
AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
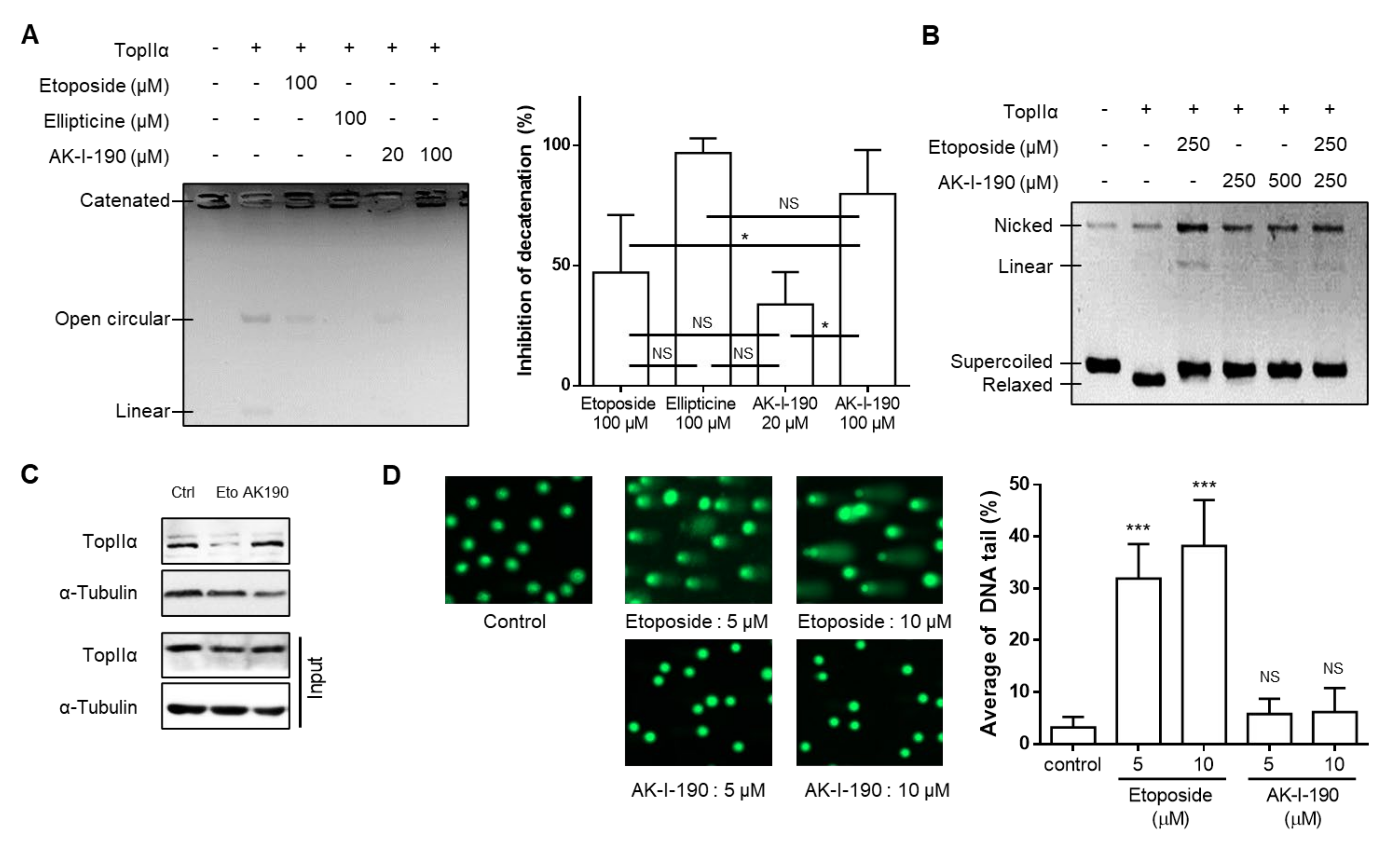
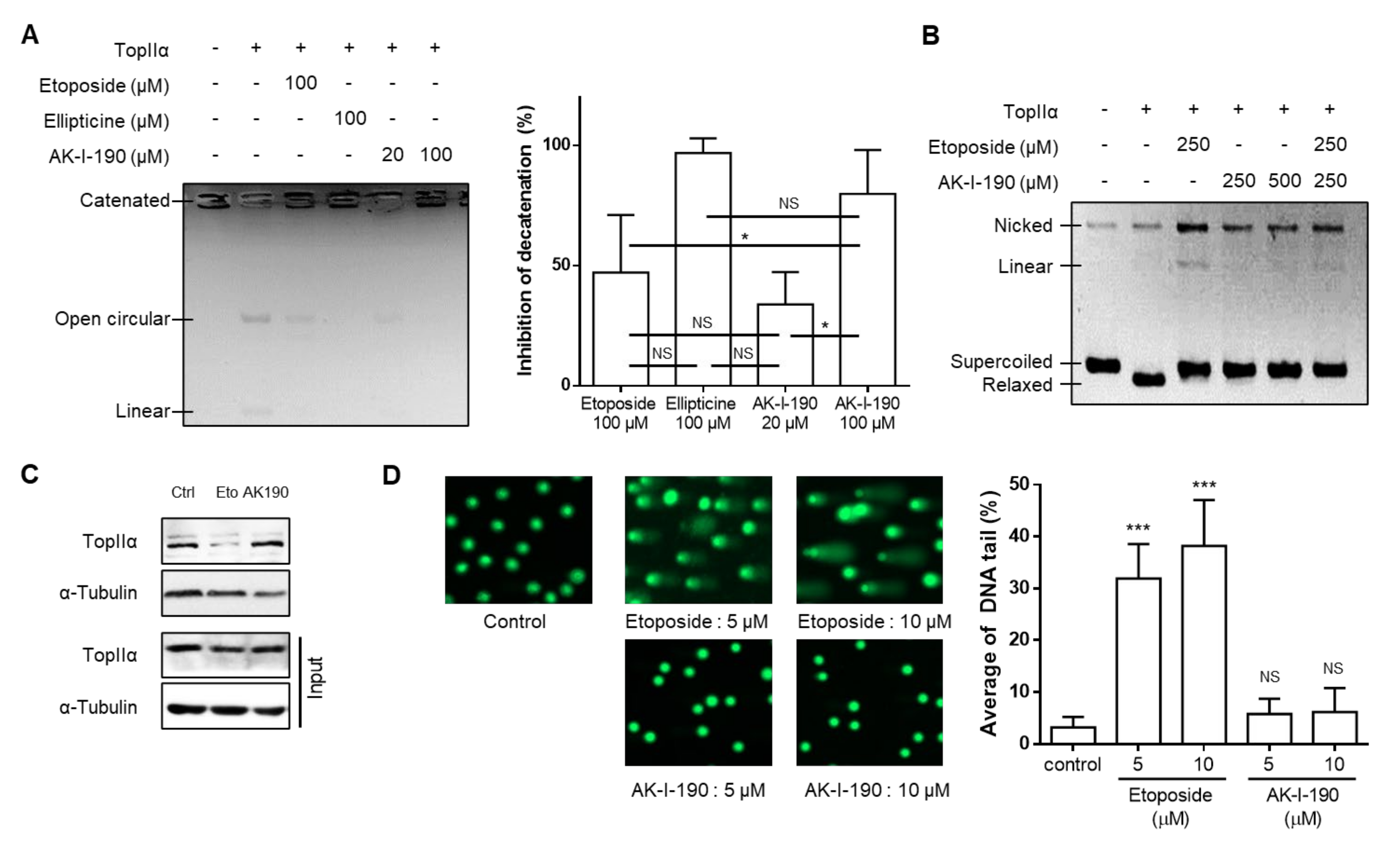
2.1. Topoisomerase II Inhibitory Activity of AK-I-190 as a Catalytic Inhibitor
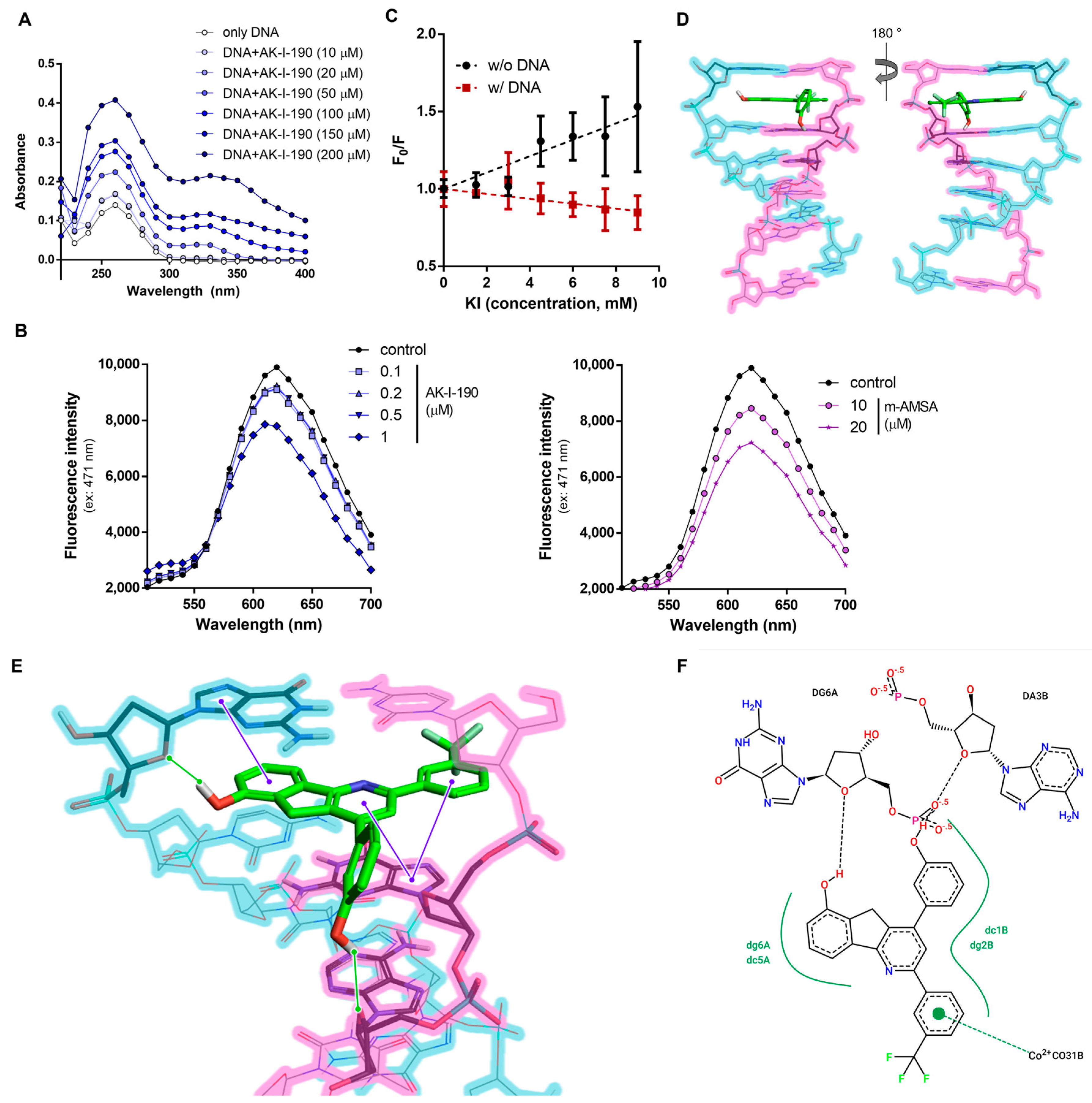
2.2. DNA Intercalation of AK-I-190
2.3. G1 Arrest and Apoptotic Induction of AK-I-190
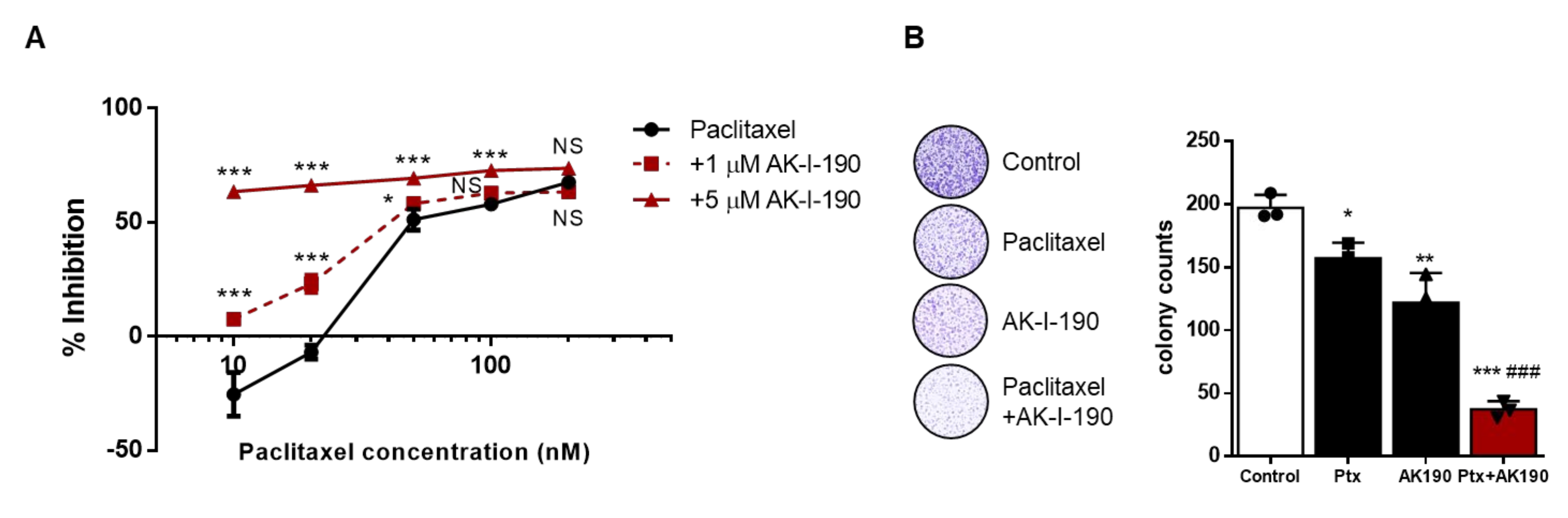
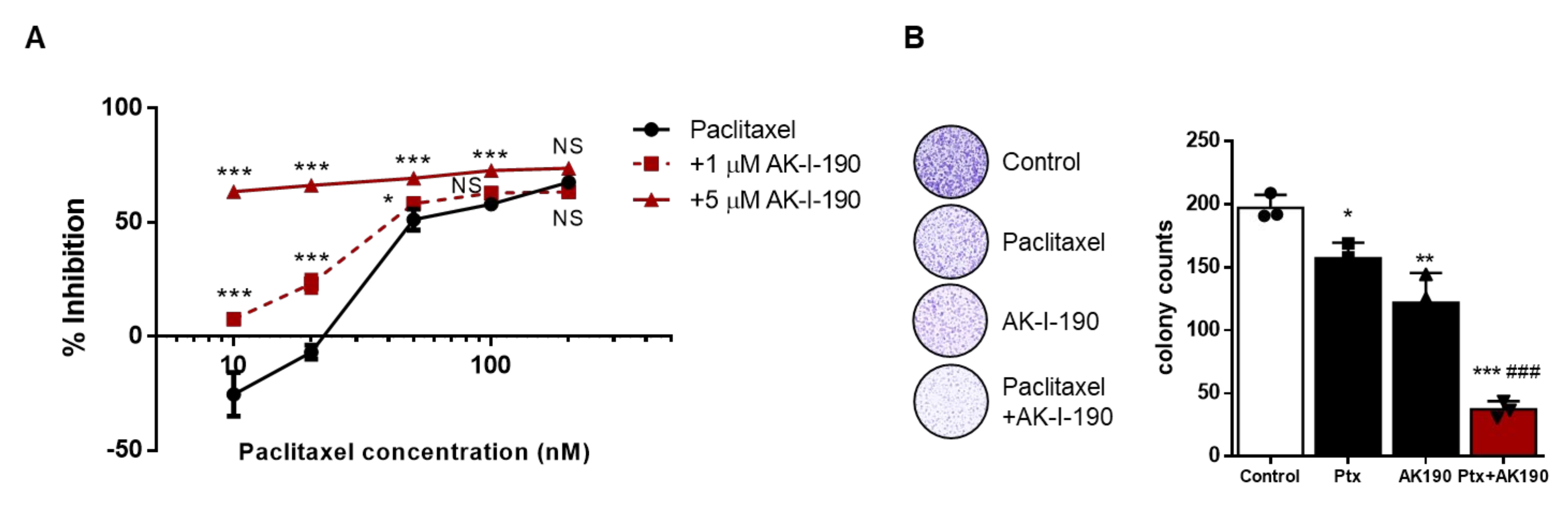
2.4. AK-I-190 in Combination with Paclitaxel Inhibits Cellular Proliferation of a CRPC Cell Line
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Viability Assay
4.2. Western Blot Analysis
4.3. In Vitro DNA Topoisomerase II Relaxation Assay
4.4. kDNA Decatenation Assay
4.5. Topoisomerase II Cleavage Complex Assay
4.6. Band Depletion Assay
4.7. Alkaline Comet Assay
4.8. Potassium Iodide (KI) Quenching Assay
4.9. Competitive EtBr Displacement Assay
4.10. In Silico Docking Study
4.11. Cell Cycle Analysis
4.12. Apoptosis Assay with Annexin V and PI Double Staining
4.13. Clonogenic Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef]
- Chi, K.N.; Bjartell, A.; Dearnaley, D.; Saad, F.; Schröder, F.H.; Sternberg, C.; Tombal, B.; Visakorpi, T. Castration-resistant prostate cancer: From new pathophysiology to new treatment targets. Eur. Urol. 2009, 56, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, M.; Hirst, C.; Crawford, E.D. Characterising the castration-resistant prostate cancer population: A systematic review. Int. J. Clin. Pract. 2011, 65, 1180–1192. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Buonerba, C.; Federico, P.; Bosso, D.; Puglia, L.; Policastro, T.; Izzo, M.; Perri, F.; Vittoria Scarpati, G.D.; Ferro, M.; Cobelli, O.D.; et al. Carboplatin plus etoposide in heavily pretreated castration-resistant prostate cancer patients. Future Oncol. 2014, 10, 1353–1360. [Google Scholar] [CrossRef]
- Laber, D.A.; Chen, M.B.; Jaglal, M.; Patel, A.; Visweshwar, N. Phase 2 Study of Cyclophosphamide, Etoposide, and Estramustine in Patients With Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2018, 16, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kadayat, T.M.; Park, S.; Shrestha, A.; Jo, H.; Hwang, S.Y.; Katila, P.; Shrestha, R.; Nepal, M.R.; Noh, K.; Kim, S.K.; et al. Discovery and Biological Evaluations of Halogenated 2,4-Diphenyl Indeno[1,2-b]pyridinol Derivatives as Potent Topoisomerase IIα-Targeted Chemotherapeutic Agents for Breast Cancer. J. Med. Chem. 2019, 62, 8194–8234. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.H.; Shrestha, A.; Jang, H.J.; Kim, J.A.; Sheen, N.; Seo, M.; Lee, E.S.; Kwon, Y. Anticancer Activity of Indeno[1,2-b]-Pyridinol Derivative as a New DNA Minor Groove Binding Catalytic Inhibitor of Topoisomerase IIα. Biomol. Ther. 2021, 29, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Li, Y.C.; Wang, Y.R.; Li, T.K.; Chan, N.L. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 2013, 41, 10630–10640. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Saldana, C.; Majidipur, A.; Beaumont, E.; Huet, E.; de la Taille, A.; Vacherot, F.; Firlej, V.; Destouches, D. Extracellular Vesicles in Advanced Prostate Cancer: Tools to Predict and Thwart Therapeutic Resistance. Cancers 2021, 13, 3791. [Google Scholar] [CrossRef]
- Wong, Y.N.; Ferraldeschi, R.; Attard, G.; de Bono, J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol. 2014, 11, 365–376. [Google Scholar] [CrossRef]
- Pendleton, M.; Lindsey, R.H., Jr.; Felix, C.A.; Grimwade, D.; Osheroff, N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef] [Green Version]
- Willman, J.H.; Holden, J.A. Immunohistochemical staining for DNA topoisomerase II-alpha in benign, premalignant, and malignant lesions of the prostate. Prostate 2000, 42, 280–286. [Google Scholar] [CrossRef]
- Hughes, C.; Murphy, A.; Martin, C.; Fox, E.; Ring, M.; Sheils, O.; Loftus, B.; O’Leary, J. Topoisomerase II-alpha expression increases with increasing Gleason score and with hormone insensitivity in prostate carcinoma. J. Clin. Pathol. 2006, 59, 721–724. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Rubin, M.A.; Maher, C.A.; Chinnaiyan, A.M. Common gene rearrangements in prostate cancer. J. Clin. Oncol. 2011, 29, 3659–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, W.G.; Haffner, M.C.; Yegnasubramanian, S. The structure of the nucleus in normal and neoplastic prostate cells: Untangling the role of type 2 DNA topoisomerases. Am. J. Clin. Exp. Urol. 2018, 6, 107–113. [Google Scholar] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef]
- Seth, A.; Watson, D.K. ETS transcription factors and their emerging roles in human cancer. Eur. J. Cancer 2005, 41, 2462–2478. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.S.; Sinding, J.; Andersen, A.H.; Alsner, J.; Jensen, P.B.; Westergaard, O. Mode of action of topoisomerase II-targeting agents at a specific DNA sequence. Uncoupling the DNA binding, cleavage and religation events. J. Mol. Biol. 1992, 228, 778–786. [Google Scholar] [CrossRef]
- Gormley, N.A.; Orphanides, G.; Meyer, A.; Cullis, P.M.; Maxwell, A. The interaction of coumarin antibiotics with fragments of DNA gyrase B protein. Biochemistry 1996, 35, 5083–5092. [Google Scholar] [CrossRef]
- Roca, J.; Ishida, R.; Berger, J.M.; Andoh, T.; Wang, J.C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl. Acad. Sci. USA 1994, 91, 1781–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Hwang, S.Y.; Shin, J.; Jo, H.; Na, Y.; Kwon, Y. A chromenone analog as an ATP-competitive, DNA non-intercalative topoisomerase II catalytic inhibitor with preferences toward the alpha isoform. Chem. Commun. 2019, 55, 12857–12860. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Thapa Magar, T.B.; Kadayat, T.M.; Lee, H.J.; Bist, G.; Shrestha, A.; Lee, E.S.; Kwon, Y. Rational design, synthesis, and evaluation of novel 2,4-Chloro- and Hydroxy-Substituted diphenyl Benzofuro[3,2-b]Pyridines: Non-intercalative catalytic topoisomerase I and II dual inhibitor. Eur. J. Med. Chem. 2017, 127, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.H.; Park, C.; Kadayat, T.M.; Shrestha, A.; Lee, E.S.; Kwon, Y. A novel indeno[1,2-b]pyridinone derivative, a DNA intercalative human topoisomerase IIα catalytic inhibitor, for caspase 3-independent anticancer activity. Chem. Commun. 2017, 53, 6864–6867. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, K.-H.; Park, S.; Jang, H.J.; Hwang, S.-Y.; Shrestha, A.; Lee, E.-S.; Kwon, Y. AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells. Int. J. Mol. Sci. 2021, 22, 11246. https://doi.org/10.3390/ijms222011246
Jeon K-H, Park S, Jang HJ, Hwang S-Y, Shrestha A, Lee E-S, Kwon Y. AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells. International Journal of Molecular Sciences. 2021; 22(20):11246. https://doi.org/10.3390/ijms222011246
Chicago/Turabian StyleJeon, Kyung-Hwa, Seojeong Park, Hae Jin Jang, Soo-Yeon Hwang, Aarajana Shrestha, Eung-Seok Lee, and Youngjoo Kwon. 2021. "AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells" International Journal of Molecular Sciences 22, no. 20: 11246. https://doi.org/10.3390/ijms222011246
APA StyleJeon, K.-H., Park, S., Jang, H. J., Hwang, S.-Y., Shrestha, A., Lee, E.-S., & Kwon, Y. (2021). AK-I-190, a New Catalytic Inhibitor of Topoisomerase II with Anti-Proliferative and Pro-Apoptotic Activity on Androgen-Negative Prostate Cancer Cells. International Journal of Molecular Sciences, 22(20), 11246. https://doi.org/10.3390/ijms222011246