction of Autoimmune Connective Tissue Disease in an At-Risk Cohort: Prognostic Value of a Novel Two-Score System for Interferon Status. Ann. Rheum. Dis. 2018, 77, 1432–1439. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Tzeng, H.T.; Chen, J.Y. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells 2019, 8, 963. [Google Scholar] [CrossRef] [Green Version]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High Levels of Circulating Interferons Type I, Type II And Type III Associate with Distinct Clinical Features of Active Systemic Lupus Erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Nehar-Belaid, D.; Hong, S.; Marches, R.; Chen, G.; Bolisetty, M.; Baisch, J.; Walters, L.; Punaro, M.; Rossi, R.J.; Chung, C.-H.; et al. Mapping Systemic Lupus Erythematosus Heterogeneity at The Single-Cell Level. Nat. Immunol. 2020, 21, 1094–1106. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, T.; Mao, K.; Shao, X.; Xu, Y.; Zhu, M.; Zhou, H.; Wang, Q.; Li, Z.; Xie, Y.; et al. A Single-Cell Survey of The Human Glomerulonephritis. J. Cell. Mol. Med. 2021, 25, 4684–4695. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Fan, W.; Jensen, M.A.; Dorschner, J.M.; Bonadurer, G.F., 3rd; Vsetecka, D.M.; Amin, S.; Makol, A.; Ernste, F.; Osborn, T.; et al. Single-cell gene expression patterns in lupus monocytes independently indicate disease activity, interferon and therapy. Lupus Sci. Med. 2017, 4, e000202. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zheng, Y.; Li, D.; Hong, Q.; Zhang, M.; Li, Q.; Fu, B.; Wu, L.; Wang, X.; Shen, W.; et al. Expression characteristics of interferon-stimulated genes and possible regulatory mechanisms in lupus patients using transcriptomics analyses. EBioMedicine 2021, 70, 103477. [Google Scholar] [CrossRef] [PubMed]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabothu, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular Cell and Keratinocyte Single-Cell Transcriptomics Applied to Lupus Nephritis Reveal Type I IFN and Fibrosis Relevant Pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef]
- Harley, J.B.; Alarcón-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; Nath, S.K.; et al. Genome-Wide Association Scan in Women with Systemic Lupus Erythematosus Identifies Susceptibility Variants in ITGAM, PXK, KIAA1542 and Other Loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Sandling, J.K.; Pucholt, P.; Hultin Rosenberg, L.; Farias, F.H.G.; Kozyrev, S.V.; Eloranta, M.-L.; Alexsson, A.; Bianchi, M.; Padyukov, L.; Bengtsson, C.; et al. Molecular Pathways in Patients with Systemic Lupus Erythematosus Revealed by Gene-Centred DNA sequencing. Ann. Rheum. Dis. 2021, 80, 109–117. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.-F.; Zhang, Y.; Lin, Z.; Cao, Y.; Zhang, H.; Liu, Z.-Y.; Morris, D.L.; Sheng, Y.; Cui, Y.; et al. Independent Replication on Genome-Wide Association Study Signals Identifies IRF3 as a Novel Locus for Systemic Lupus Erythematosus. Front. Genet. 2020, 11, 600. [Google Scholar] [CrossRef] [PubMed]
- He, Y.L.; Yang, J.; Zeng, Z.N.; Shi, X. Interaction of miR-181b and IFNA1 Polymorphisms on the Risk of Systemic Lupus Erythematosus. Biomed. Res. Int. 2020, 2020, 4757065. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Guo, Y.; Qian, Q.; Fu, M.; Lei, S.; Zhang, Y.; Zhang, H. Mendelian Randomization Analysis Revealed Potential Causal Factors for Systemic Lupus Erythematosus. Immunology 2020, 159, 279–288. [Google Scholar] [CrossRef]
- Langefeld, C.D.; Ainsworth, H.C.; Graham, D.S.C.; Kelly, J.A.; Comeau, M.E.; Marion, M.C.; Howard, T.D.; Ramos, P.S.; Croker, J.A.; Morris, D.L.; et al. Transancestral Mapping and Genetic Load in Systemic Lupus Erythematosus. Nat. Commun. 2017, 8, 16021. [Google Scholar] [CrossRef] [PubMed]
- Vajgel, G.; Lima, S.C.; Santana, D.J.S.; Oliveira, C.B.L.; Costa, D.M.N.; Hicks, P.J.; Cavalcante, M.; Langefeld, C.D.; Valente, L.M.; Crovella, S.; et al. Effect of a Single Apolipoprotein L1 Gene Nephropathy Variant on the Risk of Advanced Lupus Nephritis in Brazilians. J. Rheumatol. 2020, 47, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 1548–1550. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Martorell-Marugán, J.; Goldman, D.; Petri, M.; Carmona-Sáez, P.; Alarcón-Riquelme, M.E. Stratification of Systemic Lupus Erythematosus Patients Into Three Groups of Disease Activity Progression According to Longitudinal Gene Expression. Arthritis Rheumatol. 2018, 70, 2025–2035. [Google Scholar] [CrossRef] [Green Version]
- Panwar, B.; Schmiedel, B.J.; Liang, S.; White, B.; Rodriguez, E.; Kalunian, K.; McKnight, A.J.; Soloff, R.; Seumois, G.; Vijayanand, P.; et al. Multi-cell type gene coexpression network analysis reveals coordinated interferon response and cross-cell type correlations in systemic lupus erythematosus. Genome Res. 2021, 31, 659–676. [Google Scholar] [CrossRef]
- Adel, Y.; Sadeq, Y. Impact of IL-34, IFN-α and IFN-λ1 on Activity of Systemic Lupus Erythematosus in Egyptian Patients. Reumatologia 2020, 58, 221–230. [Google Scholar] [CrossRef]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular Accumulation of Plasmacytoid Dendritic Cells in Active Lupus Nephritis: Role of Interleukin-18. Arthritis Rheum. 2008, 58, 251–262. [Google Scholar] [CrossRef]
- Feng, X.; Wu, H.; Grossman, J.M.; Hanvivadhanakul, P.; FitzGerald, J.D.; Park, G.S.; Dong, X.; Chen, W.; Kim, M.H.; Weng, H.H.; et al. Association of Increased Interferon-Inducible Gene Expression with Disease Activity and Lupus Nephritis in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 2006, 54, 2951–2962. [Google Scholar] [CrossRef] [PubMed]
- Abdirama, D.; Tesch, S.; Grießbach, A.S.; von Spee-Mayer, C.; Humrich, J.Y.; Stervbo, U.; Babel, N.; Meisel, C.; Alexander, T.; Biesen, R.; et al. Nuclear Antigen-Reactive CD4(+) T Cells Expand in Active Systemic Lupus Erythematosus, Produce Effector Cytokines, and Invade the Kidneys. Kidney Int. 2021, 99, 238–246. [Google Scholar] [CrossRef]
- Braunstein, I.; Klein, R.; Okawa, J.; Werth, V.P. The Interferon-Regulated Gene Signature is Elevated in Subacute Cutaneous Lupus Erythematosus and Discoid Lupus Erythematosus and Correlates with the Cutaneous Lupus Area and Severity Index Score. Br. J. Dermatol. 2012, 166, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Hile, G.A.; Kahlenberg, J.M. Immunopathogenesis of Skin Injury in Systemic Lupus Erythematosus. Curr. Opin. Rheumatol. 2021, 33, 173–180. [Google Scholar] [CrossRef]
- Merola, J.F.; Wang, W.; Wager, C.G.; Hamann, S.; Zhang, X.; Thai, A.; Roberts, C.; Lam, C.; Musselli, C.; Marsh, G.; et al. RNA Tape Sampling in Cutaneous Lupus Erythematosus Discriminates Affected From Unaffected and Healthy Volunteer Skin. Lupus Sci. Med. 2021, 8, e000428. [Google Scholar] [CrossRef]
- Sarkar, M.K.; Hile, G.A.; Tsoi, L.C.; Xing, X.; Liu, J.; Liang, Y.; Berthier, C.C.; Swindell, W.R.; Patrick, M.T.; Shao, S.; et al. Photosensitivity and Type I IFN Responses in Cutaneous Lupus are Driven by Epidermal-Derived Interferon Kappa. Ann. Rheum. Dis. 2018, 77, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Thacker, S.; Mehta, H.; Somers, E.C.; Dodick, T.; Barrat, F.J.; McCune, W.J.; Kaplan, M.J. Interferon-α Promotes Abnormal Vasculogenesis in Lupus: A Potential Pathway for Premature Atherosclerosis. Blood 2007, 110, 2907–2915. [Google Scholar] [CrossRef] [Green Version]
- Somers, E.C.; Zhao, W.; Lewis, E.E.; Wang, L.; Wing, J.J.; Sundaram, B.; Kazerooni, E.A.; McCune, W.J.; Kaplan, M.J. Type I Interferons are Associated with Subclinical Markers of Cardiovascular Disease in a Cohort of Systemic Lupus Erythematosus Patients. PLoS ONE 2012, 7, e37000. [Google Scholar] [CrossRef] [Green Version]
- Casey, K.A.; Smith, M.A.; Sinibaldi, D.; Seto, N.L.; Playford, M.P.; Wang, X.; Carlucci, P.M.; Wang, L.; Illei, G.; Yu, B.; et al. Modulation of Cardiometabolic Disease Markers by Type I Interferon Inhibition in Systemic Lupus Erythematosus. Arthritis Rheum. 2021, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K. Type I Interferon in Organ-Targeted Autoimmune and Inflammatory Diseases. Arthritis Res. Ther. 2010, 12 (Suppl. 1). [Google Scholar] [CrossRef] [Green Version]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 Update of the Eular Recommendations for the Management of Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vollenhoven, R.F.; Mosca, M.; Bertsias, G.; Isenberg, D.; Kuhn, A.; Lerstrøm, K.; Aringer, M.; Bootsma, H.; Boumpas, D.; Bruce, I.N.; et al. Treat-to-Target in Systemic Lupus Erythematosus: Recommendations From an International Task Force. Ann. Rheum. Dis. 2014, 73, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Mathian, A.; Mouries-Martin, S.; Dorgham, K.; Devilliers, H.; Yssel, H.; Garrido Castillo, L.; Cohen-Aubart, F.; Haroche, J.; Hié, M.; Pineton de Chambrun, M.; et al. Ultrasensitive Serum Interferon-A Quantification During SLE Remission Identifies Patients at Risk for Relapse. Ann. Rheum. Dis. 2019, 78, 1669–1676. [Google Scholar] [CrossRef]
- Lu, R.; Guthridge, J.M.; Chen, H.; Bourn, R.L.; Kamp, S.; Munroe, M.E.; Macwana, S.R.; Bean, K.; Sridharan, S.; Merrill, J.T.; et al. Immunologic Findings Precede Rapid Lupus Flare After Transient Steroid Therapy. Sci. Rep. 2019, 9, 8590. [Google Scholar] [CrossRef] [Green Version]
- Ranade, K.; Wang, L.; Brohawn, P.; Greth, W.; Drappa, J.; Illei, G.G. High Interferon Gene Signature is Associated With Increased Disease Activity, Reduced Complement C3 And C4, and Increased Oral Corticosteroid Use in Systemic Lupus Erythematosus (SLE). Arthritis Rheum. 2015, 67 (Suppl. 10). [Google Scholar]
- Cesaroni, M.; Seridi, L.; Loza, M.J.; Schreiter, J.; Sweet, K.; Franks, C.; Ma, K.; Orillion, A.; Campbell, K.; Gordon, R.M.; et al. Suppression of Serum Interferon-γ Levels as a Potential Measure of Response to Ustekinumab Treatment in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 2021, 73, 472–477. [Google Scholar] [CrossRef]
- Shigesaka, M.; Ito, T.; Inaba, M.; Imai, K.; Yamanaka, H.; Azuma, Y.; Tanaka, A.; Amuro, H.; Nishizawa, T.; Son, Y.; et al. Mycophenolic Acid, the Active Form of Mycophenolate Mofetil, Interferes With IRF7 Nuclear Translocation and Type I IFN Production by Plasmacytoid Dendritic Cells. Arthritis Res. Ther. 2020, 22, 264. [Google Scholar] [CrossRef]
- Bodewes, I.L.A.; Gottenberg, J.E.; van Helden-Meeuwsen, C.G.; Mariette, X.; Versnel, M.A. Hydroxychloroquine Treatment Downregulates Systemic Interferon Activation in Primary Sjögren’s Syndrome in the JOQUER Randomized Trial. Rheumatology 2020, 59, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Lambers, W.M.; Westra, J.; Bootsma, H.; de Leeuw, K. Hydroxychloroquine Suppresses Interferon-Inducible Genes and B Cell Activating Factor in Patients With Incomplete and New-Onset Systemic Lupus Erythematosus. J. Rheumatol. 2021, 48, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Åkerström, E.; Sjöwall, C.; Sohrabian, A.; Jönsen, A.; Gomez, A.; Frodlund, M.; Zickert, A.; Bengtsson, A.A.; Rönnelid, J.; et al. Autoantibody and Cytokine Profiles During Treatment with Belimumab in Patients with Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2020, 21, 3463. [Google Scholar] [CrossRef]
- GlaxoSmithKline. Benlysta Packaging Insert. Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2012/125370s016lbl.pdf (accessed on 13 August 2021).
- Riggs, J.M.; Hanna, R.N.; Rajan, B.; Zerrouki, K.; Karnell, J.L.; Sagar, D.; Vainshtein, I.; Farmer, E.; Rosenthal, K.; Morehouse, C.; et al. Characterisation of Anifrolumab, a Fully Human Anti-Interferon Receptor Antagonist Antibody for the Treatment of Systemic Lupus Erythematosus. Lupus Sci. Med. 2018, 5, e000261. [Google Scholar] [CrossRef]
- Peng, L.; Oganesyan, V.; Wu, H.; Dall’Acqua, W.F.; Damschroder, M.M. Molecular Basis for Antagonistic Activity of Anifrolumab, an Anti-Interferon-α Receptor 1 Antibody. MAbs 2015, 7, 428–439. [Google Scholar] [CrossRef] [Green Version]
- AstraZeneca. Saphnelo Package Insert. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761123s000lbl.pdf (accessed on 13 August 2021).
- AstraZeneca. Saphnelo Approved in Japan for Systemic Lupus Erythematosus [press release]. 2021. Available online: https://www.astrazeneca.com/media-centre/press-releases/2021/saphnelo-approved-in-japan-for-sle.html (accessed on 7 October 2021).
- Ducreux, J.; Houssiau, F.A.; Vandepapelière, P.; Jorgensen, C.; Lazaro, E.; Spertini, F.; Colaone, F.; Roucairol, C.; Laborie, M.; Croughs, T.; et al. Interferon α Kinoid Induces Neutralizing Anti-Interferon α Antibodies That Decrease the Expression of Interferon-Induced and B Cell Activation Associated Transcripts: Analysis of Extended Follow-Up Data From the Interferon A Kinoid Phase I/II Study. Rheumatology 2016, 55, 1901–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauwerys, B.R.; Hachulla, E.; Spertini, F.; Lazaro, E.; Jorgensen, C.; Mariette, X.; Haelterman, E.; Grouard-Vogel, G.; Fanget, B.; Dhellin, O.; et al. Down-Regulation of Interferon Signature in Systemic Lupus Erythematosus Patients by Active Immunization with Interferon α-Kinoid. Arthritis Rheum. 2013, 65, 447–456. [Google Scholar] [CrossRef]
- Houssiau, F.A.; Thanou, A.; Mazur, M.; Ramiterre, E.; Gomez Mora, D.A.; Misterska-Skora, M.; Perich-Campos, R.A.; Smakotina, S.A.; Cerpa Cruz, S.; Louzir, B.; et al. IFN-α Kinoid in Systemic Lupus Erythematosus: Results From a Phase IIb, Randomised, Placebo-Controlled Study. Ann. Rheum. Dis. 2020, 79, 347–355. [Google Scholar] [CrossRef]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and Pharmacodynamics of Rontalizumab in Patients With Systemic Lupus Erythematosus: Results of a Phase I, Placebo-Controlled, Double-Blind, Dose-Escalation Study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C., Jr.; Kennedy, W.P. A Phase II Study of the Efficacy and Safety of Rontalizumab (RhumMAb Interferon-α) in Patients With Systemic Lupus Erythematosus (ROSE). Ann. Rheum. Dis. 2016, 75, 196–202. [Google Scholar] [CrossRef]
- Karnell, J.L.; Wu, Y.; Mittereder, N.; Smith, M.A.; Gunsior, M.; Yan, L.; Casey, K.A.; Henault, J.; Riggs, J.M.; Nicholson, S.M.; et al. Depleting Plasmacytoid Dendritic Cells Reduces Local Type I Interferon Responses and Disease Activity in Patients With Cutaneous Lupus. Sci. Transl. Med. 2021, 13, eabf8442. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. A Phase 2 of VIB7734 for the Treatment of Moderate to Severely Active SLE (RECAST SLE). Available online: https://clinicaltrials.gov/ct2/show/NCT04925934 (accessed on 13 August 2021).
- Furie, R.; Werth, V.P.; Merola, J.F.; Stevenson, L.; Reynolds, T.L.; Naik, H.; Wang, W.; Christmann, R.; Gardet, A.; Pellerin, A.; et al. Monoclonal Antibody Targeting BDCA2 Ameliorates Skin Lesions in Systemic Lupus Erythematosus. J. Clin. Invest. 2019, 129, 1359–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellerin, A.; Otero, K.; Czerkowicz, J.M.; Kerns, H.M.; Shapiro, R.I.; Ranger, A.M.; Otipoby, K.L.; Taylor, F.R.; Cameron, T.O.; Viney, J.L.; et al. Anti-BDCA2 Monoclonal Antibody Inhibits Plasmacytoid Dendritic Cell Activation Through Fc-Dependent and Fc-Independent Mechanisms. EMBO Mol. Med. 2015, 7, 464–476. [Google Scholar] [CrossRef]
- Furie, R.; van Vollenhoven, R.; Kalunian, K.; Navarra, S.; Romero-Díaz, J.; Werth, V.; Huang, X.; Carroll, H.; Meyers, A.; Musselli, C.; et al. Efficacy and Safety Results from A Phase 2, Randomized, Double-Blind Trial of BIIB059, an Anti-Blood Dendritic Cell Antigen 2 Antibody, in SLE. Arthritis Rheum. 2020, 72 (Suppl. 10). [Google Scholar]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Temesgen-Oyelakin, Y.; Carlucci, P.M.; Wang, X.; Naqi, M.; Playford, M.P.; Goel, R.R.; et al. Phase 1 Double-Blind Randomized Safety Trial of the Janus Kinase Inhibitor Tofacitinib in Systemic Lupus Erythematosus. Nat. Commun. 2021, 12, 3391. [Google Scholar] [CrossRef]
- Pin, A.; Tesser, A.; Pastore, S.; Moressa, V.; Valencic, E.; Arbo, A.; Maestro, A.; Tommasini, A.; Taddio, A. Biological and Clinical Changes in a Pediatric Series Treated with Off-Label JAK Inhibitors. Int. J. Mol. Sci. 2020, 21, 7767. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Tanaka, Y.; Petri, M.A.; Smolen, J.S.; Wallace, D.J.; Dow, E.R.; Higgs, R.E.; Rocha, G.; Crowe, B.; Benschop, R.J.; et al. Baricitinib-Associated Changes in Global Gene Expression During a 24-Week Phase II Clinical Systemic Lupus Erythematosus Trial Implicates a Mechanism of Action Through Multiple Immune-Related Pathways. Lupus Sci. Med. 2020, 7, e000424. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.K.; Lee, J.Y.; Park, S.H.; Cho, M.L.; Min, S.Y.; Park, S.H.; Kim, H.Y.; Cho, Y.G. Dysfunctional Interferon-Alpha Production by Peripheral Plasmacytoid Dendritic Cells Upon Toll-Like Receptor-9 Stimulation in Patients with Systemic Lupus Erythematosus. Arthritis Res. Ther. 2008, 10, R29. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Arruza, I.; Ugarte, A.; Cabezas-Rodriguez, I.; Medina, J.A.; Moran, M.A.; Ruiz-Irastorza, G. Glucocorticoids and Irreversible Damage in Patients with Systemic Lupus Erythematosus. Rheumatology 2014, 53, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Irastorza, G.; Danza, A.; Khamashta, M. Glucocorticoid Use and Abuse in SLE. Rheumatology 2012, 51, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zonana-Nacach, A.; Barr, S.G.; Magder, L.S.; Petri, M. Damage in Systemic Lupus Erythematosus and its Association with Corticosteroids. Arthritis Rheum. 2000, 43, 1801–1808. [Google Scholar] [CrossRef]
- Lever, E.; Alves, M.R.; Isenberg, D.A. Towards Precision Medicine in Systemic Lupus Erythematosus. Pharmgenomics Pers. Med. 2020, 13, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Medina-Quiñones, C.V.; Ramos-Merino, L.; Ruiz-Sada, P.; Isenberg, D. Analysis of Complete Remission in Systemic Lupus Erythematosus Patients Over a 32-Year Period. Arthritis Care Res. 2016, 68, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A Phase III, Randomized, Placebo-Controlled Study of Belimumab, a Monoclonal Antibody that Inhibits B Lymphocyte Stimulator, in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.K.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.C.; Santiago, M.B.; et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.; Henderson, R.B.; Jones-Leone, A.R.; Flint, S.M.; Lennon, M.; Levy, R.A.; Ji, B.; Bass, D.L.; Roth, D. The Role of Baseline BLyS levels and Type 1 Interferon-Inducible Gene Signature Status in Determining Belimumab Response in Systemic Lupus Erythematosus: A Post Hoc Meta-Analysis. Arthritis Res. Ther. 2020, 22, 102. [Google Scholar] [CrossRef]
- Higgs, B.W.; Zhu, W.; Morehouse, C.; White, W.I.; Brohawn, P.; Guo, X.; Rebelatto, M.; Le, C.; Amato, A.; Fiorentino, D.; et al. A Phase 1b Clinical Trial Evaluating Sifalimumab, an Anti-IFN-α Monoclonal Antibody, Shows Target Neutralisation of a Type I IFN Signature in Blood of Dermatomyositis and Polymyositis Patients. Ann. Rheum Dis. 2014, 73, 256–262. [Google Scholar] [CrossRef]
- Gatto, M.; Saccon, F.; Zen, M.; Bettio, S.; Iaccarino, L.; Punzi, L.; Doria, A. Success and Failure of Biological Treatment in Systemic Lupus Erythematosus: A Critical Analysis. J. Autoimmun. 2016, 74, 94–105. [Google Scholar] [CrossRef]
- Casey, K.A.; Guo, X.; Smith, M.A.; Wang, S.; Sinibaldi, D.; Sanjuan, M.A.; Wang, L.; Illei, G.G.; White, W.I. Type I Interferon Receptor Blockade with Anifrolumab Corrects Innate and Adaptive Immune Perturbations of SLE. Lupus Sci. Med. 2018, 5, e000286. [Google Scholar] [CrossRef]
- Guo, X.; Higgs, B.W.; Bay-Jensen, A.C.; Karsdal, M.A.; Yao, Y.; Roskos, L.K.; White, W.I. Suppression of T Cell Activation and Collagen Accumulation by an Anti-IFNAR1 mAb, Anifrolumab, in Adult Patients with Systemic Sclerosis. J. Invest. Dermatol. 2015, 135, 2402–2409. [Google Scholar] [CrossRef] [Green Version]
- Chia, Y.L.; Tummala, R.; Mai, T.; Rouse, T.; White, W.; Morand, E.F.; Furie, R. POS0688 Characterization of PK/PD of Anifrolumab in Patients with Moderate to Severe SLE. Ann. Rheum. Dis. 2021, 80, 590–591. [Google Scholar] [CrossRef]
- Ajeganova, S.; Hafström, I.; Frostegård, J. Patients with SLE Have Higher Risk of Cardiovascular Events and Mortality in Comparison with Controls with the Same Levels of Traditional Risk Factors and Intima-Media Measures, Which is Related to Accumulated Disease Damage and Antiphospholipid Syndrome: A Case-Control Study Over 10 Years. Lupus Sci. Med. 2021, 8, e000454. [Google Scholar] [CrossRef] [PubMed]
- Werth, V.; Furie, R.; Romero-Díaz, J.; Navarra, S.; Kalunian, K.; van Vollenhoven, R.; Nyberg, F.; Kaffenberger, B.; Sheikh, S.; Radunovic, G.; et al. BIIB059, a Humanized Monoclonal Antibody Targeting Blood Dendritic Cell Antigen 2 on Plasmacytoid Dendritic Cells, Shows Dose-Related Efficacy in a Phase 2 Study in Participants With Active Cutaneous Lupus Erythematosus. Arthritis Rheum. 2020, 72 (Suppl. 10). [Google Scholar]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vétizou, M.; Daillère, R.; et al. Sustained Type I Interferon Signaling as a Mechanism of Resistance to PD-1 Blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef]
- Van Vollenhoven, R. String of Successful Trials in SLE: Have we Cracked the Code? Lupus Sci. Med. 2020, 7, e000380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.J. The Evolution of Drug Discovery in Systemic Lupus Erythematosus. Nat. Rev. Rheumatol. 2015, 11, 616–620. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard Therapies | ||||
|---|---|---|---|---|
| Therapy | Impact on Type I Interferon | Treatment Recommendations | ||
| Immunosuppressives | One recent study reported that MPA, a metabolite of MMF, inhibited production of IFN-α by pDCs from healthy donors in a dose-dependent manner, which was confirmed by transcript levels [98]; further investigation is required to see if the same effect occurs in patients with SLE | High intensity recommended for treatment of flares, followed by longer period of less intensive therapy to consolidate response and prevent relapse [92] | ||
| Antimalarials | Multiple studies have reported inhibition of IFN-α production or IFNGS expression in patients with SLE following HCQ treatment [25,99,100] | Recommended for all patients with SLE unless contraindicated [92] | ||
| Glucocorticoids | - | Pulses of intravenous methylprednisolone recommended for short term; oral glucocorticoids should be tapered to 7.5 mg/day and, where possible, withdrawn [92] | ||
| Approved Biologics | ||||
| Therapy | Impact on Type I Interferon | Treatment Recommendations | ||
| Belimumab [101], anti-BLyS mAb | Belimumab treatment reduced levels of both IFN-α protein and circulating autoantibodies in patients with SLE [101] | Indicated for the treatment of patients aged 5 years or older with active, autoantibody-positive SLE who are receiving standard therapy, and adult patients with active LN who are receiving standard therapy [102] | ||
| Anifrolumab; anti-IFNAR1 mAb [103,104] | Blocks signaling of all type I IFNs, suppresses IFNGS [103,104] | Indicated for the treatment of adult patients with moderate to severe SLE who are receiving standard therapy [105,106] | ||
| Other IFN-Targeting (Direct and Indirect) Therapeutics in Clinical Development | ||||
| Target | Clinical Trial Phase | Impact on Type I Interferon | Primary Efficacy Outcome | Efficacy Results |
| IFN-κ; therapeutic vaccine composed of inactivated IFNα2b coupled to carrier protein [107,108] | Phase 2b (discontinued) [109] | Induces production of neutralizing polyclonal anti-IFN-α-antibodies leading to normalization of the IFNGS [107,108] | Coprimary endpoints: Neutralization of IFNGS, and modified BICLA response (requiring glucocorticoid taper) at Week 36 [109] | BICLA primary endpoint not met.More patients attained LLDAS or glucocorticoid tapering with IFN-K than with placebo [109] |
| Sifalimumab, anti-IFN-α mAb [53] | Phase 2 (discontinued) | Neutralizes IFN-α, suppresses IFNGS in blood and skin [7] | SRI(4) response at Week 52 [53] | Primary and secondary endpoints were met, with greater proportions of patients achieving an SRI(4) response at Week 52 with sifalimumab versus placebo, accompanied by other improvements in skin disease activity and reduction in swollen and tender joint counts [53] |
| Rontalizumab; anti-IFN-α mAb [110] | Phase 2 (discontinued) | Neutralizes IFN-α levels and suppresses IFNGS [110] | BILAG-2004 reduction at Week 24 [111] | Primary and secondary (SRI [4] response) endpoints not met [111] |
| pDC Inhibition/Depletion Strategies | ||||
| Target | Clinical Trial Phase | Impact on Type I Interferon | Primary Efficacy Outcome | Efficacy Results |
| VIB7734, pDC-depleting anti-ILT7 mAb [112] | Phase 1 [112] (phase 2 trial recruiting [113]) | Depleted levels of circulating and tissue-resident pDCs and decreased local type I IFN production [112] | Safety and tolerability [112] | Improvement from baseline in skin disease activity with VIB7734 versus placebo [112] |
| BIIB059, pDC-inhibitory anti-BDCA2 mAb [114,115] | Phase 2 [116] | BIIB059 inhibits IFN production from pDCs, leading to decreased IFNGS expression in blood and normalized IFN response proteins in affected skin [114,115] | Change in total active joint count from baseline to Week 24 [116] | BIIB059 treatment was associated with reduced active joint counts and higher SRI(4) response rates compared with placebo in patients with SLE [116] |
| Small molecule JAK inhibitors (multiple) | Various | Suppress IFNGS expression [117,118,119] | Various | Tofacitinib treatment was associated with fewer low-density granulocytes (signs of dysregulated neutrophil function) and improvements in cardiometabolic parameters and vascular function [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramaswamy, M.; Tummala, R.; Streicher, K.; Nogueira da Costa, A.; Brohawn, P.Z. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 11286. https://doi.org/10.3390/ijms222011286
Ramaswamy M, Tummala R, Streicher K, Nogueira da Costa A, Brohawn PZ. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. International Journal of Molecular Sciences. 2021; 22(20):11286. https://doi.org/10.3390/ijms222011286
Chicago/Turabian StyleRamaswamy, Madhu, Raj Tummala, Katie Streicher, Andre Nogueira da Costa, and Philip Z. Brohawn. 2021. "The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases" International Journal of Molecular Sciences 22, no. 20: 11286. https://doi.org/10.3390/ijms222011286
APA StyleRamaswamy, M., Tummala, R., Streicher, K., Nogueira da Costa, A., & Brohawn, P. Z. (2021). The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. International Journal of Molecular Sciences, 22(20), 11286. https://doi.org/10.3390/ijms222011286