
Trimethylamine n-Oxide (TMAO) Modulates the Expression of Cardiovascular Disease-Related microRNAs and Their Targets

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
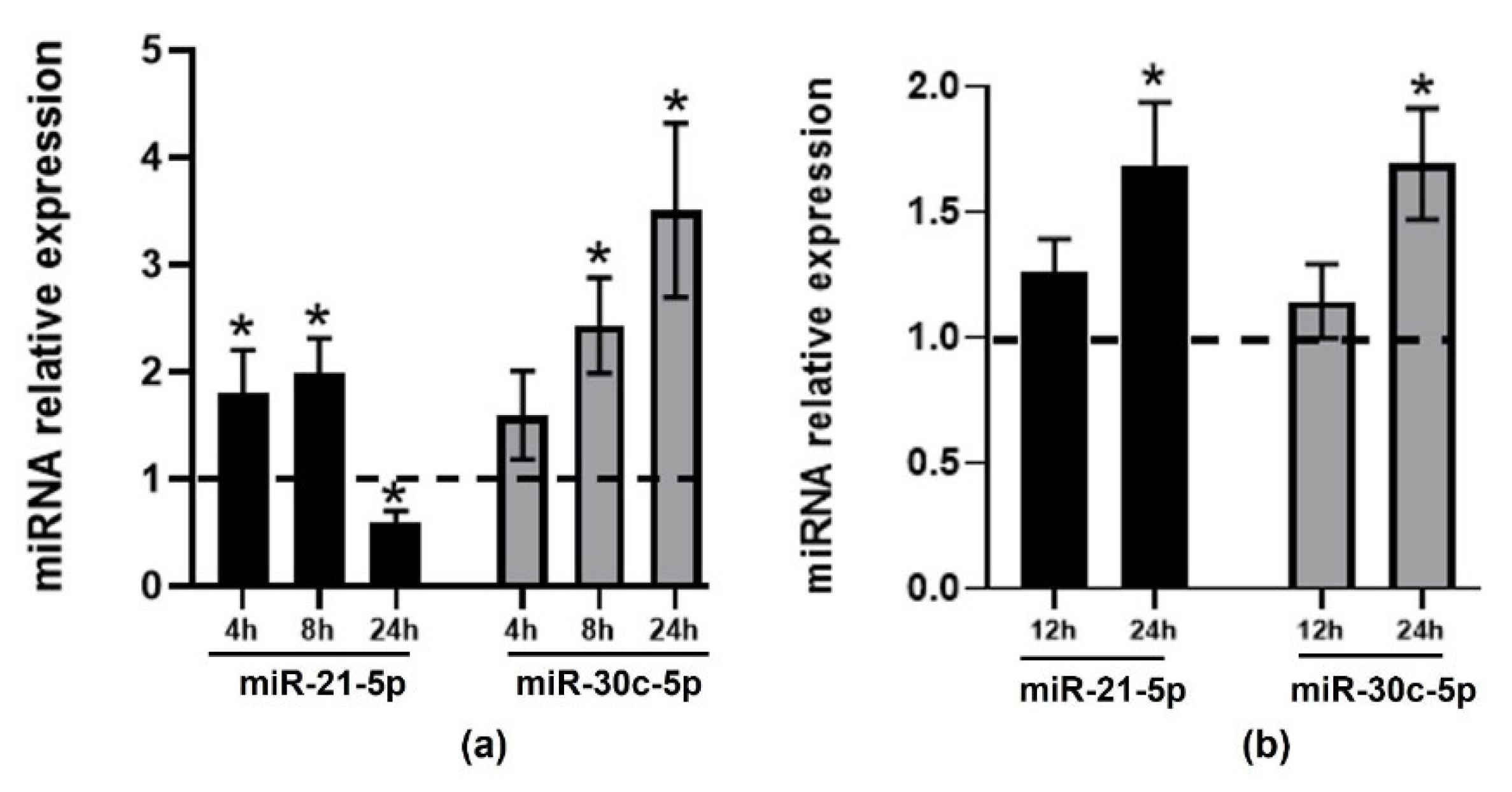
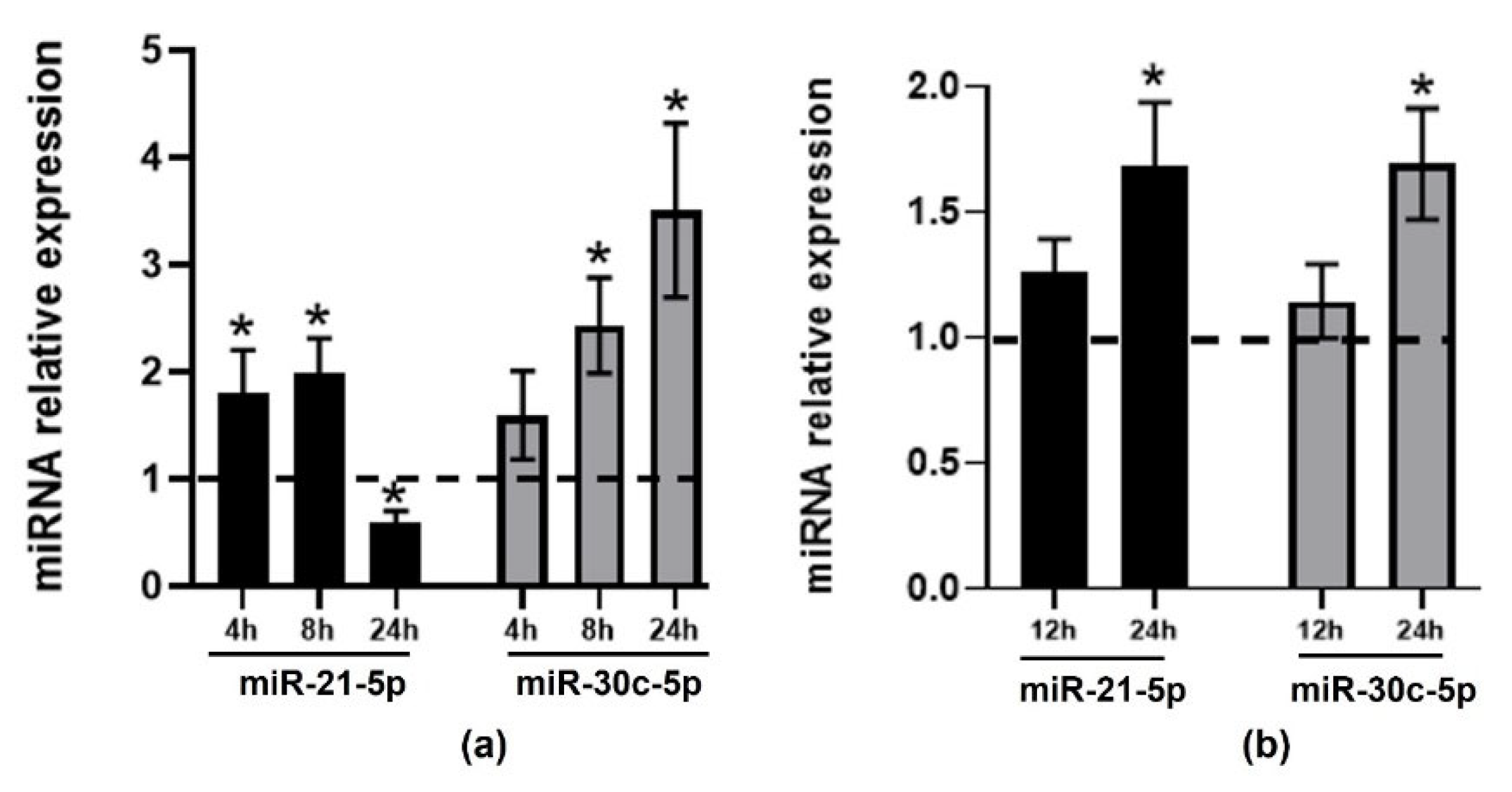
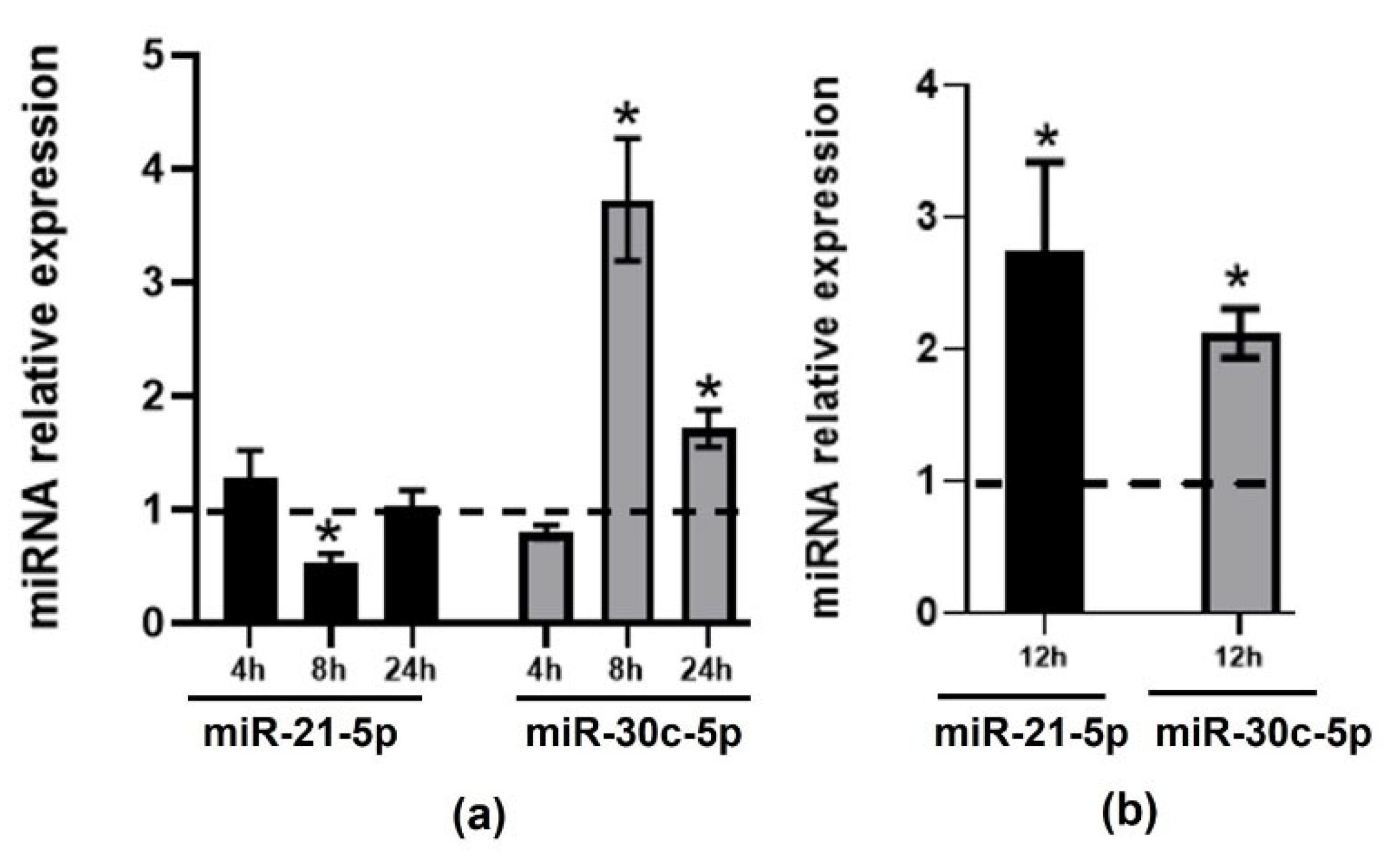
2.1. Effect of TMAO on the Expression of CVD-Related miRNAs in HEPG-2 and THP-1 Cells
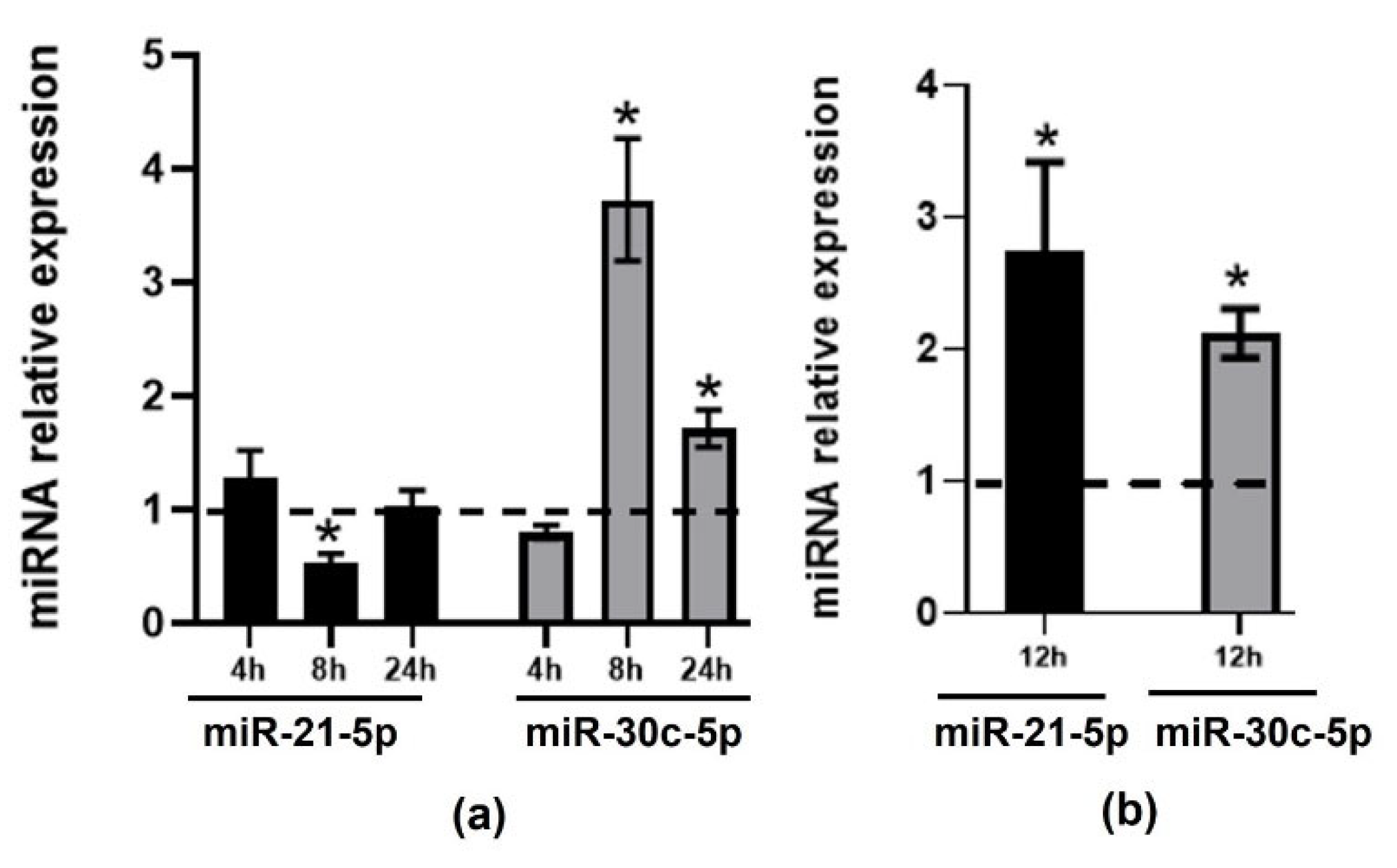
2.2. Validation of Changes in miRNAs Expression in Murine Hepatic Organoids and in Human Primary Macrophages
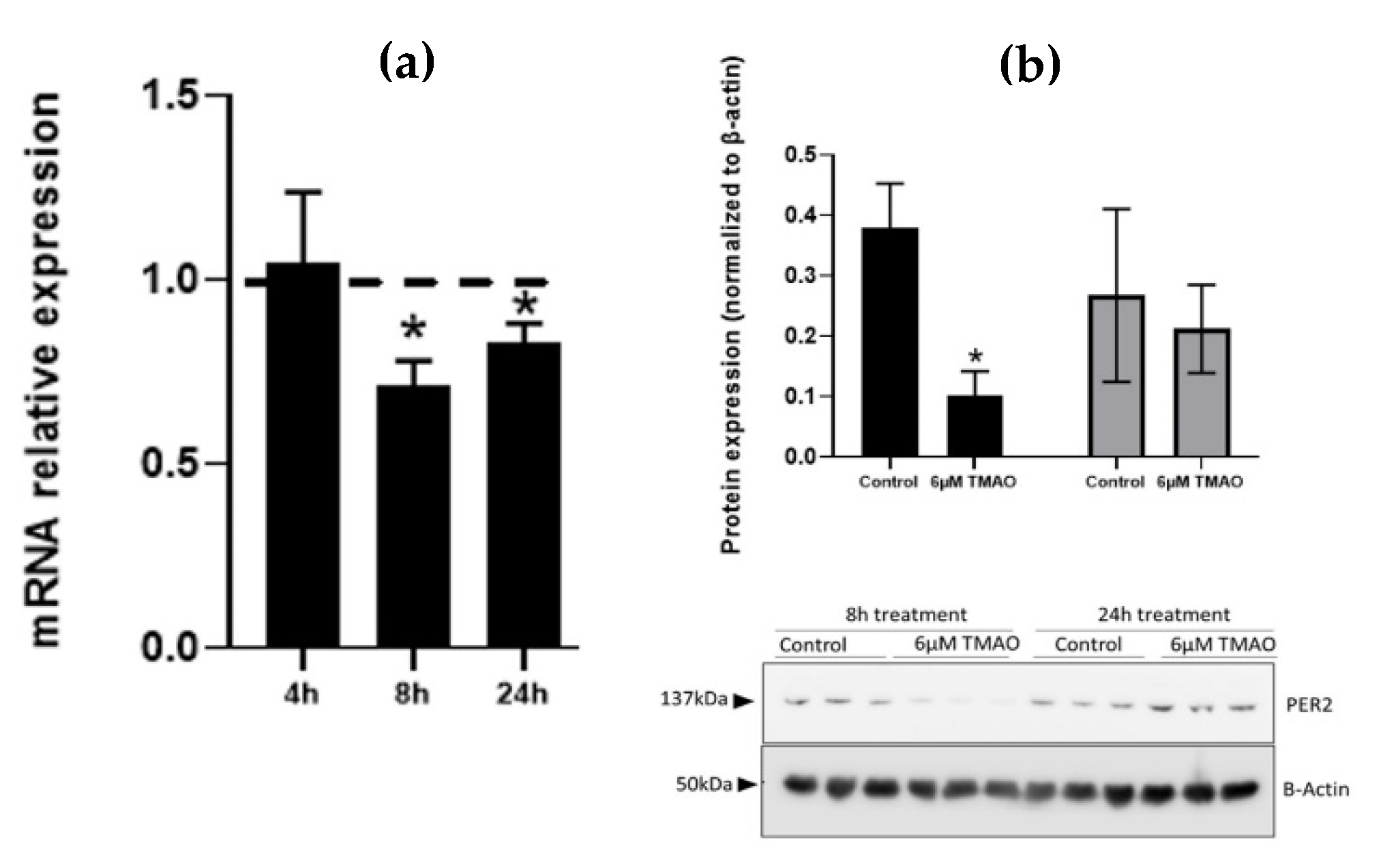
2.3. Effect of TMAO on the Expression of miRNAs Target Genes and Proteins in HEPG-2 and THP-1
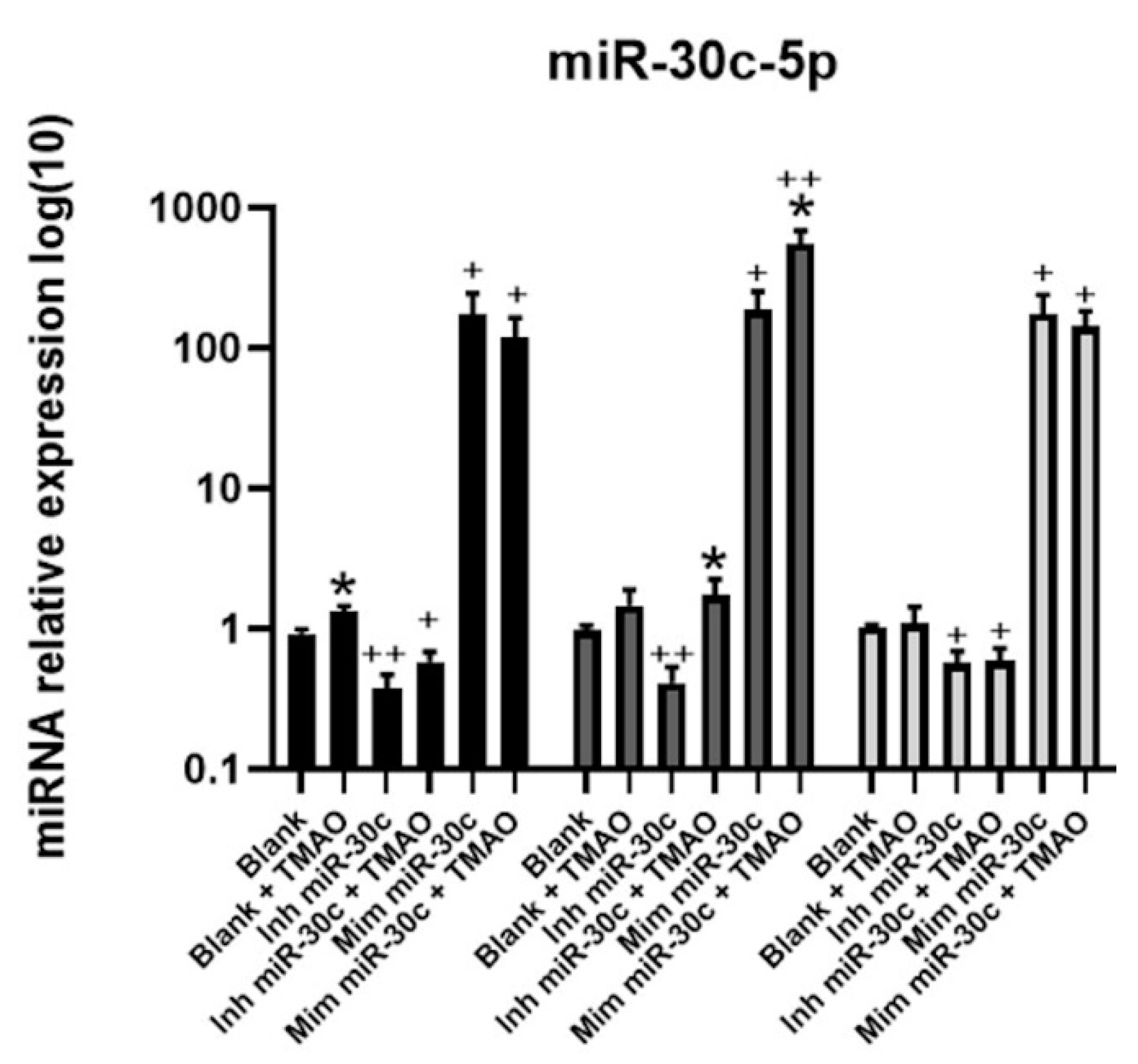
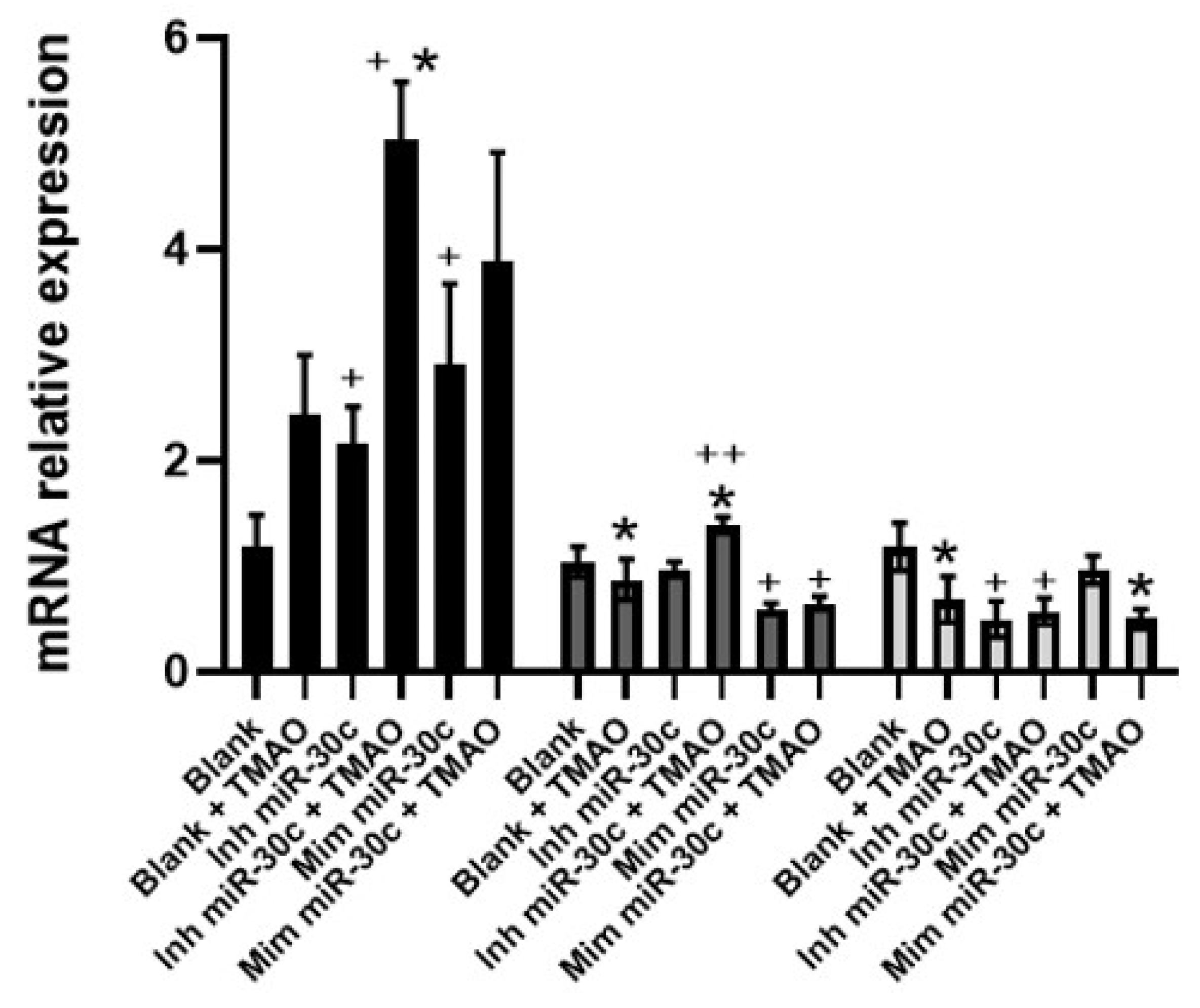
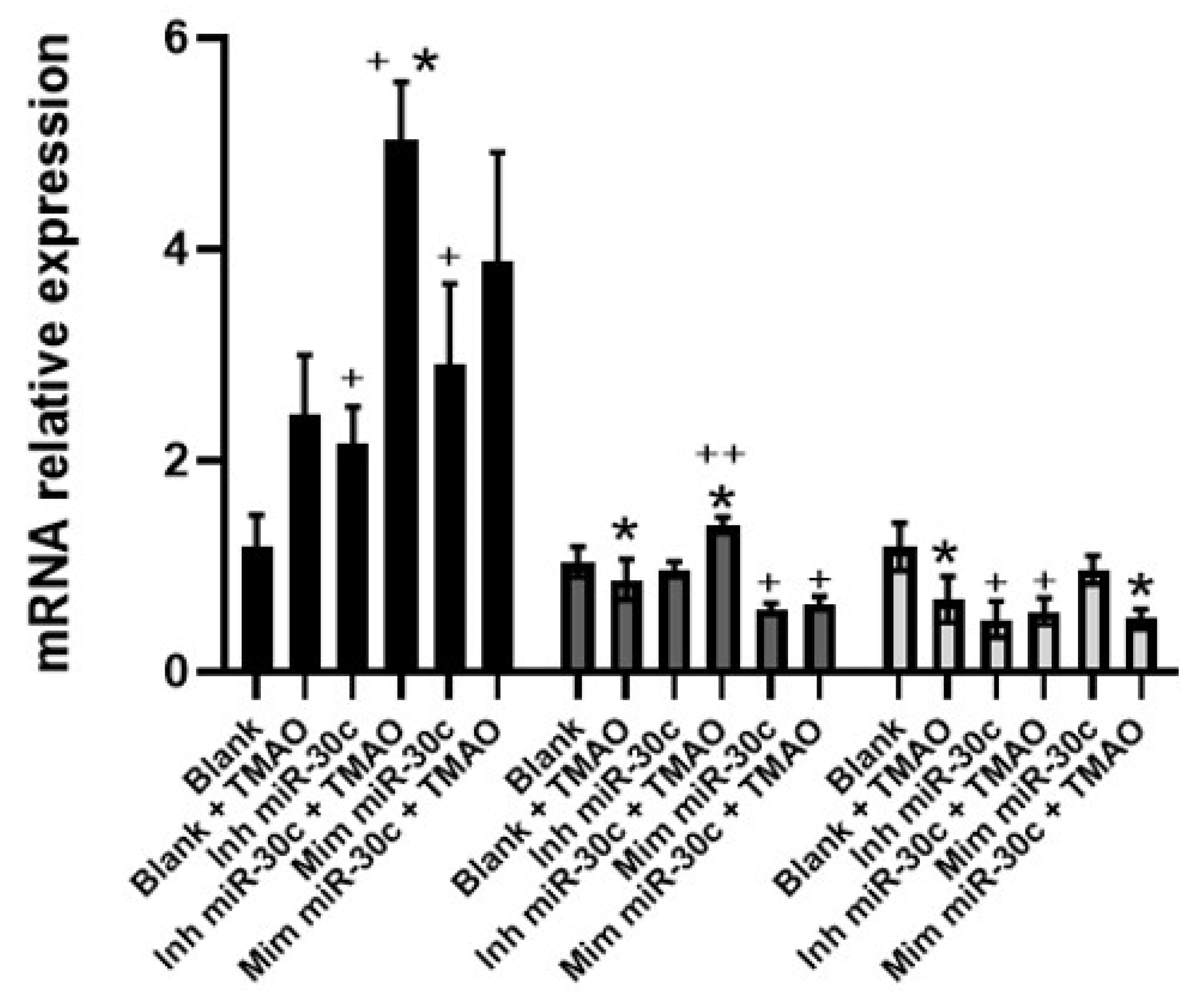
2.4. Effect of Anti-miRNA-30c and Syn-miRNA-30c with TMAO on the Expression of miR-30c-5p Target Genes and Protein Expression in HEPG-2
3. Discussion
4. Materials and Methods
4.1. Immortalized Cell Lines
4.2. Primary Human Macrophages
4.3. Mouse Hepatic Organoids
4.4. Treatments of Cells with TMAO
4.5. miRNA-Enriched RNA Isolation
4.6. miRNA Panel Selection
4.7. Determination of Gene and miRNA Expression
4.8. Functional Analysis and Target Genes Selection
4.9. Protein Quantification and Analysis
4.10. miRNA Inhibitors and Mimics Transfection
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Eurostat. Cardiovascular Diseases Statistics. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php/Cardiovascular_diseases_statistics (accessed on 5 May 2021).
- Escaño-Marín, R.; Pérez-Belmonte, L.M.; Rodríguez de la Cruz, E.; Gómez-Doblas, J.J.; de Teresa-Galván, E.; Jiménez-Navarro, M. Cardiovascular Disease and Gross Domestic Product in Spain: Correlation Analysis by Autonomous Communities. Rev. Esp. Cardiol. 2017, 70, 210–212. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; Kurpad, A.; Narayan, K.M.V. Global Non-Communicable Diseases-The Nutrition Conundrum. Front. Public Health 2018, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.; Sun, Q.; Bernstein, A.M.; Schulze, M.B.; Manson, J.E.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Red meat consumption and mortality: Results from 2 prospective cohort studies. Arch. Intern. Med. 2012, 172, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeraatkar, D.; Han, M.A.; Guyatt, G.H.; Vernooij, R.W.M.; El Dib, R.; Cheung, K.; Milio, K.; Zworth, M.; Bartoszko, J.J.; Valli, C.; et al. Red and Processed Meat Consumption and Risk for All-Cause Mortality and Cardiometabolic Outcomes: A Systematic Review and Meta-analysis of Cohort Studies. Ann. Intern. Med. 2019, 171, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernooij, R.W.M.; Zeraatkar, D.; Han, M.A.; El Dib, R.; Zworth, M.; Milio, K.; Sit, D.; Lee, Y.; Gomaa, H.; Valli, C.; et al. Patterns of Red and Processed Meat Consumption and Risk for Cardiometabolic and Cancer Outcomes: A Systematic Review and Meta-analysis of Cohort Studies. Ann. Intern. Med. 2019, 171, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Romaine, S.P.; Tomaszewski, M.; Condorelli, G.; Samani, N.J. MicroRNAs in cardiovascular disease: An introduction for clinicians. Heart 2015, 101, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Fernandez-Hernando, C.; Moore, K.J. MicroRNAs regulating lipid metabolism in atherogenesis. Thromb. Haemost. 2012, 107, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, H.Y.; Li, Y.; Guo, S.H.; Zhang, L.; Cai, J.H. Corrigendum: Peripheral blood miRNAs as a biomarker for chronic cardiovascular diseases. Sci. Rep. 2015, 5, 8178. [Google Scholar] [CrossRef] [Green Version]
- Canfran-Duque, A.; Rotllan, N.; Zhang, X.; Fernandez-Fuertes, M.; Ramirez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernandez-Hernando, C.; Suarez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef]
- Raitoharju, E.; Lyytikainen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kahonen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef]
- Corrigendum to: miR-30c-5p regulates macrophage-mediated inflammation and pro-atherosclerosis pathways. Cardiovasc. Res. 2018, 114, 1908. [CrossRef] [Green Version]
- Luo, M.; Wang, G.; Xu, C.; Zeng, M.; Lin, F.; Wu, J.; Wan, Q. Circulating miR-30c as a predictive biomarker of type 2 diabetes mellitus with coronary heart disease by regulating PAI-1/VN interactions. Life Sci. 2019, 239, 117092. [Google Scholar] [CrossRef]
- Soh, J.; Iqbal, J.; Queiroz, J.; Fernandez-Hernando, C.; Hussain, M.M. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med. 2013, 19, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Sit, A.; Feinberg, M.W. Role of miR-181 family in regulating vascular inflammation and immunity. Trends Cardiovasc. Med. 2014, 24, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Friedman, J.R. miR-122 regulates hepatic lipid metabolism and tumor suppression. J. Clin. Investig. 2012, 122, 2773–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, K.C.; Landstreet, S.R.; Levin, M.G.; Shoucri, B.M.; Toth, C.L.; Taylor, R.C.; Palmisano, B.T.; Tabet, F.; Cui, H.L.; Rye, K.A.; et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 14518–14523. [Google Scholar] [CrossRef] [Green Version]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramirez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Rayner, K.J.; Suarez, Y.; Davalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernandez-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.H.; Zhang, L.H.; Wijesekara, N.; de Haan, W.; Butland, S.; Bhattacharjee, A.; Hayden, M.R. Regulation of ABCA1 protein expression and function in hepatic and pancreatic islet cells by miR-145. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2724–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the miR-17-92 Cluster of miRNAs in Cancer. Front. Med. 2015, 2, 64. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, J.; Zhang, S.; Yan, S.; Wang, C.; Zhang, X. MiR-30e and miR-92a are related to atherosclerosis by targeting ABCA1. Mol. Med. Rep. 2019, 19, 3298–3304. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Suzuki, K.; Fujii, R.; Kawado, M.; Hashimoto, S.; Watanabe, Y.; Iso, H.; Fujino, Y.; Wakai, K.; Tamakoshi, A. Circulating miR-21, miR-29a, and miR-126 are associated with premature death risk due to cancer and cardiovascular disease: The JACC Study. Sci. Rep. 2021, 11, 5298. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, X.Z.; Li, F.; Ji, Q.R. MiR-128-3p accelerates cardiovascular calcification and insulin resistance through ISL1-dependent Wnt pathway in type 2 diabetes mellitus rats. J. Cell. Physiol. 2019, 234, 4997–5010. [Google Scholar] [CrossRef]
- Devaux, Y.; Dankiewicz, J.; Salgado-Somoza, A.; Stammet, P.; Collignon, O.; Gilje, P.; Gidlof, O.; Zhang, L.; Vausort, M.; Hassager, C.; et al. Association of Circulating MicroRNA-124-3p Levels with Outcomes After Out-of-Hospital Cardiac Arrest: A Substudy of a Randomized Clinical Trial. JAMA Cardiol. 2016, 1, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.K.; Ma, J. Alteration in microRNA-155 level correspond to severity of coronary heart disease. Scand. J. Clin. Lab. Investig. 2018, 78, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.Y.; Park, M.; Kim, S.; Kim, T.; Kim, J.; Choi, S.; Park, W.; Hwang, J.Y.; Choe, J.; et al. NF-kappaB-dependent miR-31/155 biogenesis is essential for TNF-alpha-induced impairment of endothelial progenitor cell function. Exp. Mol. Med. 2020, 52, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Narang, R.; Sreenivas, V.; Rastogi, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Circulatory miR-133b and miR-21 as Novel Biomarkers in Early Prediction and Diagnosis of Coronary Artery Disease. Genes 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.B.; Zhang, Z.Y.; Yuan, K.; Liu, Y.; Feng, X.; Cui, R.R.; Hu, Y.R.; Yuan, Z.S.; Gu, L.; Li, S.J.; et al. MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology 2013, 154, 3344–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubel, J.; Hauke, W.; Rump, S.; Viereck, J.; Batkai, S.; Poetzsch, J.; Rode, L.; Weigt, H.; Genschel, C.; Lorch, U.; et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: Results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur. Heart J. 2021, 42, 178–188. [Google Scholar] [CrossRef]
- Xiao, N.; Zhang, J.; Chen, C.; Wan, Y.; Wang, N.; Yang, J. miR-129-5p improves cardiac function in rats with chronic heart failure through targeting HMGB1. Mamm. Genome 2019, 30, 276–288. [Google Scholar] [CrossRef]
- Daimiel-Ruiz, L.; Klett-Mingo, M.; Konstantinidou, V.; Mico, V.; Aranda, J.F.; Garcia, B.; Martinez-Botas, J.; Davalos, A.; Fernandez-Hernando, C.; Ordovas, J.M. Dietary lipids modulate the expression of miR-107, a miRNA that regulates the circadian system. Mol. Nutr. Food Res. 2015, 59, 1865–1878. [Google Scholar] [CrossRef] [Green Version]
- Gil-Zamorano, J.; Martin, R.; Daimiel, L.; Richardson, K.; Giordano, E.; Nicod, N.; García-Carrasco, B.; Soares, S.M.; Iglesias-Gutiérrez, E.; Lasunción, M.A.; et al. Docosahexaenoic acid modulates the enterocyte Caco-2 cell expression of microRNAs involved in lipid metabolism. J. Nutr. 2014, 144, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Tomé-Carneiro, J.; Crespo, M.C.; Iglesias-Gutierrez, E.; Martín, R.; Gil-Zamorano, J.; Tomas-Zapico, C.; Burgos-Ramos, E.; Correa, C.; Gómez-Coronado, D.; Lasunción, M.A.; et al. Hydroxytyrosol supplementation modulates the expression of miRNAs in rodents and in humans. J. Nutr. Biochem. 2016, 34, 146–155. [Google Scholar] [CrossRef]
- Viswambharan, H.; Carvas, J.M.; Antic, V.; Marecic, A.; Jud, C.; Zaugg, C.E.; Ming, X.F.; Montani, J.P.; Albrecht, U.; Yang, Z. Mutation of the circadian clock gene Per2 alters vascular endothelial function. Circulation 2007, 115, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Daimiel, L.; Micó, V.; Díez-Ricote, L.; Ruiz-Valderrey, P.; Istas, G.; Rodríguez-Mateos, A.; Ordovás, J.M. Alcoholic and Non-Alcoholic Beer Modulate Plasma and Macrophage microRNAs Differently in a Pilot Intervention in Humans with Cardiovascular Risk. Nutrients 2020, 13, 69. [Google Scholar] [CrossRef]
- Stock, J. Gut microbiota: An environmental risk factor for cardiovascular disease. Atherosclerosis 2013, 229, 440–442. [Google Scholar] [CrossRef]
- Sheedy, F.J. Turning 21: Induction of MiR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Hänninen, M.; Jäntti, T.; Tolppanen, H.; Segersvärd, H.; Tarvasmäki, T.; Lassus, J.; Vausort, M.; Devaux, Y.; Sionis, A.; Tikkanen, I.; et al. Association of miR-21-5p, miR-122-5p, and miR-320a-3p with 90-Day Mortality in Cardiogenic Shock. Int. J. Mol. Sci. 2020, 21, 7925. [Google Scholar] [CrossRef]
- Mantilla-Escalante, D.C.; López de Las Hazas, M.C.; Crespo, M.C.; Martín-Hernández, R.; Tomé-Carneiro, J.; Del Pozo-Acebo, L.; Salas-Salvadó, J.; Bulló, M.; Dávalos, A. Mediterranean diet enriched in extra-virgin olive oil or nuts modulates circulating exosomal non-coding RNAs. Eur. J. Nutr. 2021. [Google Scholar] [CrossRef]
- Zhou, L.; Irani, S.; Sirwi, A.; Hussain, M.M. MicroRNAs regulating apolipoprotein B-containing lipoprotein production. Biochim. Biophys. Acta 2016, 1861, 2062–2068. [Google Scholar] [CrossRef]
- Oyama, Y.; Bartman, C.M.; Gile, J.; Eckle, T. Circadian MicroRNAs in Cardioprotection. Curr. Pharm. Des. 2017, 23, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Liu, A.; Weidenhammer, A.; Cooksey, R.C.; McClain, D.; Kim, M.K.; Aguilera, G.; Abel, E.D.; Chung, J.H. The role of mPer2 clock gene in glucocorticoid and feeding rhythms. Endocrinology 2009, 150, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Penalvo, J.L.; Mertens, E.; Munoz-Cabrejas, A.; Leon-Latre, M.; Jarauta, E.; Laclaustra, M.; Ordovas, J.M.; Casasnovas, J.A.; Uzhova, I.; Moreno-Franco, B. Work Shift, Lifestyle Factors, and Subclinical Atherosclerosis in Spanish Male Workers: A Mediation Analysis. Nutrients 2021, 13, 1077. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rios, A.; Perez-Martinez, P.; Delgado-Lista, J.; Phillips, C.M.; Gjelstad, I.M.; Wright, J.W.; Karlström, B.; Kiec-Wilk, B.; van Hees, A.M.; Helal, O.; et al. A Period 2 genetic variant interacts with plasma SFA to modify plasma lipid concentrations in adults with metabolic syndrome. J. Nutr. 2012, 142, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Andersson-Rolf, A.; Hindley, C.J.; Boj, S.F.; Clevers, H.; Koo, B.-K.; Huch, M. Culture and Establishment of Self-Renewing Human and Mouse Adult Liver and Pancreas 3D Organoids and Their Genetic Manipulation. Nat. Protoc. 2016, 11, 1724–1743. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, T.; Yang, P.-C. Western Blot: Technique, Theory, and Trouble Shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next Generation of Scientific Image Data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díez-Ricote, L.; Ruiz-Valderrey, P.; Micó, V.; Blanco-Rojo, R.; Tomé-Carneiro, J.; Dávalos, A.; Ordovás, J.M.; Daimiel, L. Trimethylamine n-Oxide (TMAO) Modulates the Expression of Cardiovascular Disease-Related microRNAs and Their Targets. Int. J. Mol. Sci. 2021, 22, 11145. https://doi.org/10.3390/ijms222011145
Díez-Ricote L, Ruiz-Valderrey P, Micó V, Blanco-Rojo R, Tomé-Carneiro J, Dávalos A, Ordovás JM, Daimiel L. Trimethylamine n-Oxide (TMAO) Modulates the Expression of Cardiovascular Disease-Related microRNAs and Their Targets. International Journal of Molecular Sciences. 2021; 22(20):11145. https://doi.org/10.3390/ijms222011145
Chicago/Turabian StyleDíez-Ricote, Laura, Paloma Ruiz-Valderrey, Víctor Micó, Ruth Blanco-Rojo, João Tomé-Carneiro, Alberto Dávalos, José M. Ordovás, and Lidia Daimiel. 2021. "Trimethylamine n-Oxide (TMAO) Modulates the Expression of Cardiovascular Disease-Related microRNAs and Their Targets" International Journal of Molecular Sciences 22, no. 20: 11145. https://doi.org/10.3390/ijms222011145
APA StyleDíez-Ricote, L., Ruiz-Valderrey, P., Micó, V., Blanco-Rojo, R., Tomé-Carneiro, J., Dávalos, A., Ordovás, J. M., & Daimiel, L. (2021). Trimethylamine n-Oxide (TMAO) Modulates the Expression of Cardiovascular Disease-Related microRNAs and Their Targets. International Journal of Molecular Sciences, 22(20), 11145. https://doi.org/10.3390/ijms222011145