
Regulation of B Cell Responses in SLE by Three Classes of Interferons

Abstract
:1. Introduction
1.1. Types of Interferons
1.2. Peripheral B Cell Subsets and Their Roles in SLE
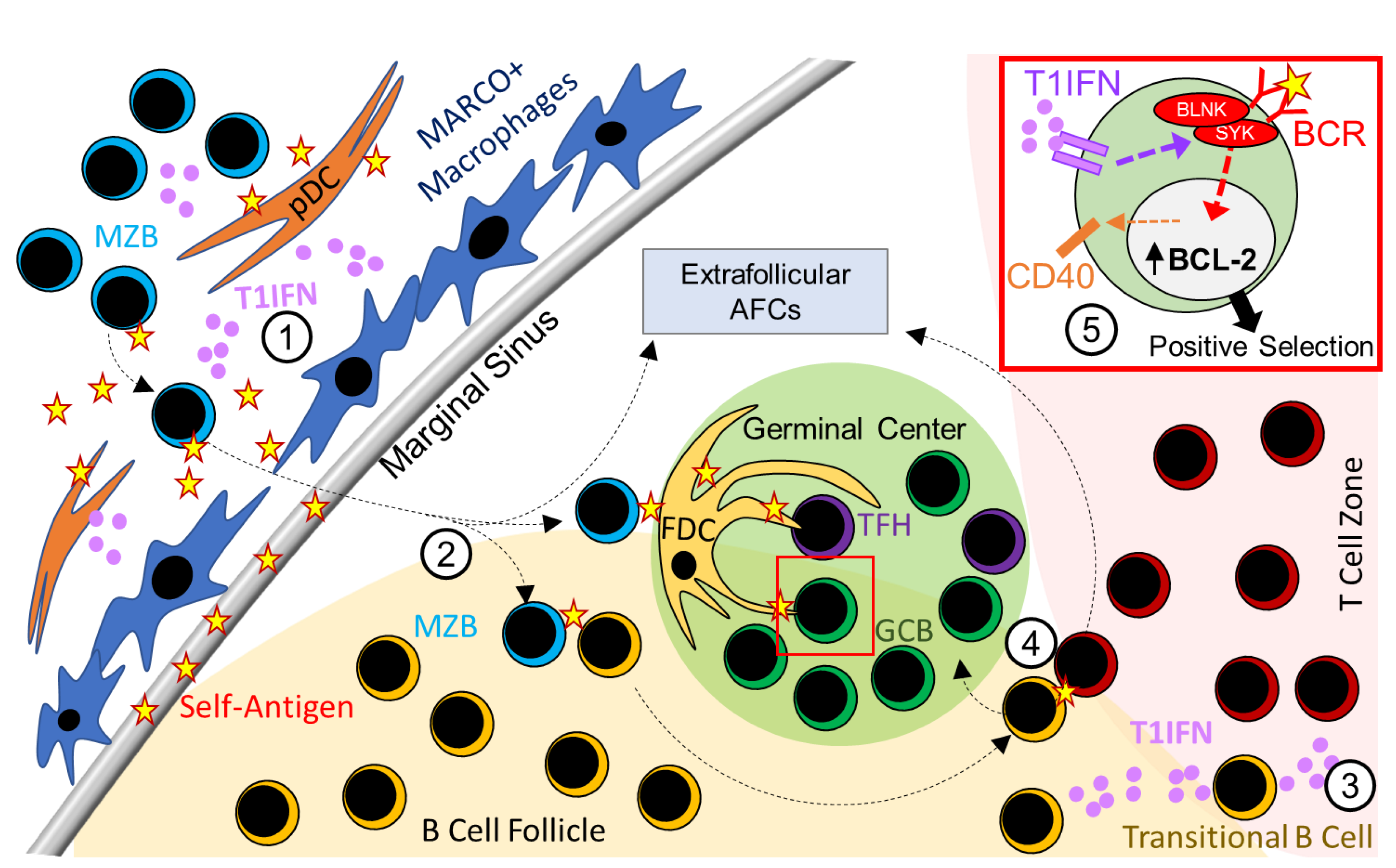
2. Type 1 Interferon Signaling in B Cells and SLE
2.1. Role of T1IFN Signaling in B-1a Cell Responses in SLE
2.2. Role of T1IFN Signaling in MZB Cell Responses in SLE
2.3. Role of T1IFN Signaling in Regulating AFC and GC Responses in SLE
2.4. Model Dependent Role of T1IFN Signaling in B Cell Responses in SLE Mouse Models
2.5. T1IFN Signaling and B Cell Responses in Type 1 Interferonopathies
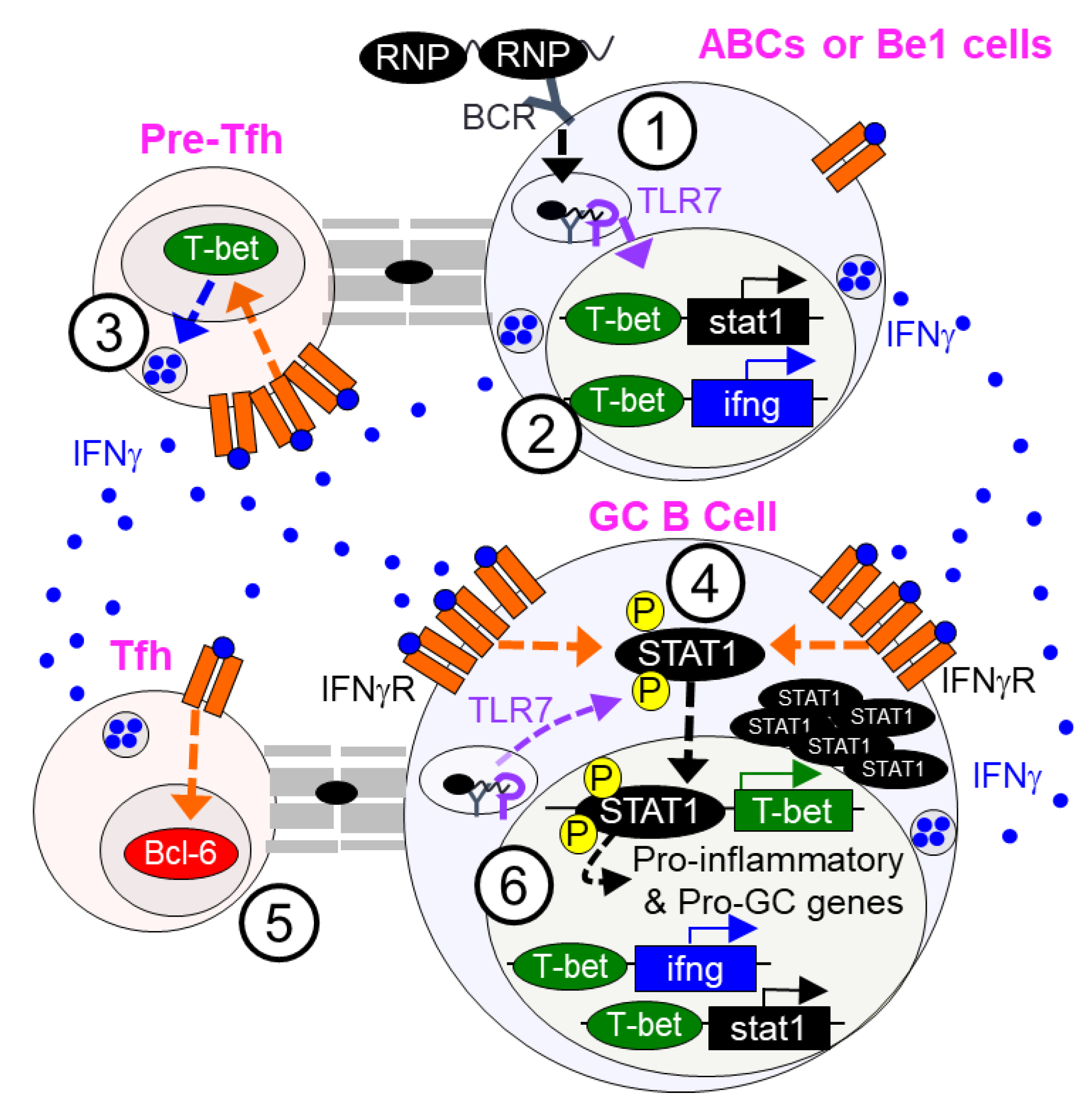
3. Type 2 Interferon Signaling in B Cells and SLE
3.1. Type 2 Interferon (T2IFN) Signaling in Regulating GC and AFC Responses in SLE
3.2. T2IFN Signaling and Age-Associated B Cells (ABCs)
3.3. T1IFN and T2IFN Signaling in the Regulation of B Effector Cell Subset
3.4. T1IFN and T2IFN Signaling in Regulatory B Cells
4. Type 3 Interferon Signaling in SLE
4.1. T3IFN in Human B Cell Activation
4.2. T3IFN Signaling in Autoimmune B Cell Responses in SLE-Prone Mice
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bennett:, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Skopelja-Gardner, S.; An, J.; Tai, J.; Tanaka, L.; Sun, X.; Hermanson, P.; Baum, R.; Kawasumi, M.; Green, R.; Gale, M.; et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci. Rep. 2020, 10, 7908. [Google Scholar] [CrossRef]
- Mak, A.; Tay, S.H. Environmental Factors, Toxicants and Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2014, 15, 16043–16056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, G.J.; Azzouz, D.F.; Alekseyenko, A.V. Systemic Lupus Erythematosus and dysbiosis in the microbiome: Cause or effect or both? Curr. Opin. Immunol. 2019, 61, 80–85. [Google Scholar] [CrossRef]
- Rönnblom, L.; Eloranta, M.-L.; Alm, G.V. Role of Natural Interferon-α Producing Cells (Plasmacytoid Dendritic Cells) in Autoimmunity. Autoimmunity 2003, 36, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2012, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.D.; Pestka, S. Evolution of the Class 2 cytokines and receptors, and discovery of new friends and relatives. Pharmacol. Ther. 2005, 106, 299–346. [Google Scholar] [CrossRef] [PubMed]
- Van Pesch, V.; Lanaya, H.; Renauld, J.C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.Y.; Solanki, H.S.; Advani, J.; Khan, A.A.; Prasad, T.S.K.; Gowda, H.; Thiyagarajan, S.; Chatterjee, A. Comprehensive network map of interferon gamma signaling. J. Cell Commun. Signal. 2018, 12, 745–751. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Bossie, A.; Vitetta, E.S. IFN-gamma enhances secretion of IgG2a from IgG2a-committed LPS-stimulated murine B cells: Implications for the role of IFN-gamma in class switching. Cell Immunol. 1991, 135, 95–104. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Tau, G.Z.; Cowan, S.N.; Weisburg, J.; Braunstein, N.S.; Rothman, P.B. Regulation of IFN-gamma signaling is essential for the cytotoxic activity of CD8(+) T cells. J. Immunol. 2001, 167, 5574–5582. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Selleri, C.; Young, N.S.; Maciejewski, J.P. Inhibition of interferon regulatory factor-1 expression results in predominance of cell growth stimulatory effects of interferon-gamma due to phosphorylation of Stat1 and Stat3. Blood 1997, 90, 4749–4758. [Google Scholar] [CrossRef] [Green Version]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-Lambda (IFN-λ) Is Expressed in a Tissue-Dependent Fashion and Primarily Acts on Epithelial Cells In Vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.; Nice, T.; Diamond, M.S. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Hemann, E.; Gale, M.J.; Savan, R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Baumgarth, N. The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 2010, 11, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.B.; Hewitt, S.L.; Heltemes-Harris, L.M.; Mandal, M.; Johnson, K.; Rajewsky, K.; Koralov, S.; Clark, M.R.; Farrar, M.A.; Skok, J.A. B-1a cells acquire their unique characteristics by bypassing the pre-BCR selection stage. Nat. Commun. 2019, 10, 4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary-Gouy, H.; Harriague, J.; Dalloul, A.; Donnadieu, E.; Bismuth, G. CD5-negative regulation of B cell receptor signaling pathways originates from tyrosine residue Y429 outside an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 2002, 168, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss, F.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Attanavanich, K.; Kearney, J.F. Marginal Zone, but Not Follicular B Cells, Are Potent Activators of Naive CD4 T Cells. J. Immunol. 2004, 172, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Nutt, S.; Hodgkin, P.; Tarlinton, D.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Vogel, L.A.; Zhang, W.; Loo, W.; Shnider, D.; Lind, E.F.; Ratliff, M.; Noelle, R.J.; Erickson, L.D. Imprinting the Fate of Antigen-Reactive B Cells through the Affinity of the B Cell Receptor. J. Immunol. 2006, 177, 7723–7732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viant, C.; Weymar, G.H.; Escolano, A.; Chen, S.; Hartweger, H.; Cipolla, M.; Gazumyan, A.; Nussenzweig, M.C. Antibody Affinity Shapes the Choice between Memory and Germinal Center B Cell Fates. Cell 2020, 183, 1298–1311.e11. [Google Scholar] [CrossRef] [PubMed]
- Schell, S.L.; Soni, C.; Fasnacht, M.J.; Domeier, P.P.; Cooper, T.K.; Rahman, Z.S.M. Mer Receptor Tyrosine Kinase Signaling Prevents Self-Ligand Sensing and Aberrant Selection in Germinal Centers. J. Immunol. 2017, 199, 4001–4015. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Atamas, S.P.; Storrer, C.E.; DaSilva, L.C.; Kelsoe, G.; Papadimitriou, J.C.; Handwerger, B.S. Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 2001, 70, 578–584. [Google Scholar]
- Domeier, P.; Schell, S.L.; Rahman, Z.S.M. Spontaneous germinal centers and autoimmunity. Autoimmunity 2017, 50, 4–18. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Wu, Q.; Yang, P.; Luo, B.; Liu, S.; Hong, H.; Li, J.; Walter, M.R.; Fish, E.N.; Hsu, H.-C.; et al. Cutting Edge: Endogenous IFN-β Regulates Survival and Development of Transitional B Cells. J. Immunol. 2017, 199, 2618–2623. [Google Scholar] [CrossRef] [Green Version]
- Giltiay, N.V.; Chappell, C.P.; Sun, X.; Kolhatkar, N.; Teal, T.H.; Wiedeman, A.; Kim, J.; Tanaka, L.; Buechler, M.; Hamerman, J.A.; et al. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J. Exp. Med. 2013, 210, 2773–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, A.M.; Reiter, K.; Mackay, M.; Aranow, C.; Hiepe, F.; Radbruch, A.; Hansen, A.; Burmester, G.-R.; Diamond, B.; Lipsky, P.E.; et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: Delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. 2008, 58, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; O’Neill, P.; Naradikian, M.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.; Morel, L. Role of B-1a cells in autoimmunity. Autoimmun. Rev. 2006, 5, 403–408. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Georg, I.; Díaz-Barreiro, A.; Varela, N.; Lauwerys, B.; Kumar, R.; Bagavant, H.; Castillo-Martín, M.; Salem, F.; Marañón, C.; et al. Concordance of Increased B1 Cell Subset and Lupus Phenotypes in Mice and Humans Is Dependent on BLK Expression Levels. J. Immunol. 2015, 194, 5692–5702. [Google Scholar] [CrossRef]
- Waffarn, E.E.; Hastey, C.J.; Dixit, N.M.; Choi, Y.S.; Cherry, S.; Kalinke, U.; Simon, S.I.; Baumgarth, N. Infection-induced type I interferons activate CD11b on B-1 cells for subsequent lymph node accumulation. Nat. Commun. 2015, 6, 8991. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.W.; Jacobs, H.; Arkatkar, T.; Dam, E.M.; Scharping, N.; Kolhatkar, N.S.; Hou, B.; Buckner, J.H.; Rawlings, D.J. B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J. Exp. Med. 2016, 213, 733–750. [Google Scholar] [CrossRef] [Green Version]
- Keller, E.J.; Patel, N.B.; Patt, M.; Nguyen, J.K.; Jørgensen, T.N. Partial Protection from Lupus-Like Disease by B-Cell Specific Type I Interferon Receptor Deficiency. Front. Immunol. 2021, 11, 616064. [Google Scholar] [CrossRef]
- Sang, A.; Zheng, Y.-Y.; Yin, Y.; Dozmorov, I.; Li, H.; Hsu, H.-C.; Mountz, J.D.; Morel, L. Dysregulated Cytokine Production by Dendritic Cells Modulates B Cell Responses in the NZM2410 Mouse Model of Lupus. PLoS ONE 2014, 9, e102151. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, J.; Xiao, C.; Han, X.; Qiu, R.; Wang, Y.; Zhou, Y.; Wu, L.; Huang, X.; Shen, N. Phenotypic and functional alterations of pDCs in lupus-prone mice. Sci. Rep. 2016, 6, 20373. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fu, Y.-X.; Wu, Q.; Zhou, Y.; Crossman, D.K.; Yang, P.; Li, J.; Luo, B.; Morel, L.M.; Kabarowski, J.H.; et al. Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J. Clin. Investig. 2015, 125, 2877–2890. [Google Scholar] [CrossRef] [Green Version]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar] [CrossRef]
- Wellmann, U.; Letz, M.; Herrmann, M.; Angermüller, S.; Kalden, J.R.; Winkler, T.H. The evolution of human anti-double-stranded DNA autoantibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 9258–9263. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, C.G.; Sanz, I.; Cook, M. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009, 9, 845–857. [Google Scholar] [CrossRef]
- Cappione, A.; Anolik, J.H.; Pugh-Bernard, A.; Barnard, J.; Dutcher, P.; Silverman, G.; Sanz, I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J. Clin. Investig. 2005, 115, 3205–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiller, T.; Kofer, J.; Kreschel, C.; Busse, C.E.; Riebel, S.; Wickert, S.; Oden, F.; Mertes, M.M.; Ehlers, M.; Wardemann, H. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J. Exp. Med. 2010, 207, 2767–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, B.; Katz, J.B.; Paul, E.; Aranow, C.; Lustgarten, D.; Scharff, M.D. The role of somatic mutation in the pathogenic anti-DNA response. Annu. Rev. Immunol. 1992, 10, 731–757. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zou, Y.R.; Goldstein, J.; Reizis, B.; Diamond, B. Tolerogenic function of Blimp-1 in dendritic cells. J. Exp. Med. 2011, 208, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Klarquist, J.; Cantrell, R.; Lehn, M.A.; Lampe, K.; Hennies, C.M.; Hoebe, K.; Janssen, E.M. Type I IFN Drives Experimental Systemic Lupus Erythematosus by Distinct Mechanisms in CD4 T Cells and B Cells. ImmunoHorizons 2020, 4, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Domeier, P.; Chodisetti, S.B.; Schell, S.L.; Kawasawa, Y.I.; Fasnacht, M.J.; Soni, C.; Rahman, Z.S. B-Cell-Intrinsic Type 1 Interferon Signaling Is Crucial for Loss of Tolerance and the Development of Autoreactive B Cells. Cell Rep. 2018, 24, 406–418. [Google Scholar] [CrossRef] [Green Version]
- Chodisetti, S.B.; Fike, A.J.; Domeier, P.; Singh, H.; Choi, N.M.; Corradetti, C.; Kawasawa, Y.I.; Cooper, T.K.; Caricchio, R.; Rahman, Z.S.M. Type II but Not Type I IFN Signaling Is Indispensable for TLR7-Promoted Development of Autoreactive B Cells and Systemic Autoimmunity. J. Immunol. 2020, 204, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Akita, K.; Yasaka, K.; Shirai, T.; Ishii, T.; Harigae, H.; Fujii, H. Interferon α Enhances B Cell Activation Associated with FOXM1 Induction: Potential Novel Therapeutic Strategy for Targeting the Plasmablasts of Systemic Lupus Erythematosus. Front. Immunol. 2021, 11, 498703. [Google Scholar] [CrossRef] [PubMed]
- Denton, A.E.; Innocentin, S.; Carr, E.J.; Bradford, B.M.; Lafouresse, F.; Mabbott, N.A.; Mörbe, U.; Ludewig, B.; Groom, J.R.; Good-Jacobson, K.L.; et al. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 2019, 216, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, Y.; Núñez, S.; Fuenzalida, M.J.; Flores-Santibáñez, F.; Sáez, P.J.; Dorner, J.; Lennon-Dumenil, A.-M.; Martínez, V.; Zorn, E.; Rosemblatt, M.; et al. Thymic B Cells Promote Germinal Center-Like Structures and the Expansion of Follicular Helper T Cells in Lupus-Prone Mice. Front. Immunol. 2020, 11, 696. [Google Scholar] [CrossRef]
- Wolf, S.J.; Theros, J.; Reed, T.J.; Liu, J.; Grigorova, I.L.; Martínez-Colón, G.; Jacob, C.O.; Hodgin, J.B.; Kahlenberg, J.M. TLR7-Mediated Lupus Nephritis Is Independent of Type I IFN Signaling. J. Immunol. 2018, 201, 393–405. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Wu, Q.; Yang, P.; Luo, B.; Liu, S.; Li, J.; Mattheyses, A.L.; Sanz, I.; Chatham, W.W.; Hsu, H.-C.; et al. Cutting Edge: Intracellular IFN-β and Distinct Type I IFN Expression Patterns in Circulating Systemic Lupus Erythematosus B Cells. J. Immunol. 2018, 201, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- Soni, C.; Perez, O.A.; Voss, W.N.; Pucella, J.N.; Serpas, L.; Mehl, J.; Ching, K.L.; Goike, J.; Georgiou, G.; Ippolito, G.C.; et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity 2020, 52, 1022–1038.e7. [Google Scholar] [CrossRef]
- Karnell, J.L.; Wu, Y.; Mittereder, N.; Smith, M.A.; Gunsior, M.; Yan, L.; Casey, K.A.; Henault, J.; Riggs, J.M.; Nicholson, S.M.; et al. Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci. Transl. Med. 2021, 13, eabf8442. [Google Scholar] [CrossRef]
- Kalunian, K.C. Interferon-targeted therapy in systemic lupus erythematosus: Is this an alternative to targeting B and T cells? Lupus 2016, 25, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Hron, J.D.; Peng, S.L. Type I IFN Protects Against Murine Lupus. J. Immunol. 2004, 173, 2134–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, M.E.; Lu, R.; Zhao, Y.D.; Fife, D.A.; Robertson, J.M.; Guthridge, J.; Niewold, T.B.; Tsokos, G.C.; Keith, M.P.; Harley, J.B.; et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 2016, 75, 2014–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Silva, D.G.; Martin, J.; Pratama, A.; Hu, X.; Chang, P.-P.; Walters, G.; Vinuesa, C.G. Interferon-γ Excess Leads to Pathogenic Accumulation of Follicular Helper T Cells and Germinal Centers. Immunity 2012, 37, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Domeier, P.; Chodisetti, S.B.; Soni, C.; Schell, S.L.; Elias, M.J.; Wong, E.B.; Cooper, T.K.; Kitamura, D.; Rahman, Z.S. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J. Exp. Med. 2016, 213, 715–732. [Google Scholar] [CrossRef] [Green Version]
- Varinou, L.; Ramsauer, K.; Karaghiosoff, M.; Kolbe, T.; Pfeffer, K.; Müller, M.; Decker, T. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity 2003, 19, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Chodisetti, S.B.; Fike, A.J.; Domeier, P.P.; Schell, S.L.; Mockus, T.E.; Choi, N.M.; Corradetti, C.; Hou, B.; Atkins, H.M.; Caricchio, R.; et al. Serine Phosphorylation of the STAT1 Transactivation Domain Promotes Autoreactive B Cell and Systemic Autoimmunity Development. J. Immunol. 2020, 204, 2641–2650. [Google Scholar] [CrossRef]
- Cebra, J.J.; Schrader, C.E.; Shroff, K.E.; Weinstein, P.D. Are Peyer’s patch germinal centre reactions different from those occurring in other lymphoid tissues? Res. Immunol. 1991, 142, 222–226. [Google Scholar] [CrossRef]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef]
- Hara, S.; Sasaki, T.; Satoh-Takayama, N.; Kanaya, T.; Kato, T.; Takikawa, Y.; Takahashi, M.; Tachibana, N.; Kim, K.S.; Surh, C.D.; et al. Dietary Antigens Induce Germinal Center Responses in Peyer’s Patches and Antigen-Specific IgA Production. Front. Immunol. 2019, 10, 2432. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, R.; Sims, G.P.; Robbins, R.; Withers, D.; Fischer, R.T.; Grammer, A.; Kuchen, S.; Lipsky, P.E. IL-21 and BAFF/BLyS Synergize in Stimulating Plasma Cell Differentiation from a Unique Population of Human Splenic Memory B Cells. J. Immunol. 2007, 178, 2872–2882. [Google Scholar] [CrossRef]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.-H.; Milner, E.C.B.; Sanz, I. A New Population of Cells Lacking Expression of CD27 Represents a Notable Component of the B Cell Memory Compartment in Systemic Lupus Erythematosus. J. Immunol. 2007, 178, 6624–6633. [Google Scholar] [CrossRef] [Green Version]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Ratliff, M.; Alter, S.; Frasca, D.; Blomberg, B.B.; Riley, R.L. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell 2013, 12, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Zumaquero, E.; Stone, S.L.; Scharer, C.D.; Jenks, S.A.; Nellore, A.; Mousseau, B.; Rosal-Vela, A.; Botta, D.; Bradley, J.E. IFNγ induces epigenetic programming of human T-bet. elife 2019, 8, e41641. [Google Scholar] [CrossRef] [PubMed]
- Herve, M.-G.D.G.D.; Durali, D.; Dembele, B.; Giuliani, M.; Tran, T.-A.; Azzarone, B.; Eid, P.; Tardieu, M.; Delfraissy, J.-F.; Taoufik, Y. Interferon-Alpha Triggers B Cell Effector 1 (Be1) Commitment. PLoS ONE 2011, 6, e19366. [Google Scholar] [CrossRef]
- Harris, D.P.; Goodrich, S.; Gerth, A.J.; Peng, S.L.; Lund, F.E. Regulation of IFN-gamma production by B effector 1 cells: Essential roles for T-bet and the IFN-gamma receptor. J. Immunol. 2005, 174, 6781–6790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fike, A.J.; Chodisetti, S.B.; Bricker, K.N.; Choi, N.M.; Chroneos, Z.C.; Kaplan, M.H.; Rahman, Z.S. STAT4 Is Largely Dispensable for Systemic Lupus Erythematosus-like Autoimmune- and Foreign Antigen-Driven Antibody-Forming Cell, Germinal Center, and Follicular Th Cell Responses. Immunohorizons 2021, 5, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Parekh, V.V.; Prasad, D.V.; Banerjee, P.P.; Joshi, B.N.; Kumar, A.; Mishra, G.C. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: Role of TGF-beta 1. J. Immunol. 2003, 170, 5897–5911. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Zekzer, D.; Hanssen, L.; Lu, Y.; Olcott, A.; Kaufman, D.L. Lipopolysaccharide-Activated B Cells Down-Regulate Th1 Immunity and Prevent Autoimmune Diabetes in Nonobese Diabetic Mice. J. Immunol. 2001, 167, 1081–1089. [Google Scholar] [CrossRef]
- Carter, N.A.; Vasconcellos, R.; Rosser, E.C.; Tulone, C.; Muñoz-Suano, A.; Kamanaka, M.; Ehrenstein, M.R.; Flavell, R.A.; Mauri, C. Mice Lacking Endogenous IL-10–Producing Regulatory B Cells Develop Exacerbated Disease and Present with an Increased Frequency of Th1/Th17 but a Decrease in Regulatory T Cells. J. Immunol. 2011, 186, 5569–5579. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Mei, Y.; Li, Z. Research Progress on Regulatory B Cells in Systemic Lupus Erythematosus. BioMed Res. Int. 2019, 2019, 7948687. [Google Scholar] [CrossRef] [PubMed]
- Chodisetti, S.B.; Fike, A.J.; Domeier, P.P.; Choi, N.M.; Soni, C.; Rahman, Z.S.M. TLR7 Negatively Regulates B10 Cells Predominantly in an IFNγ Signaling Dependent Manner. Front. Immunol. 2020, 11, 1632. [Google Scholar] [CrossRef]
- Imbrechts, M.; De Samblancx, K.; Fierens, K.; Brisse, E.; Vandenhaute, J.; Mitera, T.; Libert, C.; Smets, I.; Goris, A.; Wouters, C.; et al. IFN-γ stimulates CpG-induced IL-10 production in B cells via p38 and JNK signalling pathways. Eur. J. Immunol. 2018, 48, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.W.; Scharping, N.; Kolhatkar, N.S.; Khim, S.; Schwartz, M.A.; Li, Q.-Z.; Hudkins, K.L.; Alpers, C.E.; Liggitt, D.; Rawlings, D.J. Opposing Impact of B Cell–Intrinsic TLR7 and TLR9 Signals on Autoantibody Repertoire and Systemic Inflammation. J. Immunol. 2014, 192, 4525–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, C.; Wong, E.B.; Domeier, P.P.; Khan, T.N.; Satoh, T.; Akira, S.; Rahman, Z.S.M. B Cell–Intrinsic TLR7 Signaling Is Essential for the Development of Spontaneous Germinal Centers. J. Immunol. 2014, 193, 4400–4414. [Google Scholar] [CrossRef] [Green Version]
- Miles, K.; Heaney, J.; Sibinska, Z.; Salter, D.; Savill, J.; Gray, D.; Gray, M. A tolerogenic role for Toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Yang, Q.; Lourenco, E.; Sun, H.; Zhang, Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: Its correlation with disease activity. Arthritis Res. Ther. 2011, 13, R88. [Google Scholar] [CrossRef] [Green Version]
- Adel, Y.; Sadeq, Y. Impact of IL-34, IFN-α and IFN-λ1 on activity of systemic lupus erythematosus in Egyptian patients. Reumatologia 2020, 58, 221–230. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Q.; Sun, H.; Li, M.; Zhang, Y.; Cava, A.L. Serum IFN-λ1 is abnormally elevated in rheumatoid arthritis patients. Autoimmunity 2013, 46, 40–43. [Google Scholar] [CrossRef]
- Wang, F.; Xu, L.; Feng, X.; Guo, D.; Tan, W.; Zhang, M. Interleukin-29 modulates proinflammatory cytokine production in synovial inflammation of rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R228. [Google Scholar] [CrossRef] [Green Version]
- Apostolou, E.; Kapsogeorgou, E.K.; Konsta, O.D.; Giotakis, I.; Saridaki, M.I.; Andreakos, E.; Tzioufas, A.G. Expression of type III interferons (IFNλs) and their receptor in Sjögren’s syndrome. Clin. Exp. Immunol. 2016, 186, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metwally, M.; Thabet, K.; Bayoumi, A.; Nikpour, M.; Stevens, W.; Sahhar, J.; Zochling, J.; Roddy, J.; Tymms, K.; Strickland, G.; et al. IFNL3 genotype is associated with pulmonary fibrosis in patients with systemic sclerosis. Sci. Rep. 2019, 9, 14834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amezcua-Guerra, L.M.; Márquez-Velasco, R.; Chávez-Rueda, A.K.; Castillo-Martínez, D.; Massó, F.; Páez, A.; Colín-Fuentes, J.; Bojalil, R. Type III Interferons in Systemic Lupus Erythematosus: Association Between Interferon λ3, Disease Activity, and Anti-Ro/SSA Antibodies. J. Clin. Rheumatol. 2017, 23, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Wang, C.M.; Chen, T.D.; Wu, Y.J.; Lin, J.C.; Lu, L.Y.; Wu, J. Interferon-λ3/4 genetic variants and interferon-λ3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese. Arthritis Res. Ther. 2018, 20, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, R.R.; Kotenko, S.V.; Kaplan, M.J. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2021, 17, 349–362. [Google Scholar] [CrossRef]
- Hu, F.-Q.; Zhang, Y.-P.; Yin, J.; Tang, Z.-Q.; Han, Y.-F.; Shi, Z.-R.; Tan, G.-Z.; Wang, L. Characterization of autoantibodies and cytokines related to cutaneous lupus erythematosus. Lupus 2020, 30, 315–319. [Google Scholar] [CrossRef] [PubMed]
- de Groen, R.A.; Groothuismink, Z.M.; Liu, B.S.; Boonstra, A. IFN-λ is able to augment TLR-mediated activation and subsequent function of primary human B cells. J. Leukoc. Biol. 2015, 98, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Syedbasha, M.; Bonfiglio, F.; Linnik, J.; Stuehler, C.; Wüthrich, D.; Egli, A. Interferon-λ Enhances the Differentiation of Naive B Cells into Plasmablasts via the mTORC1 Pathway. Cell Rep. 2020, 33, 108211. [Google Scholar] [CrossRef]
- Goel, R.R.; Wang, X.; O’Neil, L.J.; Nakabo, S.; Hasneen, K.; Gupta, S.; Wigerblad, G.; Blanco, L.P.; Kopp, J.B.; Morasso, M.I.; et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc. Natl. Acad. Sci. USA 2020, 117, 5409–5419. [Google Scholar] [CrossRef]
- Hahn, W.O.; Pepper, M.; Liles, W.C. B cell intrinsic expression of IFNλ receptor suppresses the acute humoral immune response to experimental blood-stage malaria. Virulence 2020, 11, 594–606. [Google Scholar] [CrossRef]
- Rodero, M.P.; Crow, Y.J. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med. 2016, 213, 2527–2538. [Google Scholar] [CrossRef]
- Volpi, S.; Picco, P.; Caorsi, R.; Candotti, F.; Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatr. Rheumatol. 2016, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, D.M.; Di Filippo, P.; Breda, L.; Chiarelli, F. Type I Interferonopathies in Children: An Overview. Front. Pediatr. 2021, 9, 11. [Google Scholar] [CrossRef]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune Disorders Associated with Gain of Function of the Intracellular Sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, A.; Treuting, P.; Elkon, K.B.; Loo, Y.-M.; Gale, M.; Barber, G.N.; Stetson, D.B. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity 2012, 36, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Human | Mouse | |||
|---|---|---|---|---|
| Interferon Family | Receptor | Group Members | Receptor | Group Members |
| Type 1 | Primary: IFNαR1 Co-receptors: IFNαR2a IFNαR2b IFNαR2c | 14 Alphas (α), (2 pseudogenes) 1 Beta (β) 1 Omega (ω) 1 Delta (δ) 1 Nu (ν), (pseudogene) 1 Epsilon (ε) | Primary: IFNαR1 Co-receptors: IFNαR2a IFNαR2b IFNαR2c | 13 Alphas (α), (1 pseudogene) 1 Beta (β) 7 Omegas (ω), (6 pseudogenes) 1 Kappa (κ) 1 Epsilon (ε) 1 Limitin (similar to interferon δ) |
| Type 2 | IFNγR1 IFNγR2 | 1 Gamma (γ) | IFNγR1 IFNγR2 | 1 Gamma (γ) |
| Type 3 | IL28Rα IL-10Rβ | IL-29 (IFNλ1) IL-28A (IFNλ2) IL-28B (IFNλ3) IFNλ4 | IL28Rα IL-10Rβ | IFNλ2 IFNλ3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domeier, P.P.; Rahman, Z.S.M. Regulation of B Cell Responses in SLE by Three Classes of Interferons. Int. J. Mol. Sci. 2021, 22, 10464. https://doi.org/10.3390/ijms221910464
Domeier PP, Rahman ZSM. Regulation of B Cell Responses in SLE by Three Classes of Interferons. International Journal of Molecular Sciences. 2021; 22(19):10464. https://doi.org/10.3390/ijms221910464
Chicago/Turabian StyleDomeier, Phillip P., and Ziaur S. M. Rahman. 2021. "Regulation of B Cell Responses in SLE by Three Classes of Interferons" International Journal of Molecular Sciences 22, no. 19: 10464. https://doi.org/10.3390/ijms221910464
APA StyleDomeier, P. P., & Rahman, Z. S. M. (2021). Regulation of B Cell Responses in SLE by Three Classes of Interferons. International Journal of Molecular Sciences, 22(19), 10464. https://doi.org/10.3390/ijms221910464