
NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Cetuximab Induces Tumor Cell Killing by NK Cells
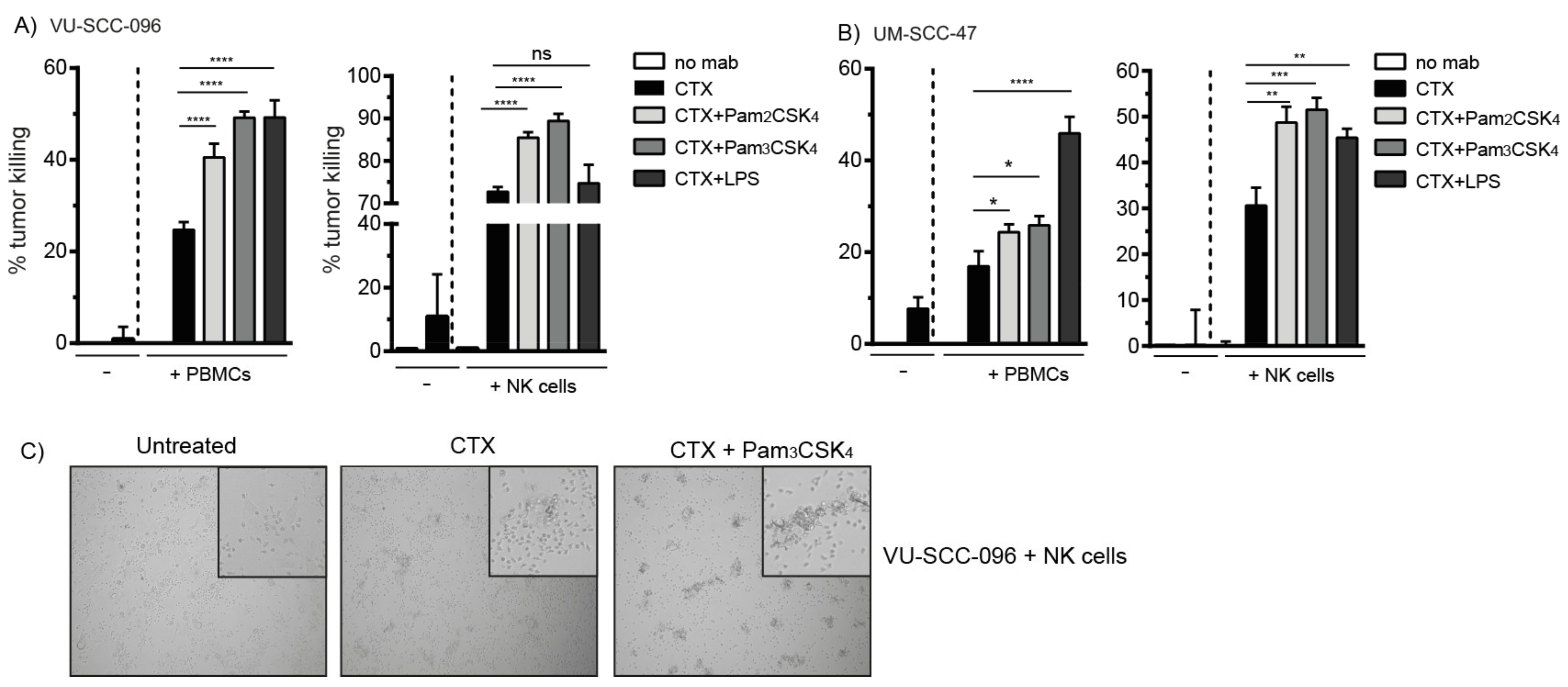
2.2. Co-Activation of FcγRs and TLR2 Enhances Tumor Cell Killing by NK Cells
2.3. TLR2 Agonists Enhance the Number of Cytotoxic NK Cells in Combination with Cetuximab
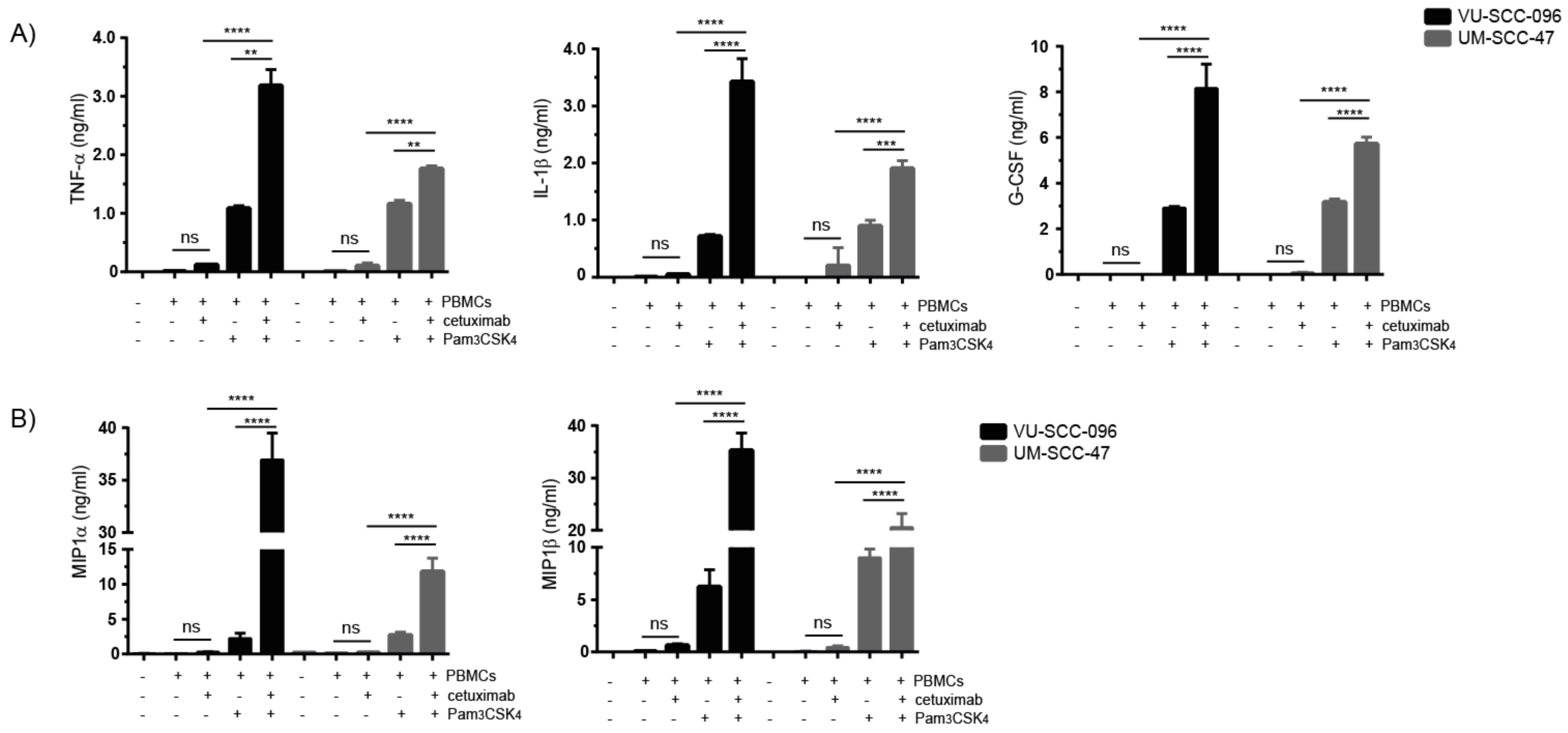
2.4. Combination of Cetuximab with TLR2 Agonists Induces Profound Pro-Inflammatory Responses by PBMCs
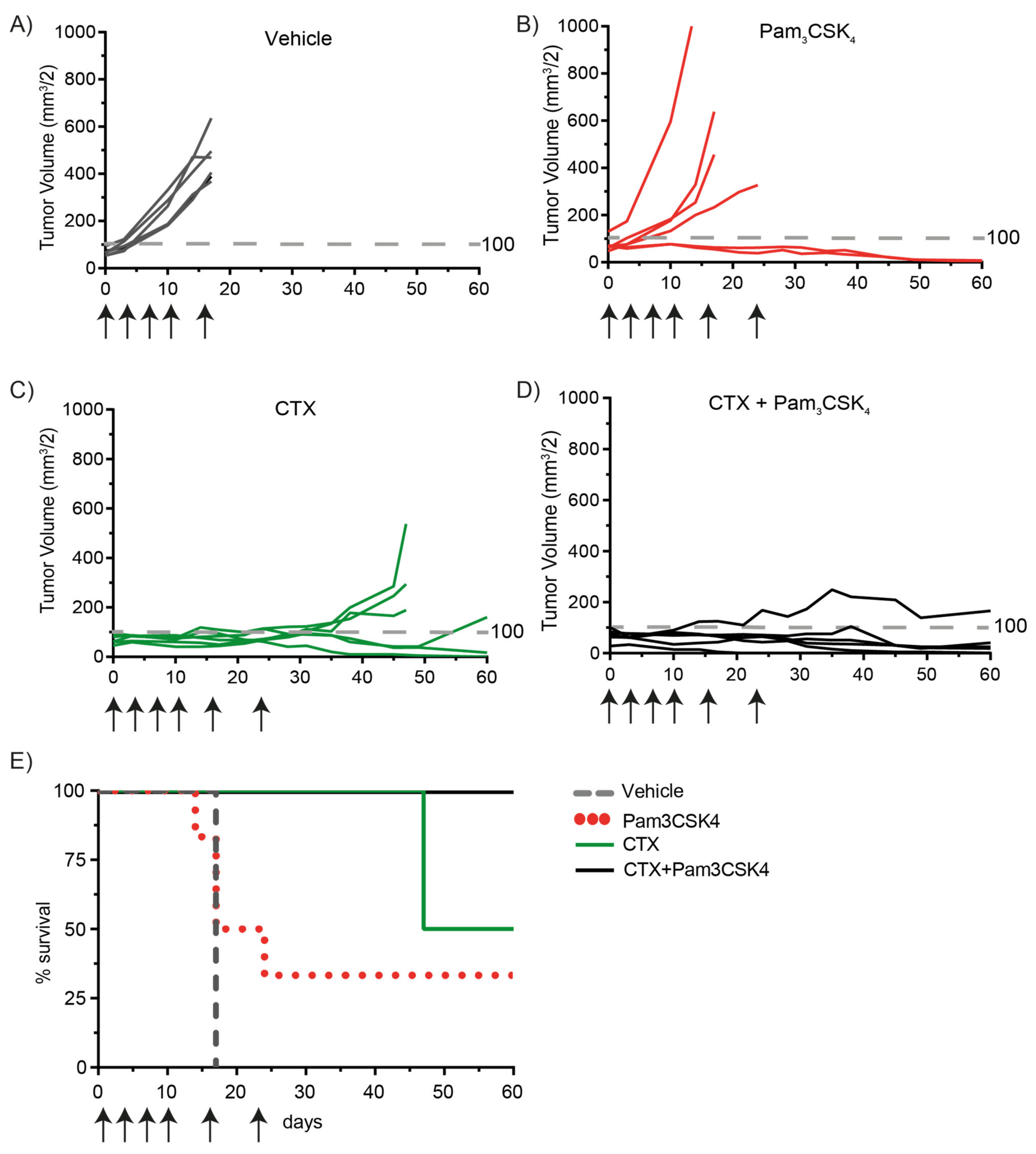
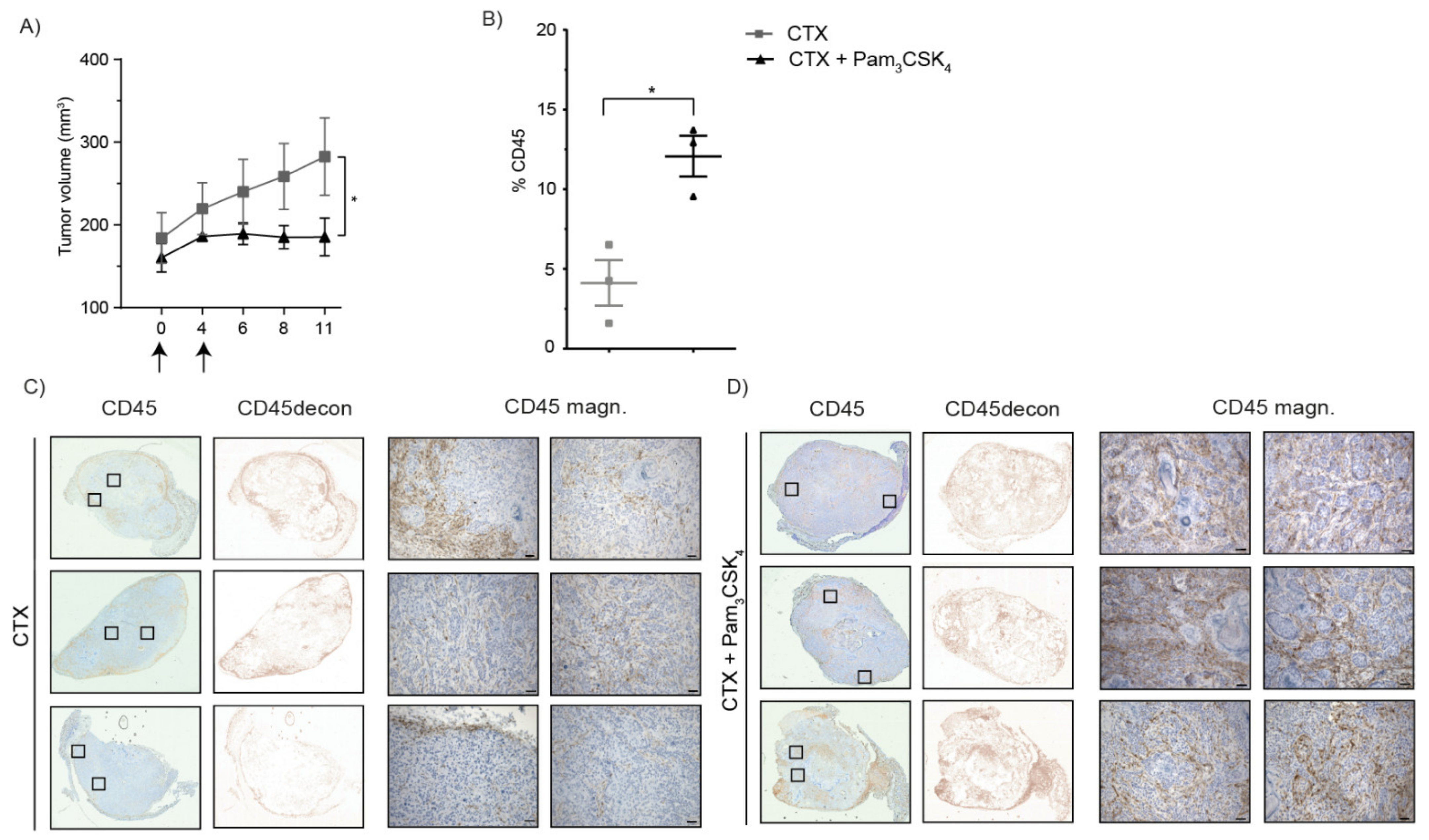
2.5. Treatment of Tumor-Bearing Mice with Cetuximab and Pam3CSK4 Results in Prolonged Survival
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.2.1. HNSCC Cell Lines
4.2.2. Immune Cells
4.3. ADCC Assay
4.4. Flow Cytometry
4.5. Multiplex Protein Analysis
4.6. In Vivo Experiments
4.7. Immunohistochemistry
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef]
- Peltanova, B.; Raudenska, M.; Masarik, M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: A systematic review. Mol. Cancer 2019, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L. Immunology and Immunotherapy of Head and Neck Cancer. J. Clin. Oncol. 2015, 33, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Wondergem, N.E.; Nauta, I.H.; Muijlwijk, T.; Leemans, C.R.; van de Ven, R. The Immune Microenvironment in Head and Neck Squamous Cell Carcinoma: On Subsets and Subsites. Curr. Oncol. Rep. 2020, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Senbabaoglu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Zandberg, D.P.; Ferris, R.L. Window Studies in Squamous Cell Carcinoma of the Head and Neck: Values and Limits. Curr. Treat. Options Oncol. 2018, 19, 68. [Google Scholar] [CrossRef]
- Jie, H.B.; Schuler, P.J.; Lee, S.C.; Srivastava, R.M.; Argiris, A.; Ferrone, S.; Whiteside, T.L.; Ferris, R.L. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res. 2015, 75, 2200–2210. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.K.; Zhao, W.; Sturgis, E.M.; Burtness, B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; Castro, G.; Psyrri, D., Jr.; Baste, A.N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Vincenzi, B.; Zoccoli, A.; Pantano, F.; Venditti, O.; Galluzzo, S. Cetuximab: From bench to bedside. Curr. Cancer Drug Targets 2010, 10, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Bakema, J.E.; van Egmond, M. Fc receptor-dependent mechanisms of monoclonal antibody therapy of cancer. Curr. Top. Microbiol. Immunol. 2014, 382, 373–392. [Google Scholar]
- Braster, R.; O’Toole, T.; van Egmond, M. Myeloid cells as effector cells for monoclonal antibody therapy of cancer. Methods 2014, 65, 28–37. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Lee, S.C.; Andrade Filho, P.A.; Lord, C.A.; Jie, H.B.; Davidson, H.C.; Lopez-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef]
- Charap, A.J.; Enokida, T.; Brody, R.; Sfakianos, J.; Miles, B.; Bhardwaj, N.; Horowitz, A. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e001523. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef]
- Van Egmond, M.; Vidarsson, G.; Bakema, J.E. Cross-talk between pathogen recognizing Toll-like receptors and immunoglobulin Fc receptors in immunity. Immunol. Rev. 2015, 268, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, N.; Estornes, Y.; Hasan, U.; Lebecque, S.; Caux, C. Targeting pattern recognition receptors in cancer immunotherapy. Target. Oncol. 2012, 7, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Kohrt, H.E.; Houot, R.; Marabelle, A.; Cho, H.J.; Osman, K.; Goldstein, M.; Levy, R.; Brody, J. Combination strategies to enhance antitumor ADCC. Immunotherapy 2012, 4, 511–527. [Google Scholar] [CrossRef]
- Bakema, J.E.; Tuk, C.W.; van Vliet, S.J.; Bruijns, S.C.; Vos, J.B.; Letsiou, S.; Dijkstra, C.D.; van Kooyk, Y.; Brenkman, A.B.; van Egmond, M. Antibody-opsonized bacteria evoke an inflammatory dendritic cell phenotype and polyfunctional Th cells by cross-talk between TLRs and FcRs. J. Immunol. 2015, 194, 1856–1866. [Google Scholar] [CrossRef]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; De Waele, J.; Peeters, M.; Pauwels, P.; Vermorken, J.B.; Smits, E.; Lardon, F.; Jacobs, J.; et al. Cetuximab-induced natural killer cell cytotoxicity in head and neck squamous cell carcinoma cell lines: Investigation of the role of cetuximab sensitivity and HPV status. Br. J. Cancer 2020, 123, 752–761. [Google Scholar] [CrossRef]
- Derer, S.; Bauer, P.; Lohse, S.; Scheel, A.H.; Berger, S.; Kellner, C.; Peipp, M.; Valerius, T. Impact of epidermal growth factor receptor (EGFR) cell surface expression levels on effector mechanisms of EGFR antibodies. J. Immunol. 2012, 189, 5230–5239. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Zarember, K.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.D.O.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar]
- Overdijk, M.B.; Verploegen, S.; Ortiz Buijsse, A.; Vink, T.; Leusen, J.H.; Bleeker, W.K.; Parren, P.W. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 2012, 189, 3430–3438. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Chan, L.-P.; Cho, S.-F. Targeting the Immune Microenvironment in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 1084. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Ming Lim, C.; Stephenson, R.; Salazar, A.M.; Ferris, R.L. TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR head and neck cancer cells. Oncoimmunology 2013, 2, e24677. [Google Scholar] [CrossRef]
- Stephenson, R.M.; Lim, C.M.; Matthews, M.; Dietsch, G.; Hershberg, R.; Ferris, R.L. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol. Immunother. 2013, 62, 1347–1357. [Google Scholar] [CrossRef]
- Shayan, G.; Kansy, B.A.; Gibson, S.P.; Srivastava, R.M.; Bryan, J.K.; Bauman, J.E.; Ohr, J.; Kim, S.; Duvvuri, U.; Clump, D.A.; et al. Phase Ib Study of Immune Biomarker Modulation with Neoadjuvant Cetuximab and TLR8 Stimulation in Head and Neck Cancer to Overcome Suppressive Myeloid Signals. Clin. Cancer Res. 2017, 24, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Saba, N.F.; Gitlitz, B.J.; Haddad, R.; Sukari, A.; Neupane, P.; Morris, J.C.; Misiukiewicz, K.; Bauman, J.E.; Fenton, M.; et al. Effect of Adding Motolimod to Standard Combination Chemotherapy and Cetuximab Treatment of Patients With Squamous Cell Carcinoma of the Head and Neck: The Active8 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M.; Morishima, C.; Eaton, K.D.; Baik, C.S.; Goulart, B.H.; Anderson, L.N.; Manjarrez, K.L.; Dietsch, G.N.; Bryan, J.K.; Hershberg, R.M.; et al. Phase Ib Trial of the Toll-like Receptor 8 Agonist, Motolimod (VTX-2337), Combined with Cetuximab in Patients with Recurrent or Metastatic SCCHN. Clin. Cancer Res. 2016, 23, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Van Pul, K.M.; Fransen, M.F.; Van de Ven, R.; De Gruijl, T.D. Immunotherapy Goes Local: The Central Role of Lymph Nodes in Driving Tumor Infiltration and Efficacy. Front. Immunol. 2021, 12, 643291. [Google Scholar] [CrossRef]
- Van den Hout, M.F.; Sluijter, B.J.; Santegoets, S.J.; Van Leeuwen, P.A.; Van den Tol, M.P.; Van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Local delivery of CpG-B and GM-CSF induces concerted activation of effector and regulatory T cells in the human melanoma sentinel lymph node. Cancer Immunol. Immunother. 2016, 65, 405–415. [Google Scholar] [CrossRef][Green Version]
- Molenkamp, B.G.; Sluijter, B.J.; Van Leeuwen, P.A.; Santegoets, S.J.; Meijer, S.; Wijnands, P.G.; Haanen, J.B.; Van den Eertwegh, A.J.; Scheper, R.J.; De Gruijl, T.D. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clin. Cancer Res. 2008, 14, 4532–4542. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Pearson, C.I.; Gregorio, J.D.; Joseph, C.G.; Justin, A.K.; Felix, J.H.; Angela, L.; Po, Y.H.; Heidi, L.; Lisa, K.B.; et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat. Rev. Cancer 2020, 2, 18–33. [Google Scholar] [CrossRef]
- Fauriat, C.; Hansen, I.S.; Visser, M.W.; Nagelkerke, S.Q.; Kuijpers, T.W.; Kapsenberg, M.L.; De Jong, E.C.; Den Dunnen, J. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef]
- Lisa, T.C.V.; Ivo, S.H.; Marijke, W.V.; Sietse, Q.N.; Taco, W.K.; Martien, L.K.; Esther, C.D.J.; Jeroen, D.D. FcgammaRIIa cross-talk with TLRs, IL-1R, and IFNgammaR selectively modulates cytokine production in human myeloid cells. Immunobiology 2015, 220, 193–199. [Google Scholar]
- Vogelpoel, L.T.; Hansen, I.S.; Rispens, T.; Muller, F.J.; Van Capel, T.M.; Turina, M.C.; Vos, J.B.; Baeten, D.L.; Kapsenberg, M.L.; De Jong, E.C.; et al. Fc gamma receptor-TLR cross-talk elicits pro-inflammatory cytokine production by human M2 macrophages. Nat. Commun. 2014, 5, 5444. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.; Vogelpoel, L.T.; Wypych, T.; Muller, F.J.; De Boer, L.; Kuijpers, T.W.; Zaat, S.A.; Kapsenberg, M.L.; De Jong, E.C. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcgammaRIIa in human dendritic cells. Blood 2012, 120, 112–121. [Google Scholar] [CrossRef]
- Heemskerk, N.; Van Egmond, M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12962. [Google Scholar] [CrossRef]
- Van Egmond, M.; Bakema, J.E. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin. Cancer Biol. 2013, 23, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Van Zeeburg, H.J.; Snijders, P.J.; Pals, G.; Hermsen, M.A.; Rooimans, M.A.; Bagby, G.; Soulier, J.; Gluckman, E.; Wennerberg, J.; Leemans, C.R.; et al. Generation and molecular characterization of head and neck squamous cell lines of fanconi anemia patients. Cancer Res. 2005, 65, 1271–1276. [Google Scholar] [CrossRef]
- Hermsen, M.A.; Joenje, H.; Arwert, F.; Braakhuis, B.J.; Baak, J.P.; Westerveld, A.; Slater, R. Assessment of chromosomal gains and losses in oral squamous cell carcinoma by comparative genomic hybridisation. Oral Oncol. 1997, 33, 414–418. [Google Scholar] [CrossRef]
- Lin, C.J.; Grandis, J.R.; Carey, T.E.; Gollin, S.M.; Whiteside, T.L.; Koch, W.M.; Ferris, R.L.; Lai, S.Y. Head and neck squamous cell carcinoma cell lines: Established models and rationale for selection. Head Neck 2007, 29, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Smeets, S.J.; Hesselink, A.T.; Speel, E.J.; Haesevoets, A.; Snijders, P.J.; Pawlita, M.; Meijer, C.J.; Braakhuis, B.J.; Leemans, C.R.; Brakenhoff, R.H. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. Int. J. Cancer 2007, 121, 2465–2472. [Google Scholar] [CrossRef]
- Martens-de Kemp, S.R.; Brink, A.; Stigter-van Walsum, M.; Damen, J.M.; Rustenburg, F.; Wu, T.; Van Wieringen, W.N.; Schuurhuis, G.J.; Braakhuis, B.J.; Slijper, M.; et al. CD98 marks a subpopulation of head and neck squamous cell carcinoma cells with stem cell properties. Stem Cell Res. 2013, 10, 477–488. [Google Scholar] [CrossRef]
- Ruifrok, A.C.; Johnston, D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001, 23, 291–299. [Google Scholar] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruijs, M.; Ganzevles, S.H.; Stigter-van Walsum, M.; van der Mast, R.; van Ostaijen-ten Dam, M.M.; Tuk, C.W.; Schilham, M.W.; Leemans, C.R.; Brakenhoff, R.H.; van Egmond, M.; et al. NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models. Int. J. Mol. Sci. 2021, 22, 11057. https://doi.org/10.3390/ijms222011057
Gruijs M, Ganzevles SH, Stigter-van Walsum M, van der Mast R, van Ostaijen-ten Dam MM, Tuk CW, Schilham MW, Leemans CR, Brakenhoff RH, van Egmond M, et al. NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models. International Journal of Molecular Sciences. 2021; 22(20):11057. https://doi.org/10.3390/ijms222011057
Chicago/Turabian StyleGruijs, Mandy, Sonja H. Ganzevles, Marijke Stigter-van Walsum, Richard van der Mast, Monique M. van Ostaijen-ten Dam, Cornelis W. Tuk, Marco W. Schilham, C. René Leemans, Ruud H. Brakenhoff, Marjolein van Egmond, and et al. 2021. "NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models" International Journal of Molecular Sciences 22, no. 20: 11057. https://doi.org/10.3390/ijms222011057
APA StyleGruijs, M., Ganzevles, S. H., Stigter-van Walsum, M., van der Mast, R., van Ostaijen-ten Dam, M. M., Tuk, C. W., Schilham, M. W., Leemans, C. R., Brakenhoff, R. H., van Egmond, M., van de Ven, R., & Bakema, J. E. (2021). NK Cell-Dependent Antibody-Mediated Immunotherapy Is Improved In Vitro and In Vivo When Combined with Agonists for Toll-like Receptor 2 in Head and Neck Cancer Models. International Journal of Molecular Sciences, 22(20), 11057. https://doi.org/10.3390/ijms222011057