
CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Querying the Genomics of Drug Sensitivity Database for Epithelioid Sarcoma
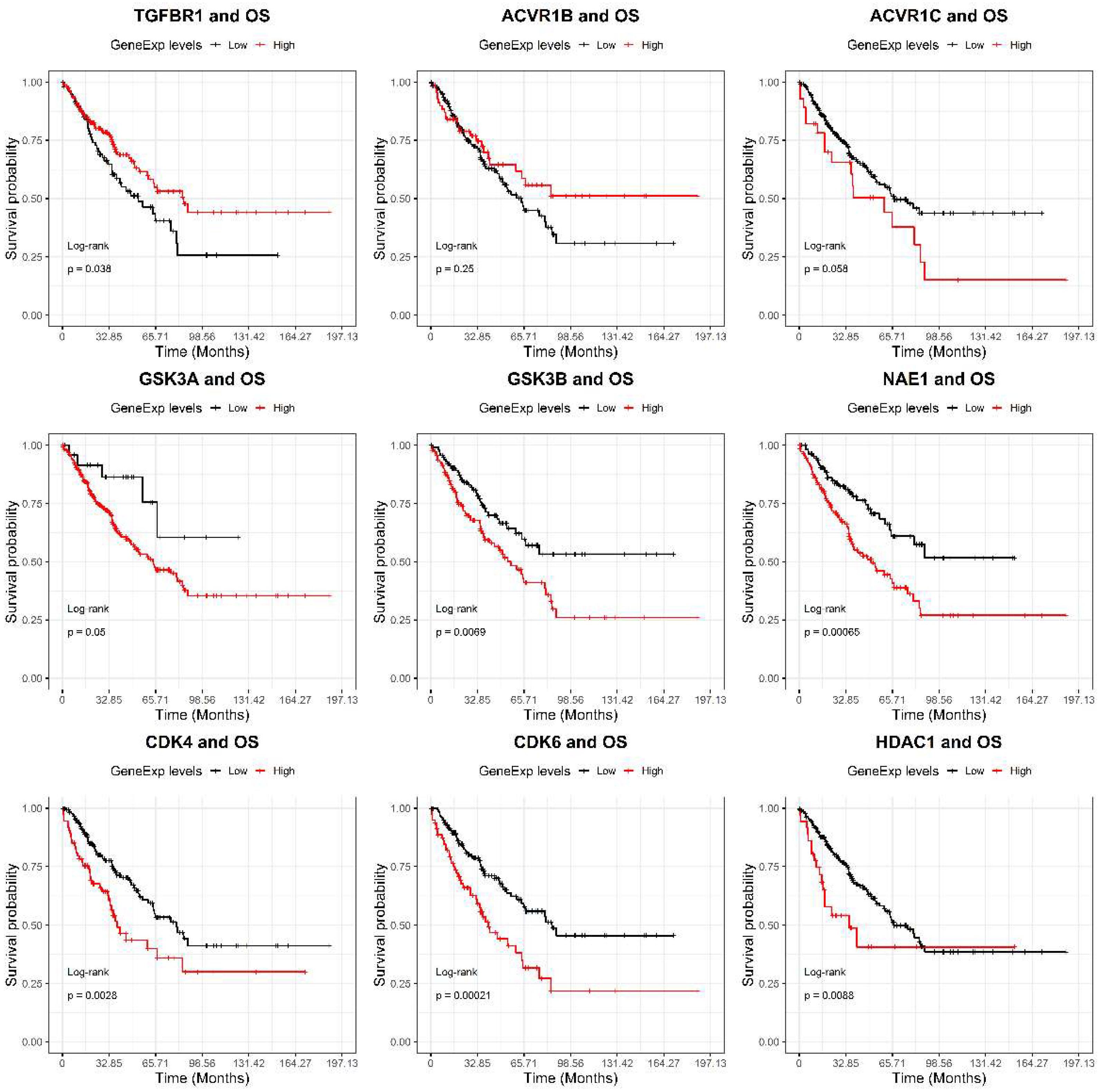
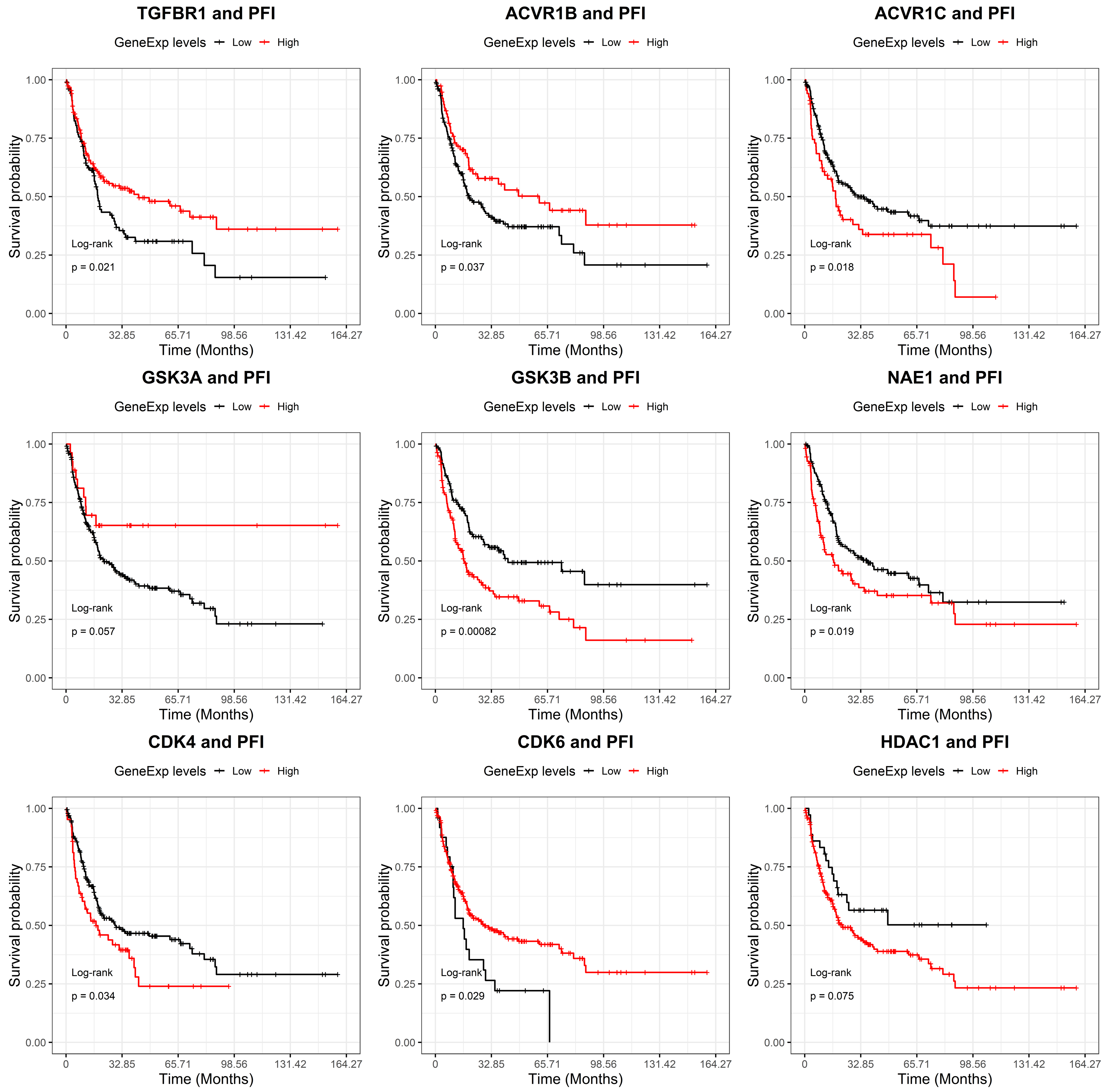
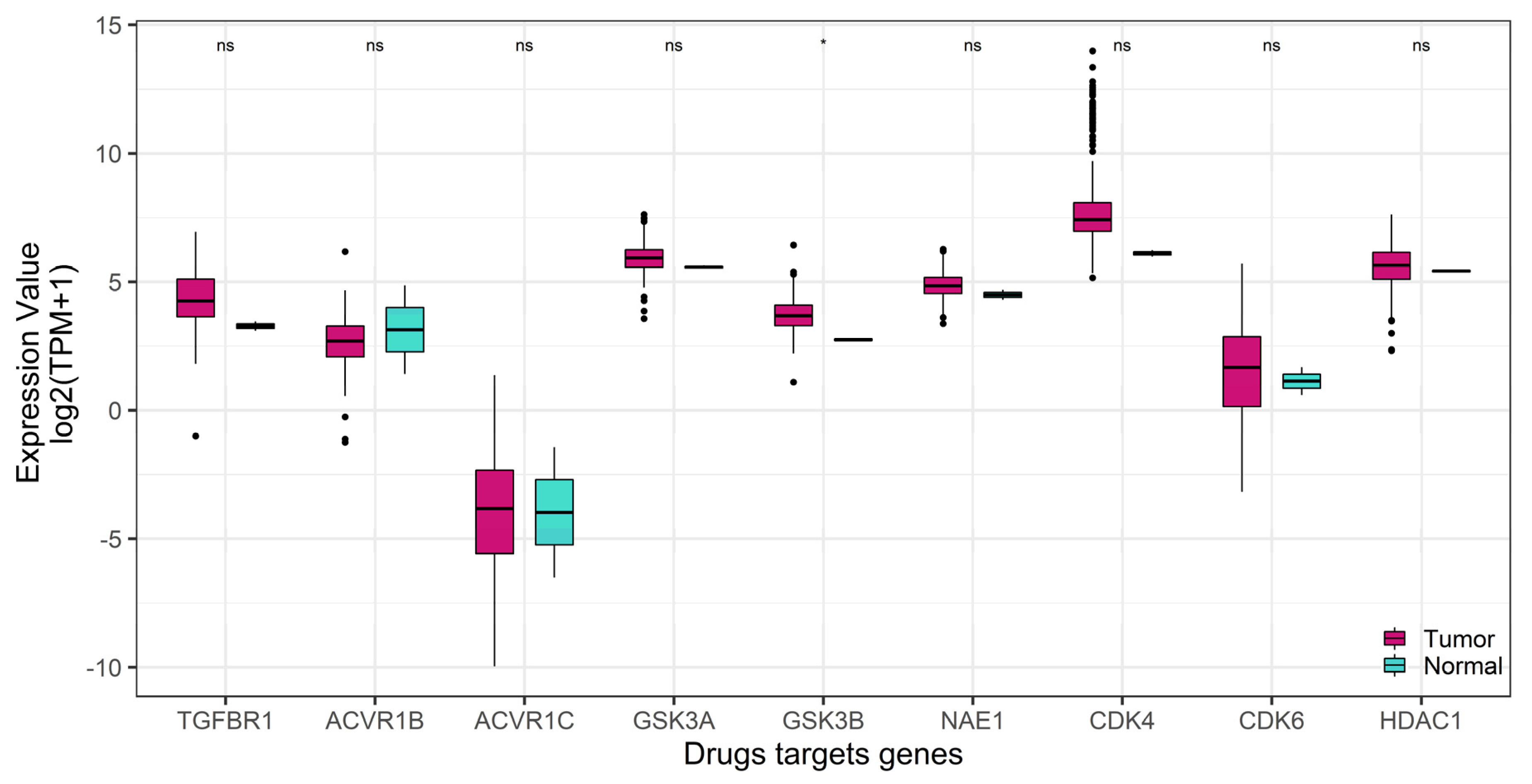
2.2. GSK-3β Was Associated with Poor Outcome and Was Overexpressed in Tumor versus Normal Sarcoma Samples
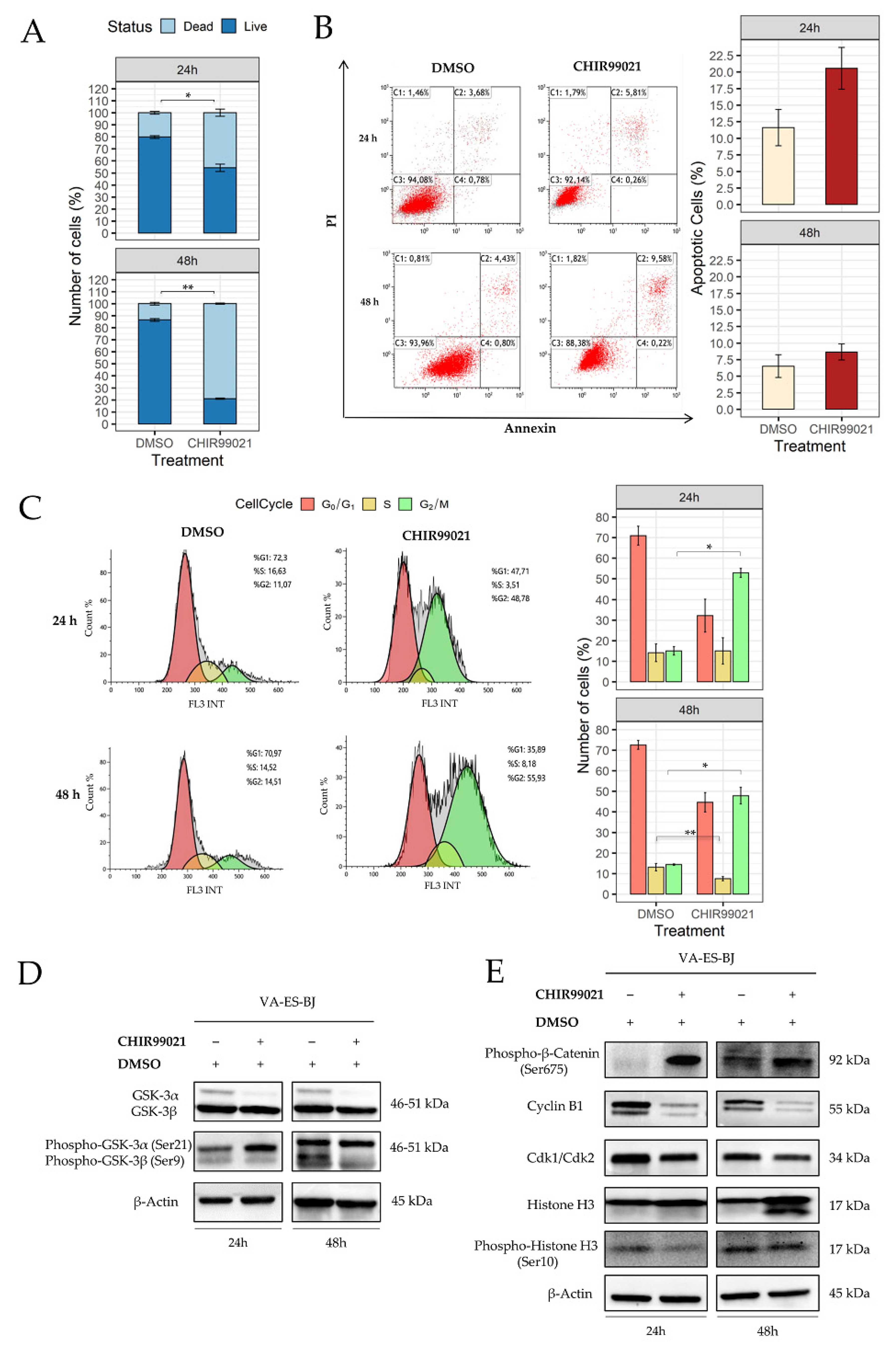
2.3. Inhibition of GSK-3β Induces an Anti-Proliferative Effect on Epithelioid Sarcoma Cells
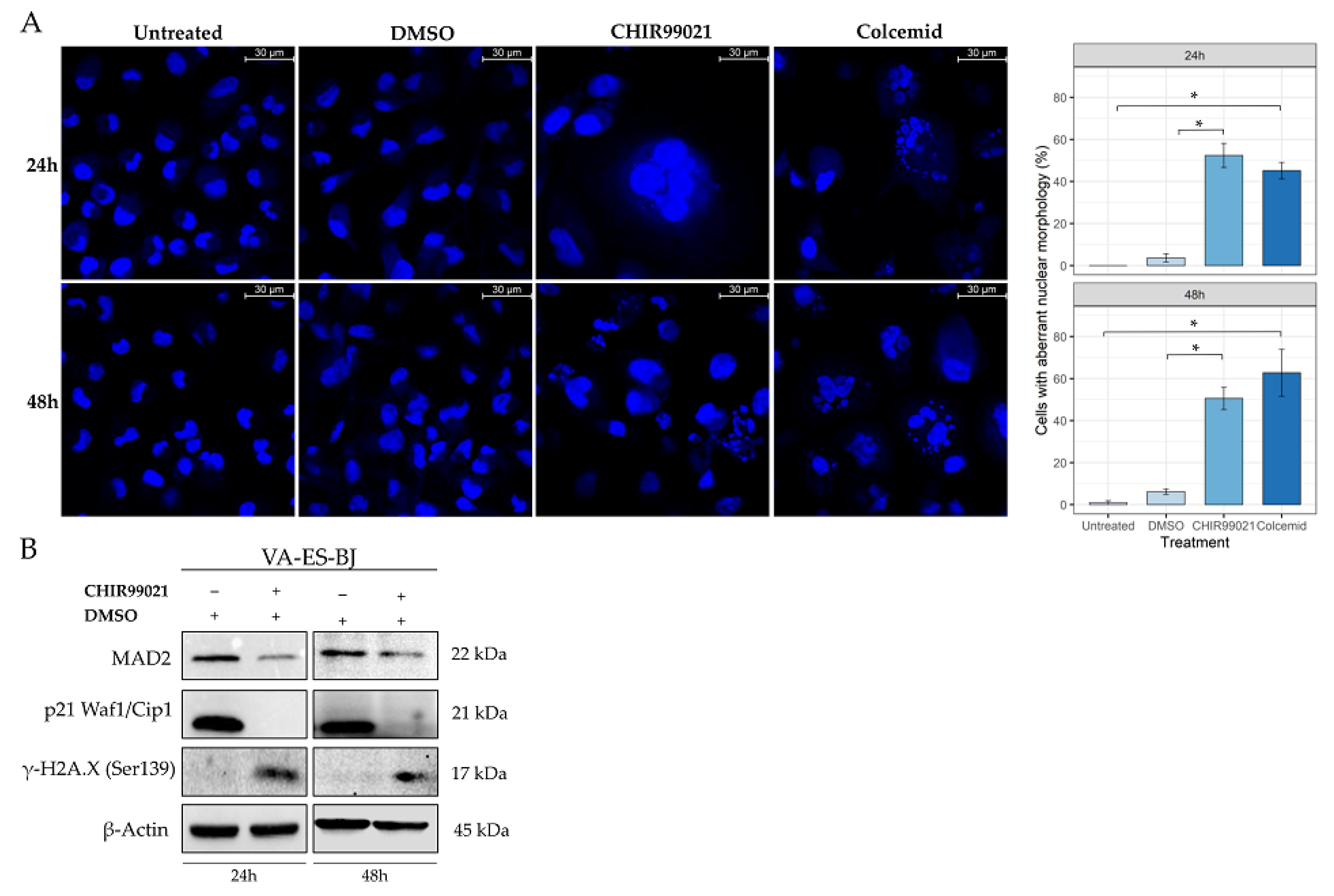
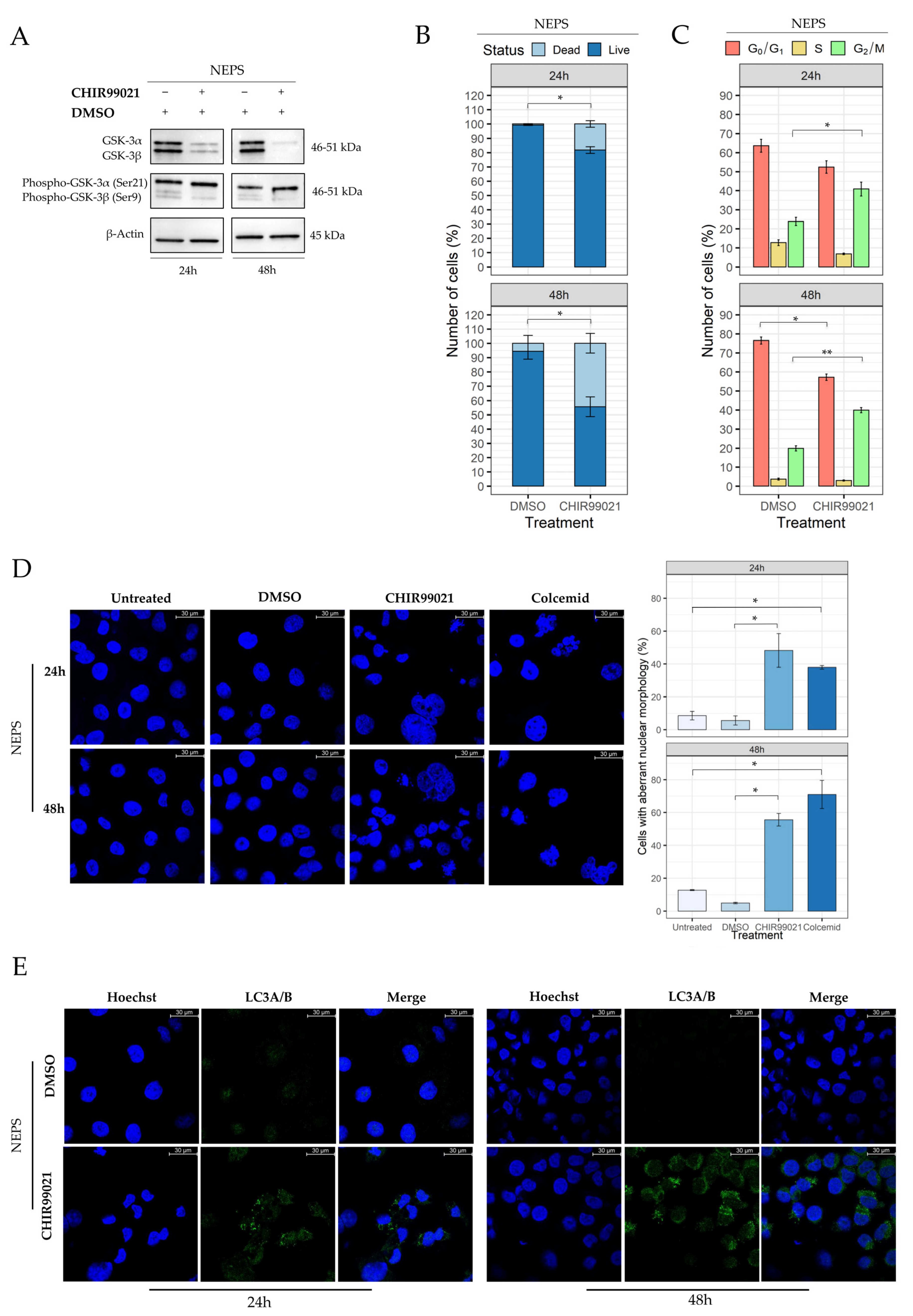
2.4. GSK-3β Inhibition Induces Cell Morphology Aberration Resembling Mitotic Catastrophe
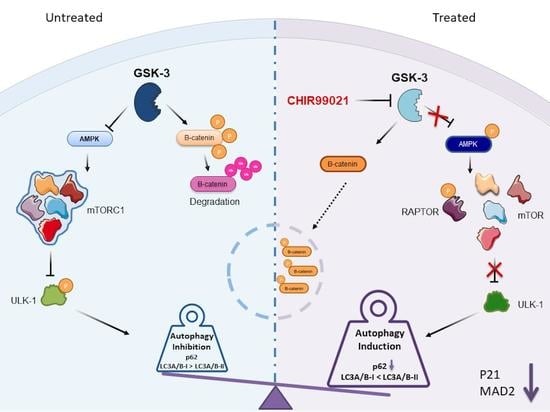
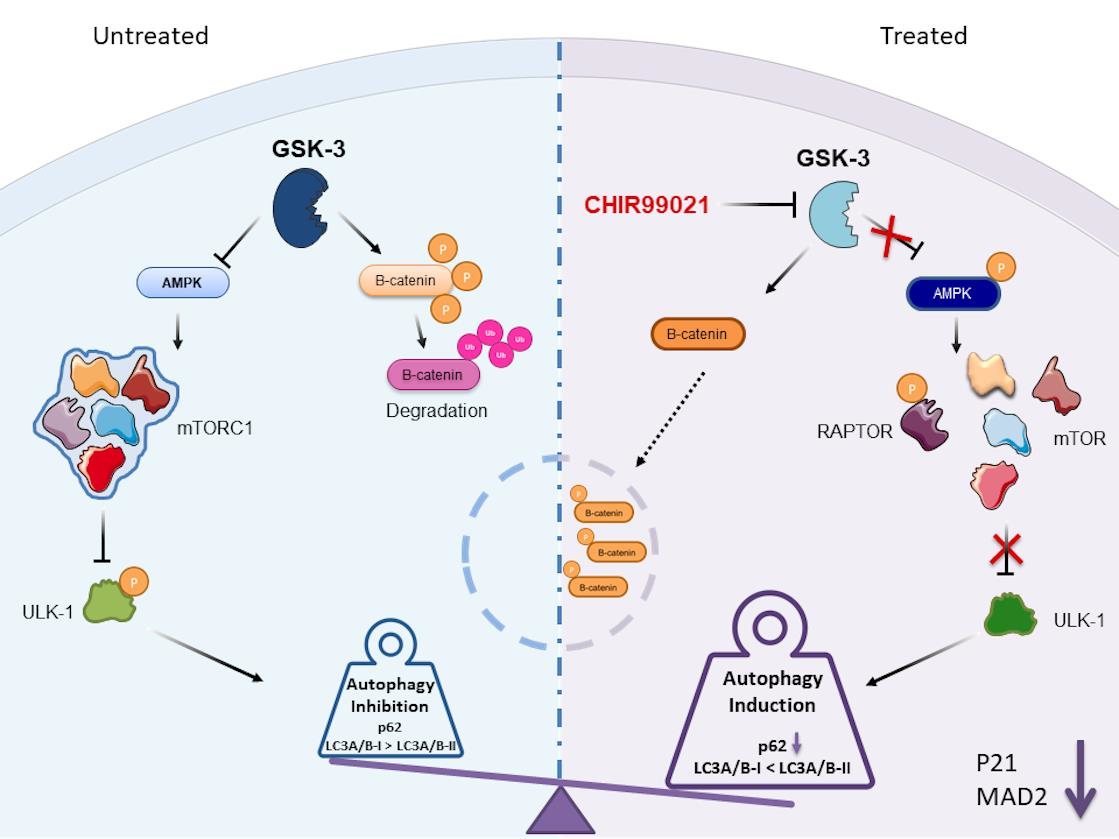
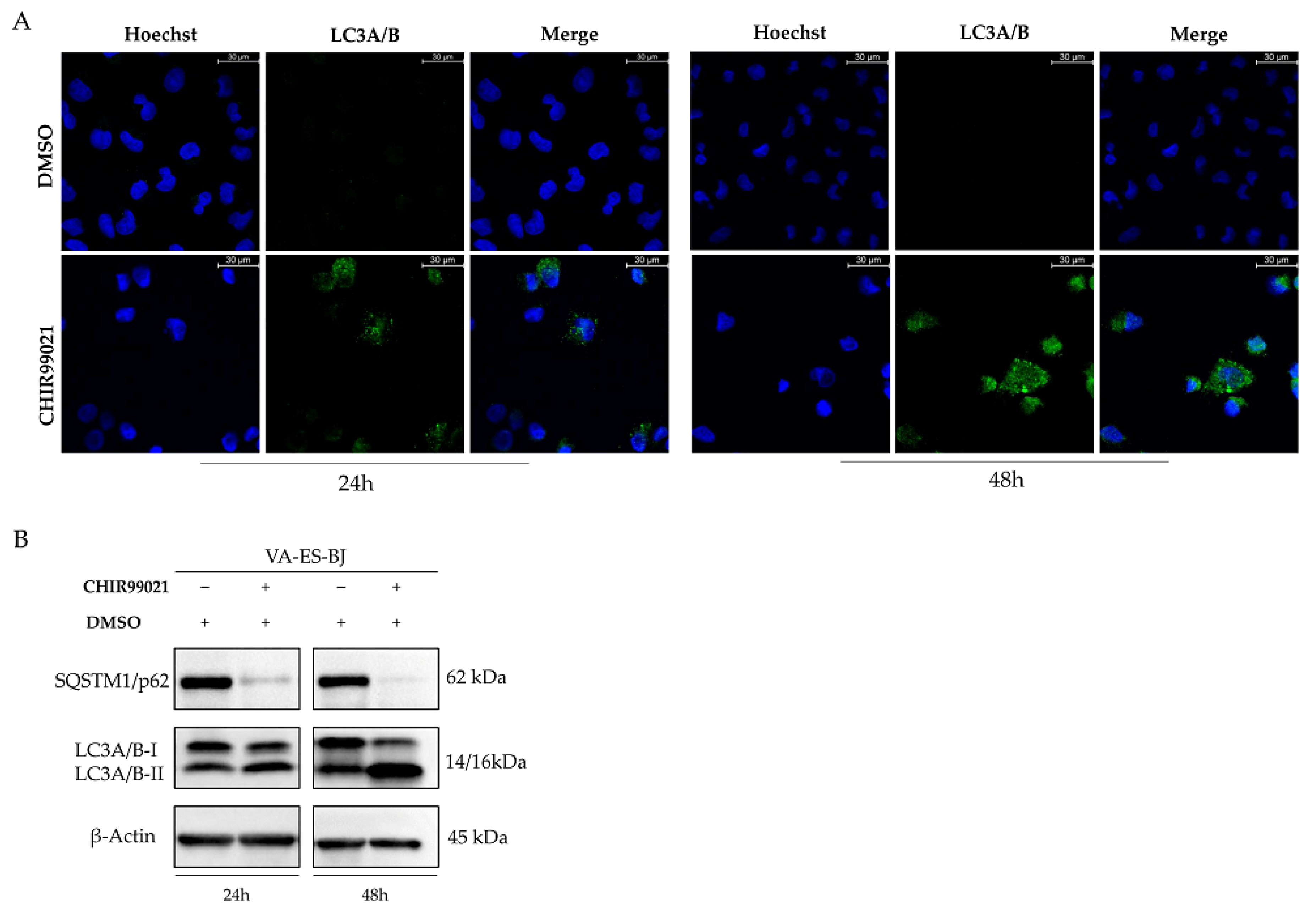
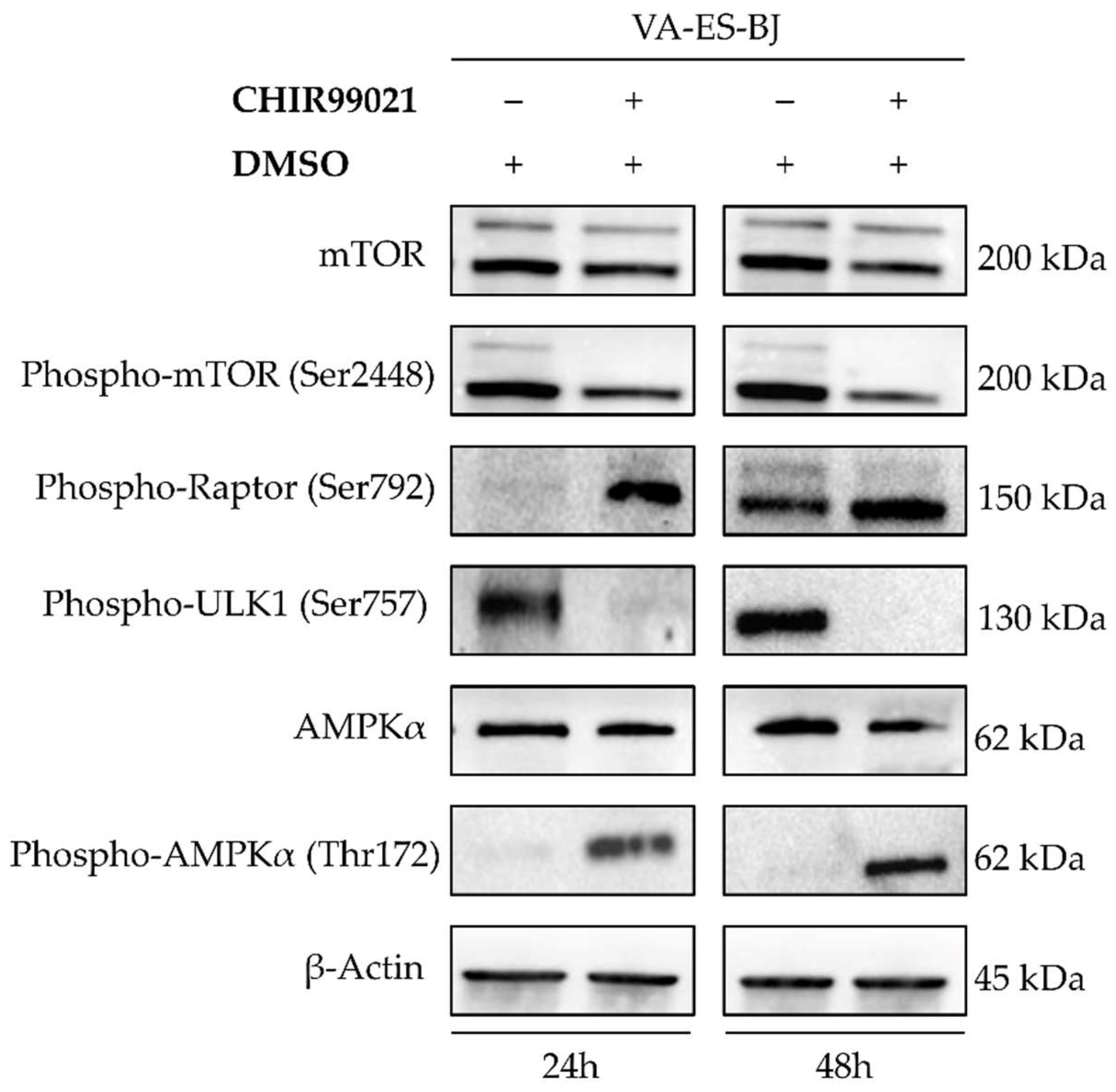
2.5. Autophagy Response as the Cell Death Mechanism Mediated by GSK3-β Inhibition
2.6. Evaluation of CHIR99021 Effects on the NEPS Cell Line
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Maintenance
4.2. Drugs Data Analysis: Inhibitors Selection
4.3. Cell Viability Assays
4.4. Cell Apoptosis Assay
4.5. Cell Cycle Assay
4.6. Western Blot Analysis
4.7. Immunofluorescence and Confocal Microscopy Analysis
4.8. Bioinformatics Analyses on Drugs Target Genes Prognostic Value and Differential Expression
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Enzinger, F.M. Epitheloid Sarcoma. A Sarcoma Simulating a Granuloma or a Carcinoma. Cancer 1970, 26, 1029–1041. [Google Scholar] [CrossRef]
- de Visscher, S.A.H.J.; van Ginkel, R.J.; Wobbes, T.; Veth, R.P.H.; Ten Heuvel, S.E.; Suurmeijer, A.J.H.; Hoekstra, H.J. Epithelioid Sarcoma: Still an Only Surgically Curable Disease. Cancer 2006, 107, 606–612. [Google Scholar] [CrossRef]
- Fisher, C. Epithelioid Sarcoma of Enzinger. Adv. Anat. Pathol. 2006, 13, 114–121. [Google Scholar] [CrossRef]
- Needs, T.; Fillman, E.P. Cancer, Epithelioid Sarcoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Guillou, L.; Wadden, C.; Coindre, J.M.; Krausz, T.; Fletcher, C.D. “Proximal-Type” Epithelioid Sarcoma, a Distinctive Aggressive Neoplasm Showing Rhabdoid Features. Clinicopathologic, Immunohistochemical, and Ultrastructural Study of a Series. Am. J. Surg. Pathol. 1997, 21, 130–146. [Google Scholar] [CrossRef]
- Chase, D.R.; Enzinger, F.M. Epithelioid Sarcoma. Diagnosis, Prognostic Indicators, and Treatment. Am. J. Surg. Pathol. 1985, 9, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Wilson, K.S.; Meehan, J.; Schultz, R.A.; Gomez, A.M. “Proximal-Type” and Classic Epithelioid Sarcomas Represent a Clinicopathologic Continuum: Case Report. Pediatr. Dev. Pathol. 2005, 8, 105–114. [Google Scholar] [CrossRef]
- Hasegawa, T.; Matsuno, Y.; Shimoda, T.; Umeda, T.; Yokoyama, R.; Hirohashi, S. Proximal-Type Epithelioid Sarcoma: A Clinicopathologic Study of 20 Cases. Mod. Pathol. 2001, 14, 655–663. [Google Scholar] [CrossRef]
- Siontis, B.L.; Chugh, R.; Schuetze, S.M. The Potential of Emerging Therapeutics for Epithelioid Sarcoma. Expert Opin. Orphan Drugs 2017, 5, 983–989. [Google Scholar] [CrossRef]
- Wolf, P.S.; Flum, D.R.; Tanas, M.R.; Rubin, B.P.; Mann, G.N. Epithelioid Sarcoma: The University of Washington Experience. Am. J. Surg. 2008, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Shimm, D.S.; Suit, H.D. Radiation Therapy of Epithelioid Sarcoma. Cancer 1983, 52, 1022–1025. [Google Scholar] [CrossRef]
- Callister, M.D.; Ballo, M.T.; Pisters, P.W.; Patel, S.R.; Feig, B.W.; Pollock, R.E.; Benjamin, R.S.; Zagars, G.K. Epithelioid Sarcoma: Results of Conservative Surgery and Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 384–391. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Sobczuk, P.; Kostrzanowski, M.; Spalek, M.; Chojnacka, M.; Szumera-Cieckiewicz, A.; Rutkowski, P. Epithelioid Sarcoma-From Genetics to Clinical Practice. Cancers 2020, 12, 2112. [Google Scholar] [CrossRef] [PubMed]
- Frezza, A.M.; Jones, R.L.; Lo Vullo, S.; Asano, N.; Lucibello, F.; Ben-Ami, E.; Ratan, R.; Teterycz, P.; Boye, K.; Brahmi, M.; et al. Anthracycline, Gemcitabine, and Pazopanib in Epithelioid Sarcoma: A Multi-Institutional Case Series. JAMA Oncol. 2018, 4, e180219. [Google Scholar] [CrossRef]
- Touati, N.; Schöffski, P.; Litière, S.; Judson, I.; Sleijfer, S.; van der Graaf, W.T.; Italiano, A.; Isambert, N.; Gil, T.; Blay, J.Y.; et al. European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Experience with Advanced/Metastatic Epithelioid Sarcoma Patients Treated in Prospective Trials: Clinical Profile and Response to Systemic Therapy. Clin. Oncol. 2018, 30, 448–454. [Google Scholar] [CrossRef]
- Jones, R.L.; Constantinidou, A.; Olmos, D.; Thway, K.; Fisher, C.; Al-Muderis, O.; Scurr, M.; Judson, I.R. Role of Palliative Chemotherapy in Advanced Epithelioid Sarcoma. Am. J. Clin. Oncol. 2012, 35, 351–357. [Google Scholar] [CrossRef]
- Tariq, Z.; Ghose, A.; Bawany, M.Z.; Saeed, B.; Mohamed, I.; Harmon, D. A Case Report of Complete Remission of Pulmonary Metastases from Epithelioid Sarcoma to Navelbine Chemotherapy. Am. J. Ther. 2012, 19, e95–e97. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Hoshi, M.; Oebisu, N.; Shimatani, A.; Takada, N.; Nakamura, H. Promising Effects of Eribulin for Cystic Lung Metastases of Epithelioid Sarcoma: A Case Report. Anticancer Drugs 2018, 29, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Pink, D.; Richter, S.; Gerdes, S.; Andreou, D.; Tunn, P.-U.; Busemann, C.; Ehninger, G.; Reichardt, P.; Schuler, M.K. Gemcitabine and Docetaxel for Epithelioid Sarcoma: Results from a Retrospective, Multi-Institutional Analysis. Oncology 2014, 87, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Schoffski, P.; Jones, R.; Agulnik, M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Italiano, A.; Demetri, G.D.; Cote, G.M.; et al. Safety and Efficacy of Tazemetostat, a First-in-Class EZH2 Inhibitor, in Patients (Pts) with Epithelioid Sarcoma (ES) (NCT02601950). JCO 2019, 37, 11003. [Google Scholar] [CrossRef]
- Elsamna, S.T.; Amer, K.; Elkattawy, O.; Beebe, K.S. Epithelioid Sarcoma: Half a Century Later. Acta Oncol. 2020, 59, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Nisar, S.; Hashem, S.; Macha, M.A.; Yadav, S.K.; Muralitharan, S.; Therachiyil, L.; Sageena, G.; Al-Naemi, H.; Haris, M.; Bhat, A.A. Exploring Dysregulated Signaling Pathways in Cancer. Curr. Pharm. Des. 2020, 26, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Sakharpe, A.; Lahat, G.; Gulamhusein, T.; Liu, P.; Bolshakov, S.; Nguyen, T.; Zhang, P.; Belousov, R.; Young, E.; Xie, X.; et al. Epithelioid Sarcoma and Unclassified Sarcoma with Epithelioid Features: Clinicopathological Variables, Molecular Markers, and a New Experimental Model. Oncologist 2011, 16, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Yasui, H.; Outani, H.; Wakamatsu, T.; Hamada, K.; Nakai, T.; Yamada, S.; Myoui, A.; Araki, N.; Ueda, T.; et al. Combined Targeting of MTOR and C-MET Signaling Pathways for Effective Management of Epithelioid Sarcoma. Mol. Cancer 2014, 13, 185. [Google Scholar] [CrossRef]
- Xie, X.; Ghadimi, M.P.H.; Young, E.D.; Belousov, R.; Zhu, Q.-S.; Liu, J.; Lopez, G.; Colombo, C.; Peng, T.; Reynoso, D.; et al. Combining EGFR and MTOR Blockade for the Treatment of Epithelioid Sarcoma. Clin. Cancer Res. 2011, 17, 5901–5912. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y. Oncogenic Roles of SMARCB1/INI1 and Its Deficient Tumors. Cancer Sci. 2017, 108, 547–552. [Google Scholar] [CrossRef]
- Mora-Blanco, E.L.; Mishina, Y.; Tillman, E.J.; Cho, Y.J.; Thom, C.S.; Pomeroy, S.L.; Shao, W.; Roberts, C.W.M. Activation of β-Catenin/TCF Targets Following Loss of the Tumor Suppressor SNF5. Oncogene 2014, 33, 933–938. [Google Scholar] [CrossRef]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the Tumor Suppressor Snf5 Leads to Aberrant Activation of the Hedgehog-Gli Pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Lopez, G.; Song, Y.; Lam, R.; Ruder, D.; Creighton, C.J.; Bid, H.K.; Bill, K.L.; Bolshakov, S.; Zhang, X.; Lev, D.; et al. HDAC Inhibition for the Treatment of Epithelioid Sarcoma: Novel Cross Talk between Epigenetic Components. Mol. Cancer Res. 2016, 14, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Noujaim, J.; Thway, K.; Bajwa, Z.; Bajwa, A.; Maki, R.G.; Jones, R.L.; Keller, C. Epithelioid Sarcoma: Opportunities for Biology-Driven Targeted Therapy. Front. Oncol. 2015, 5, 186. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective Glycogen Synthase Kinase 3 Inhibitors Potentiate Insulin Activation of Glucose Transport and Utilization in Vitro and in Vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Hino, S.; Tanji, C.; Nakayama, K.I.; Kikuchi, A. Phosphorylation of Beta-Catenin by Cyclic AMP-Dependent Protein Kinase Stabilizes Beta-Catenin through Inhibition of Its Ubiquitination. Mol. Cell. Biol. 2005, 25, 9063–9072. [Google Scholar] [CrossRef]
- Olmeda, D.; Castel, S.; Vilaró, S.; Cano, A. Beta-Catenin Regulation during the Cell Cycle: Implications in G2/M and Apoptosis. Mol. Biol. Cell 2003, 14, 2844–2860. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mandal, C.; Sangwan, R.; Mandal, C. Coupling G2/M Arrest to the Wnt/β-Catenin Pathway Restrains Pancreatic Adenocarcinoma. Endocr. Relat. Cancer 2014, 21, 113–125. [Google Scholar] [CrossRef]
- Sorokina, I.V.; Denisenko, T.V.; Imreh, G.; Tyurin-Kuzmin, P.A.; Kaminskyy, V.O.; Gogvadze, V.; Zhivotovsky, B. Involvement of Autophagy in the Outcome of Mitotic Catastrophe. Sci. Rep. 2017, 7, 14571. [Google Scholar] [CrossRef]
- Miyashita, K.; Nakada, M.; Shakoori, A.; Ishigaki, Y.; Shimasaki, T.; Motoo, Y.; Kawakami, K.; Minamoto, T. An Emerging Strategy for Cancer Treatment Targeting Aberrant Glycogen Synthase Kinase 3 Beta. Anticancer Agents Med. Chem. 2009, 9, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen Synthase Kinases: Moonlighting Proteins with Theranostic Potential in Cancer. Semin. Cancer Biol. 2019, 56, 25–36. [Google Scholar] [CrossRef]
- Mai, W.; Kawakami, K.; Shakoori, A.; Kyo, S.; Miyashita, K.; Yokoi, K.; Jin, M.; Shimasaki, T.; Motoo, Y.; Minamoto, T. Deregulated GSK3β Sustains Gastrointestinal Cancer Cells Survival by Modulating Human Telomerase Reverse Transcriptase and Telomerase. Clin. Cancer Res. 2009, 15, 6810–6819. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Yamamoto, N.; Domoto, T.; Bolidong, D.; Hayashi, K.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Inatani, H.; Aoki, Y.; et al. Glycogen Synthase Kinase 3β as a Potential Therapeutic Target in Synovial Sarcoma and Fibrosarcoma. Cancer Sci. 2020, 111, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Cerulo, L.; Tagliaferri, D.; Marotta, P.; Zoppoli, P.; Russo, F.; Mazio, C.; DeFelice, M.; Ceccarelli, M.; Falco, G. Identification of a Novel Gene Signature of ES Cells Self-Renewal Fluctuation through System-Wide Analysis. PLoS ONE 2014, 9, e83235. [Google Scholar] [CrossRef]
- Shimozaki, S.; Yamamoto, N.; Domoto, T.; Nishida, H.; Hayashi, K.; Kimura, H.; Takeuchi, A.; Miwa, S.; Igarashi, K.; Kato, T.; et al. Efficacy of Glycogen Synthase Kinase-3β Targeting against Osteosarcoma via Activation of β-Catenin. Oncotarget 2016, 7, 77038–77051. [Google Scholar] [CrossRef][Green Version]
- Tang, Q.-L.; Xie, X.-B.; Wang, J.; Chen, Q.; Han, A.-J.; Zou, C.-Y.; Yin, J.-Q.; Liu, D.-W.; Liang, Y.; Zhao, Z.-Q.; et al. Glycogen Synthase Kinase-3β, NF-ΚB Signaling, and Tumorigenesis of Human Osteosarcoma. J. Natl. Cancer Inst. 2012, 104, 749–763. [Google Scholar] [CrossRef]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-Cyclin B1 Initiates Its Cdc20-Dependent Destruction during Mitotic Arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Ohi, R.; Gould, K.L. Regulating the Onset of Mitosis. Curr. Opin. Cell Biol. 1999, 11, 267–273. [Google Scholar] [CrossRef]
- Liokatis, S.; Stützer, A.; Elsässer, S.J.; Theillet, F.-X.; Klingberg, R.; van Rossum, B.; Schwarzer, D.; Allis, C.D.; Fischle, W.; Selenko, P. Phosphorylation of Histone H3 Ser10 Establishes a Hierarchy for Subsequent Intramolecular Modification Events. Nat. Struct. Mol. Biol. 2012, 19, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell Death by Mitotic Catastrophe: A Molecular Definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Jha, M.N.; Bamburg, J.R.; Bedford, J.S. Cell Cycle Arrest by Colcemid Differs in Human Normal and Tumor Cells. Cancer Res. 1994, 54, 5011–5015. [Google Scholar]
- Rieder, C.L.; Palazzo, R.E. Colcemid and the Mitotic Cycle. J. Cell Sci. 1992, 102 Pt 3, 387–392. [Google Scholar] [CrossRef]
- Dash, B.C.; El-Deiry, W.S. Phosphorylation of P21 in G2/M Promotes Cyclin B-Cdc2 Kinase Activity. Mol. Cell. Biol. 2005, 25, 3364–3387. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Wang, F.-F. Mechanisms Underlying the Pro-Survival Pathway of P53 in Suppressing Mitotic Death Induced by Adriamycin. Cell. Signal. 2008, 20, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Ishioka, C. Inhibition of Glycogen Synthase Kinase-3 Beta Induces Apoptosis and Mitotic Catastrophe by Disrupting Centrosome Regulation in Cancer Cells. Sci. Rep. 2015, 5, 13249. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy in Higher Eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. In Autophagy in Mammalian Systems, Part B; Methods in Enzymology; Elsevier: San Diego, CA, USA, 2009; Volume 452, pp. 181–197. ISBN 9780123745477. [Google Scholar]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK Catabolic Action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of MTOR Complex 1 (MTORC1) by Raptor Ser863 and Multisite Phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Melick, C.H.; Jewell, J.L. Regulation of MTORC1 by Upstream Stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Wong, P.-M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 Complex: Sensing Nutrient Signals for Autophagy Activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Mamais, A.; Dihanich, S.; Soutar, M.P.M.; Plun-Favreau, H.; Bandopadhyay, R.; Abeti, R.; Giunti, P.; Hardy, J.; Cookson, R.M.; et al. MTOR Independent Alteration in ULK1 Ser758 Phosphorylation Following Chronic LRRK2 Kinase Inhibition. Biosci. Rep. 2018, 38, BSR20171669. [Google Scholar] [CrossRef]
- Hoshino, M.; Kawashima, H.; Ogose, A.; Kudo, N.; Ariizumi, T.; Hotta, T.; Umezu, H.; Hatano, H.; Morita, T.; Nishio, J.; et al. Serum CA 125 expression as a tumor marker for diagnosis and monitoring the clinical course of epithelioid sarcoma. J. Cancer Res. Clin. Oncol. 2010, 136, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Hothorn, T.; Lausen, B. On the Exact Distribution of Maximally Selected Rank Statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Therneau, T.M. A Package for Survival Analysis in R. 2021. R Package Version 3.2-13. Available online: https://CRAN.R-project.org/package=survival (accessed on 1 September 2021).
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, L.; Shnyder, S.D.; Cooper, P.A.; Cross, S.J.; Wakefield, J.G.; Pardo, O.E.; Seckl, M.J.; Tavaré, J.M. Tumor growth suppression using a combination of taxol-based therapy and GSK3 inhibition in non-small cell lung cancer. PLoS ONE 2019, 14, e0214610. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Targets | Z Score |
|---|---|---|
| SB505124 | TGFBR1, ACVR1B, ACVR1C | −4.42779 |
| CHIR99021 | GSK-3α, GSK-3β | −3.67765 |
| Pevonedistat | NAE1 | −2.39559 |
| palbociclib | CDK4, CDK6 | −1.36358 |
| parthenolide | HDAC1 | −0.92757 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russi, S.; Sgambato, A.; Bochicchio, A.M.; Zoppoli, P.; Aieta, M.; Capobianco, A.M.L.; Ruggieri, V.; Zifarone, E.; Falco, G.; Laurino, S. CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy. Int. J. Mol. Sci. 2021, 22, 11147. https://doi.org/10.3390/ijms222011147
Russi S, Sgambato A, Bochicchio AM, Zoppoli P, Aieta M, Capobianco AML, Ruggieri V, Zifarone E, Falco G, Laurino S. CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy. International Journal of Molecular Sciences. 2021; 22(20):11147. https://doi.org/10.3390/ijms222011147
Chicago/Turabian StyleRussi, Sabino, Alessandro Sgambato, Anna Maria Bochicchio, Pietro Zoppoli, Michele Aieta, Alba Maria Lucia Capobianco, Vitalba Ruggieri, Emanuela Zifarone, Geppino Falco, and Simona Laurino. 2021. "CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy" International Journal of Molecular Sciences 22, no. 20: 11147. https://doi.org/10.3390/ijms222011147
APA StyleRussi, S., Sgambato, A., Bochicchio, A. M., Zoppoli, P., Aieta, M., Capobianco, A. M. L., Ruggieri, V., Zifarone, E., Falco, G., & Laurino, S. (2021). CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy. International Journal of Molecular Sciences, 22(20), 11147. https://doi.org/10.3390/ijms222011147