
Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications

 , and
, and
Abstract
1. Introduction
2. Epigenetics and Carcinogenesis
2.1. Metabolism Pathways as Epigenetic Regulators
2.1.1. Oxidative Stress as an Epigenetic Regulator
2.2. Methyltransferases
2.3. Demethylases
2.4. Histone Acetylation
2.5. Mechanisms of DNA Hyper or Hypomethylation
2.5.1. DNA Hypermethylation
2.5.2. DNA Hypomethylation
Consequences of Global Genome DNA Hypomethylation
Global Genome DNA Hypomethylation and Chromosome Instability
3. Positioning Epigenetics in Chemical Hazard Assessment
3.1. Epigenetics in Adverse Outcome Pathways to Overcome the Multiplicity of NGTxC Modes of Action
3.2. Adverse Epigenetic Effects vs. Normal Epigenetic Regulation and Disturbances
3.3. Dose/Concentration–Response, Response Threshold and Magnitude, and Assay Duration
3.4. Assay Robustness and Reproducibility
3.5. Extrapolation from In Vitro Culture Systems
3.6. Extrapolation of Experimental Model to Human
4. Epigenetic Disturbances Induced by Genotoxic and Non-Genotoxic Carcinogens
4.1. Arsenicals
4.1.1. Oxidative Stress
4.1.2. DNMT Expression and Activities
4.1.3. Ten Eleven Translocation (TET) Enzymes
4.1.4. Micro RNAs
4.1.5. Histone Variants
4.1.6. Histone Post-Translational Modifications (HPTM)
4.1.7. DNA Repair
4.1.8. Mitochondrial Biogenesis
4.1.9. In Utero Exposure
4.1.10. Human Blood Measurements Confounding Factors
4.2. Nickel
4.2.1. Heterochromatization and H3K9 Methylation
4.2.2. Inhibition of Dioxygenases
4.2.3. Hypoxia and Oxidative Stress
4.2.4. Ubiquitination/Deubiquitination Machinery
4.2.5. Interference with the Zn2+ Finger Protein CTCF
4.2.6. Long-Term Exposure and Persistent Effects in the Absence of Exposure
4.3. Phenobarbital
4.3.1. Mechanisms of Cell Proliferation and DNA Demethylation
4.3.2. 5-Hydroxymetylcytosine (5hmC) as an Early Marker of NGTxC
4.3.3. NcRNAs from the Dlk1-Dio3 Locus as Marker of Carcinogenesis and Epigenetic Deregulation
4.3.4. DNA Methylation Enzymes and L1 vs. Apical Endpoint
4.4. Irradiation
4.5. H4K20 Methylation in Target vs. Non-Target Tissues, and Effects of Other Chemicals
5. Overview of Potential Epigenetic Assay Types Adaptable to Chemical Hazard Assessment
5.1. Epigenetic Enzyme Screening Assays (Cell-Free Biochemistry, Enzymology Assay, High-Content Image Analyses, Reporter Systems, and QSAR Approaches)
5.2. Changes in Global Genome Epigenetic Marks and in Repeated DNA Sequences
5.3. Consideration of Multiple DMR/Genes in Parallel as Markers of Key Events
6. Strategy for Evaluators to Assess the Importance of Cancer Epigenetic Data in Chemical Hazard Assessment
7. Conclusions
8. Types of Relevant Epigenetic Assays
8.1. Cell-Free Biochemistry/Enzymology Assays
8.2. High-Content Image Analysis (HCA) In Situ Assays
8.3. Antibody Requirement for Enzyme-Specific Assays
8.4. Reporter Systems in the Cellular Environment
8.5. Global Genome Changes in Histone Modifications and DNA Methylation
9. Markers of Specific Key Events for Consideration in Multiplex DNA Methylation Assays
9.1. Epigenetic Impairment of Detoxification and DNA Repair Pathway; MGMT, BRCA1, and GSTP1
9.1.1. BRCA1, H2AX, and H4K20
9.1.2. GSTP1
9.2. Cell Cycle Regulation Breakdown, the Ink4b/Arf/Ink4a Locus
9.3. Inflammation/Immune Response Disruption
9.3.1. Major Histocompatibility Complex-I and -II
9.3.2. T Cell Immune Checkpoints, NK Cell NKG2D Receptor, and Their Ligands
9.3.3. Cancer-Testis Antigen Gene Families as Epigenetic Markers of Carcinogenicity
9.4. The Cytoskeleton; Relevance to Global Genome Epigenetic Marks, E-Cadherin, MYO10
9.5. Senescence Bypass and Telomerase Reverse Transcriptase (TERT) Regulation
9.6. Angiogenesis and Thrombospondin-1
9.7. The Homeobox (HOX) Genes
10. Transcriptomic Biomarkers and Genome-Wide Signatures
10.1. Transcriptomic Biomarkers
10.2. Transcription Factors, Enhancers, and Other List of Endpoints for Targeted NGS Assay Development
11. Technical Considerations for Improving the Regulatory Value of Epigenetic Data
11.1. DNA Methylation Assay Robustness and Performance Comparisons
11.2. Methodology to Distinguish 5mC from 5hmC
11.3. Extrapolation of Experimental Models to Human
11.4. Considerations in the Extrapolation from In Vitro Cultures, Cell Lines and Cell Types
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
Abbreviations
| 5aCdR | 5-aza-2′-deoxycytidine. |
| 5hmC | 5-hydroxymethylcytosine. |
| 5hmC | hydroxymethyl cytosine. |
| 5mC | 5-methylcytosine. |
| AE | adverse event. |
| ALT | alternative lengthening of telomeres pathway. |
| Alu | arthrobacter luteus DNA repeats. |
| AML | acute myeloid leukemia. |
| AOP | adverse outcome pathway. |
| APC | adenomatous polyposis coli protein. |
| B[a]P | benzo[a]pyrene. |
| BCAT1 | branched-chain-amino-acid aminotransferase, cytosolic. |
| BEAS-2B | human bronchial epithelial SV-40 immortalised cell line. |
| BMD | benchmark-doses. |
| BMP3 | bone morphogenic protein 3. |
| BPA | bisphenol-A. |
| CAN | copy number alterations. |
| CAR | constitutive androstane receptor. |
| cGAS | cyclic GMP-AMP synthase. |
| CTA | cell transformation assay. |
| CTAG | cancer-testis antigens. |
| CTCF | CCCTC-binding factor. |
| DNAm | DNA methylation. |
| DNMT1, 2, 3a, 3b, 3L | DNA methyltransferases. |
| Ecad | E-cadherin. |
| EMT | epithelial to mesenchymal transition. |
| GGD | global genome DNA. |
| GGDHo | global genome DNA hypomethylation. |
| GGDm | global genome DNA methylation. |
| GSH | glutathione. |
| GSTP1 | glutathione S-transferase Pi1. |
| GTxC | genotoxic carcinogens. |
| HA | hazard assessment. |
| HaCaT | human keratinocyte immortalized cell line. |
| HBE | human bronchial epithelial cell line. |
| HCA | high-content analysis. |
| HDAC | histone deacetylase. |
| HDM | histone demethylase. |
| hESC | human embryonic stem cells. |
| HMEC | human mammary epithelial cells. |
| HMT | histone methyltransferase. |
| HPTM | histone post-translational modifications. |
| HTS | high-throughput screening. |
| HUC1 | human urothelial immortalised cell line. |
| IARC | International Agency for the Research on Cancer. |
| IATA | integrated approach to the testing and assessment. |
| IDH | isocitrate dehydrogenase. |
| IKZF1 | DNA-binding protein Ikaros. |
| KDM | lysine demethylase. |
| KE | key event. |
| KER | key event relationship. |
| KMT | lysine methyltransferase. |
| L-02 | human hepatic cell line. |
| Line | long interspersed nuclear element. |
| LncRNA | long non-coding RNA. |
| LOAELs | lowest-observable-adverse-effect levels. |
| LTR | long terminal repeat. |
| MCF7 | breast cancer cell line #7 from the Michigan Cancer Foundation. |
| mESC | mouse embryonic stem cells. |
| MGMT | methylated DNA-protein-cysteine methyltransferase. |
| MHC | major histocompatibility complex. |
| MIE | molecular initiating event. |
| MS | mass spectrometry. |
| NAM | novel approach methodologies. |
| ncRNA | non-coding RNA. |
| NDRG4 | N-myc downregulated gene 4. |
| NGS | next-generation sequencing. |
| NGTxC | non-genotoxic carcinogens. |
| NOAELs | no-observable-adverse- effect levels. |
| OECD | Organization for Economic Cooperation and Development, |
| OGG1 | 8-oxoguanine glycosylase. |
| ONECUT2 | one cut domain family member 2. |
| OTX1 | homeobox protein OTX1. |
| PBMC | peripheral blood mononuclear cells. |
| PCB | polychlorinated biphenyls. |
| PCNA | proliferative cell nuclear antigen. |
| PHD | prolyl hydroxylase dioxygenase. |
| PND | postnatal day. |
| PoD | point of departure. |
| RA | risk assessment. |
| RASSF1 | ras association domain-containing protein 1. |
| RCB | two-year rodent cancer bioassay. |
| RRBS | reduced-representation bisulphite sequencing. |
| SAHA | suberoylanilide hydroxamic acid. |
| SAM | s-adenosylmethionine. |
| 09-Sep | septin-9. |
| SHOX2 | short stature homeobox protein 2. |
| Sine | short interspersed nuclear element. |
| SUV39H1 | suppressor of variegation 3–9 homolog 1; catalyzes H3K9me3. |
| TCA | tricarboxylic acid cycle. |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin. |
| TDG | thymine-DNA glycosylase. |
| TET-1, -2, -3 | ten eleven translocation enzymes. |
| TSG | tumour suppressor gene. |
| TSS | transcription start sites. |
| TWIST1 | twist-related protein 1. |
| UHRF1 | ubiquitin-like PHD and RING finger domain 1. |
| WGBS | whole-genome bisulphite sequencing. |
| WoE | weight of evidence. |
References
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Leung, D.; Jung, I.; Rajagopal, N.; Schmitt, A.; Selvaraj, S.; Lee, A.Y.; Yen, C.A.; Lin, S.; Lin, Y.; Qiu, Y.; et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 2015, 518, 350–354. [Google Scholar] [CrossRef]
- Hanna, C.W.; Demond, H.; Kelsey, G. Epigenetic regulation in development: Is the mouse a good model for the human? Hum. Reprod. Update 2018, 24, 556–576. [Google Scholar] [CrossRef]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Cummings-Lorbetskie, C.; Li, N.; Xiao, G.H.; Marro, L.; Khan, N.; Leingartner, K. Sodium bisulfite pyrosequencing revealed that developmental exposure to environmental contaminant mixtures does not affect DNA methylation of DNA repeats in Sprague-Dawley rats. J. Toxicol. Environ. Health A 2016, 149, 1204–1225. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.F.; Santamarina, P.; Tejedor, J.R.; Urdinguio, R.G.; Alvarez-Pitti, J.; Redon, P.; Fernandez, A.F.; Fraga, M.F.; Lurbe, E. Longitudinal genome-wide DNA methylation analysis uncovers persistent early-life DNA methylation changes. J. Transl. Med. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, E.L.; Jacobs, M.N.; Gant, T.W. Environmentally induced epigenetic toxicity: Potential public health concerns. Crit. Rev. Toxicol. 2016, 46, 676–700. [Google Scholar] [CrossRef]
- Heflich, R.H.; Johnson, G.E.; Zeller, A.; Marchetti, F.; Douglas, G.R.; Witt, K.L.; Gollapudi, B.B.; White, P.A. Mutation as a toxicological endpoint for regulatory decision-making. Environ. Mol. Mutagen. 2020, 61, 34–41. [Google Scholar] [CrossRef]
- Luijten, M.; Corvi, R.; Mehta, J.; Corvaro, M.; Delrue, N.; Felter, S.; Haas, B.; Hewitt, N.J.; Hilton, G.; Holmes, T.; et al. A comprehensive view on mechanistic approaches for cancer risk assessment of non-genotoxic agrochemicals. Regul. Toxicol. Pharmacol. 2020, 118, 104789. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.N.; Colacci, A.; Corvi, R.; Vaccari, M.; Aguila, M.C.; Corvaro, M.; Delrue, N.; Desaulniers, D.; Ertych, N.; Jacobs, A.; et al. Chemical carcinogen safety testing: OECD expert group international consensus on the development of an integrated approach for the testing and assessment of chemical non-genotoxic carcinogens. Arch. Toxicol. 2020, 8, 2899–2923. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- OECD Guidance Document for the Use of Adverse Outcome Pathways in developing Integrated Approaches to Testing and Assessment (IATA); ENV/JM/MONO(2016)67; Series on Testing and Assessment No 260. 2016, pp. 1–25. Available online: http://www.oecd.org/chemicalsafety/testing/ (accessed on 7 October 2021).
- OECD. OECD Overview of Concepts and Available Guidance Related to Integrated Approaches to Testing and Assessment (IATA); OECD Series on Testing and Assessment, No 329; Environment, Health and Safety, Environment Directorate; OECD: Paris, France, 2020; pp. 1–55. [Google Scholar]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; Rusyn, I.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Persp. 2015, 124, 713–721. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Ehrlich, K.C. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics 2014, 6, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Turker, M.S. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 2002, 21, 5388–5393. [Google Scholar] [CrossRef] [PubMed]
- Jost, D.; Vaillant, C. Epigenomics in 3D: Importance of long-range spreading and specific interactions in epigenomic maintenance. Nucleic Acids Res. 2018, 46, 2252–2264. [Google Scholar] [CrossRef]
- Klein, B.J.; Simithy, J.; Wang, X.; Ahn, J.; Andrews, F.H.; Zhang, Y.; Cote, J.; Shi, X.; Garcia, B.A.; Kutateladze, T.G. Recognition of histone H3K14 acylation by MORF. Structure 2017, 25, 650–654. [Google Scholar] [CrossRef]
- Raurell-Vila, H.; Bosch-Presegue, L.; Gonzalez, J.; Kane-Goldsmith, N.; Casal, C.; Brown, J.P.; Marazuela-Duque, A.; Singh, P.B.; Serrano, L.; Vaquero, A. An HP1 isoform-specific feedback mechanism regulates Suv39h1 activity under stress conditions. Epigenetics 2017, 12, 166–175. [Google Scholar] [CrossRef]
- Rebollo, R.; Miceli-Royer, K.; Zhang, Y.; Farivar, S.; Gagnier, L.; Mager, D.L. Epigenetic interplay between mouse endogenous retroviruses and host genes. Genome Biol. 2012, 13, R89. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Shivalila, C.S.; Soldner, F.; Markoulaki, S.; Jaenisch, R. Tracing dynamic changes of DNA methylation at single-cell resolution. Cell 2015, 163, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Tomkova, M.; Schuster-Bockler, B. DNA modifications: Naturally more error prone? Trends Genet. 2018, 34, 627–638. [Google Scholar] [CrossRef]
- Tsuda, M.; Fukuda, A.; Kawai, M.; Araki, O.; Seno, H. The role of the SWI/SNF chromatin remodeling complex in pancreatic ductal adenocarcinoma. Cancer Sci. 2021, 112, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef]
- Kadoch, C. Diverse compositions and functions of chromatin remodeling machines in cancer. Sci. Transl. Med. 2019, 11, aay1018. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Primary role of the nucleosome. Mol. Cell 2020, 79, 371–375. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef]
- Noberini, R.; Restellini, C.; Savoia, E.O.; Raimondi, F.; Ghiani, L.; Jodice, M.G.; Bertalot, G.; Bonizzi, G.; Capra, M.; Maffini, F.A.; et al. Profiling of epigenetic features in clinical samples reveals novel widespread changes in cancer. Cancers 2019, 11, 723. [Google Scholar] [CrossRef]
- LaRocca, J.; Johnson, K.J.; Lebaron, M.J.; Rasoulpour, R.J. The interface of epigenetics and toxicology in product safety assessment. Curr. Opin. Toxicol. 2017, 6, 87–92. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 2013, 754, 215–232. [Google Scholar]
- Salemi, R.; Marconi, A.; Di Salvatore, V.; Franco, S.; Rapisarda, V.; Libra, M. Epigenetic alterations and occupational exposure to benzene, fibers, and heavy metals associated with tumor development (Review). Mol. Med. Rep. 2017, 15, 3366–3371. [Google Scholar] [CrossRef]
- Lebaron, M.J.; Rasoulpour, R.J.; Klapacz, J.; Ellis-Hutchings, R.G.; Hollnagel, H.M.; Gollapudi, B.B. Epigenetics and chemical safety assessment. Mutat. Res. 2010, 705, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Chappell, G.; Pogribny, I.P.; Guyton, K.Z.; Rusyn, I. Epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens: A systematic literature review. Mutat. Res. Rev. Mutat. Res. 2016, 768, 27–45. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Marczylo, E.L.; Guerrero-Bosagna, C.; Ruegg, J. Marked for life: Epigenetic effects of endocrine disrupting chemicals. Annu. Rev. Environ. Resour. 2017, 42, 105–160. [Google Scholar] [CrossRef]
- Mahna, D.; Puri, S.; Sharma, S. DNA methylation modifications: Mediation to stipulate pesticide toxicity. Int. J. Environ. Sci. Technol. 2021, 18, 531–544. [Google Scholar] [CrossRef]
- Chung, F.F.; Herceg, Z. The promises and challenges of toxico-epigenomics: Environmental chemicals and their impacts on the epigenome. Environ. Health Perspect. 2020, 128, 15001. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenet. 2019, 11, 131. [Google Scholar] [CrossRef]
- Breton, C.V.; Marsit, C.J.; Faustman, E.; Nadeau, K.; Goodrich, J.M.; Dolinoy, D.C.; Herbstman, J.; Holland, N.; LaSalle, J.M.; Schmidt, R.; et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: The Children’s Environmental Health and Disease Prevention Research Center’s Epigenetics Working Group. Environ. Health Perspect. 2017, 125, 511–526. [Google Scholar] [CrossRef]
- Motta, V.; Bonzini, M.; Grevendonk, L.; Iodice, S.; Bollati, V. Epigenetics applied to epidemiology: Investigating environmental factors and lifestyle influence on human health. Med. Lav 2017, 108, 10–23. [Google Scholar] [PubMed]
- Isaevska, E.; Moccia, C.; Asta, F.; Cibella, F.; Gagliardi, L.; Ronfani, L.; Rusconi, F.; Stazi, M.A.; Richiardi, L. Exposure to ambient air pollution in the first 1000 days of life and alterations in the DNA methylome and telomere length in children: A systematic review. Environ. Res. 2021, 193, 110504. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental influences on the epigenome: Exposure-associated DNA Methylation in human populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Halbritter, F.; Carmona, F.J.; Tierling, S.; Datlinger, P.; Assenov, Y.; Berdasco, M.; Bergmann, A.K.; Booher, K.; Busato, F.; et al. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726–737. [Google Scholar]
- Goeppert, B.; Toth, R.; Singer, S.; Albrecht, T.; Lipka, D.B.; Lutsik, P.; Brocks, D.; Baehr, M.; Muecke, O.; Assenov, Y.; et al. Integrative analysis defines distinct prognostic subgroups of intrahepatic cholangiocarcinoma. Hepatology 2019, 69, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Kaminska, K.; Nalejska, E.; Kubiak, M.; Wojtysiak, J.; Zolna, L.; Kowalewski, J.; Lewandowska, M.A. Prognostic and predictive epigenetic biomarkers in oncology. Mol. Diagn. Ther. 2018, 23, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Arif, K.M.T.; Elliott, E.K.; Haupt, L.M.; Griffiths, L.R. Regulatory mechanisms of epigenetic miRNA relationships in human cancer and potential as therapeutic targets. Cancers 2020, 12, 2922. [Google Scholar] [CrossRef]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Hernandez, L.G.; van Steeg, H.; Luijten, M.; van Benthem, J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res. 2009, 682, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Weisz, J.; Kuemmerle, N.B.; Salzberg, A.C.; Berg, A.; Brown, D.G.; Kubik, L.; Palorini, R.; Al-Mulla, F.; Al-Temaimi, R.; et al. Metabolic reprogramming and dysregulated metabolism: Cause, consequence and/or enabler of environmental carcinogenesis? Carcinogenesis 2015, 36 (Suppl. S1), S203–S231. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande, V.J.; Blyth, K.; et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Burr, S.; Caldwell, A.; Chong, M.; Beretta, M.; Metcalf, S.; Hancock, M.; Arno, M.; Balu, S.; Kropf, V.L.; Mistry, R.K.; et al. Oxygen gradients can determine epigenetic asymmetry and cellular differentiation via differential regulation of Tet activity in embryonic stem cells. Nucleic Acids Res. 2018, 46, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, C. Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicology 2017, 391, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quiros, P.M.; Auwerx, J. Mitochondria and epigenetics—Crosstalk in homeostasis and stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Dellino, G.I.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Guadagnini, R.; Halamoda, K.B.; Walker, L.; Pojana, G.; Magdolenova, Z.; Bilanicova, D.; Saunders, M.; Juillerat-Jeanneret, L.; Marcomini, A.; Huk, A.; et al. Toxicity screenings of nanomaterials: Challenges due to interference with assay processes and components of classic in vitro tests. Nanotoxicology 2015, 9 (Suppl. S1), 13–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, R.; Burwinkel, B.; Breitling, L.P.; Brenner, H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ. Health Perspect. 2014, 122, 131–137. [Google Scholar] [CrossRef]
- Parkinson, A. Biotransformation of xenobiotics. In Casarett and Doull’s Toxicology: The Basic Science of Poisons; Klaassen, C.D., Amdur, M.O., Doull, J., Eds.; McGraw-Hill: New York, NY, USA, 1996; pp. 113–186. [Google Scholar]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Cyr, A.R.; Domann, F.E. The redox basis of epigenetic modifications: From mechanisms to functional consequences. Antioxid. Redox Signal. 2011, 15, 551–589. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kamendulis, L.M.; Zhang, H.; Wang, Y.; Klaunig, J.E. Morphological transformation and oxidative stress induced by cyanide in Syrian hamster embryo (SHE) cells. Toxicol. Sci. 2002, 68, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kamendulis, L.M.; Klaunig, J.E. Mechanisms for the induction of oxidative stress in Syrian hamster embryo cells by acrylonitrile. Toxicol. Sci. 2002, 67, 247–255. [Google Scholar] [CrossRef][Green Version]
- Berwald, Y.; Sachs, L. In vitro cell transformation with chemical carcinogens. Nature 1963, 200, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Pienta, R.J.; Poiley, J.A.; Lebherz, W.B., III. Morphological transformation of early passage golden Syrian hamster embryo cells derived from cryopreserved primary cultures as a reliable in vitro bioassay for identifying diverse carcinogens. Int. J. Cancer 1977, 19, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Friso, S.; Udali, S.; De, S.D.; Choi, S.W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metab. 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [CrossRef]
- Dominguez-Salas, P.; Moore, S.E.; Cole, D.; da Costa, K.A.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Innis, S.M.; Waterland, R.A.; Zeisel, S.H.; et al. DNA methylation potential: Dietary intake and blood concentrations of one-carbon metabolites and cofactors in rural African women. Am. J. Clin. Nutr. 2013, 97, 1217–1227. [Google Scholar] [CrossRef]
- Leal, J.; Ferrer, I.; Blanco-Aparicio, C.; Hernandez-Losa, J.; Ramon, Y.C.; Carnero, A.; Lleonart, M. S-Adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis 2008, 29, 2089–2095. [Google Scholar] [CrossRef]
- Landkocz, Y.; Poupin, P.; Atienzar, F.; Vasseur, P. Transcriptomic effects of di-(2-ethylhexyl)-phthalate in Syrian hamster embryo cells: An important role of early cytoskeleton disturbances in carcinogenesis? BMC Genom. 2011, 12, 524. [Google Scholar] [CrossRef]
- Lehman-McKeeman, L.D.; Gamsky, E.A. Choline supplementation inhibits diethanolamine-induced morphological transformation in syrian hamster embryo cells: Evidence for a carcinogenic mechanism. Toxicol. Sci. 2000, 55, 303–310. [Google Scholar] [CrossRef][Green Version]
- Desaulniers, D.; Xiao, G.H.; Lian, H.; Feng, Y.L.; Zhu, J.; Nakai, J.; Bowers, W.J. Effects of mixtures of polychlorinated biphenyls, methylmercury, and organochlorine pesticides on hepatic DNA methylation in prepubertal female Sprague-Dawley rats. Int. J. Toxicol. 2009, 28, 294–307. [Google Scholar] [CrossRef]
- Fling, R.R.; Doskey, C.M.; Fader, K.A.; Nault, R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) dysregulates hepatic one carbon metabolism during the progression of steatosis to steatohepatitis with fibrosis in mice. Sci. Rep. 2020, 10, 14831. [Google Scholar] [CrossRef]
- Park, I.Y.; Chowdhury, P.; Tripathi, D.N.; Powell, R.T.; Dere, R.; Terzo, E.A.; Rathmell, W.K.; Walker, C.L. Methylated alpha-tubulin antibodies recognize a new microtubule modification on mitotic microtubules. MAbs 2016, 8, 1590–1597. [Google Scholar] [CrossRef]
- Park, I.Y.; Powell, R.T.; Tripathi, D.N.; Dere, R.; Ho, T.H.; Blasius, T.L.; Chiang, Y.C.; Davis, I.J.; Fahey, C.C.; Hacker, K.E.; et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell 2016, 166, 950–962. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Kienhofer, S.; Musheev, M.U.; Stapf, U.; Helm, M.; Schomacher, L.; Niehrs, C.; Schafer, A. GADD45a physically and functionally interacts with TET1. Differentiation 2015, 90, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gu, T.P.; Weber, A.R.; Shen, J.Z.; Li, B.Z.; Xie, Z.G.; Yin, R.; Guo, F.; Liu, X.; Tang, F.; et al. Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 2015, 43, 3986–3997. [Google Scholar] [CrossRef]
- Feng, W.; Chen, S.; Wang, J.; Wang, X.; Chen, H.; Ning, W.; Zhang, Y. DHX33 recruits Gadd45a to cause DNA demethylation and regulates a subset of gene transcription. Mol. Cell. Biol. 2020, 40, e00460-19. [Google Scholar] [CrossRef] [PubMed]
- Pietrasik, S.; Zajac, G.; Morawiec, J.; Soszynski, M.; Fila, M.; Blasiak, J. Interplay between BRCA1 and GADD45A and its potential for nucleotide excision repair in breast cancer pathogenesis. Int. J. Mol. Sci. 2020, 21, 870. [Google Scholar] [CrossRef]
- Lopez, V.; Fernandez, A.F.; Fraga, M.F. The role of 5-hydroxymethylcytosine in development, aging and age-related diseases. Ageing Res. Rev. 2017, 37, 28–38. [Google Scholar] [CrossRef]
- Munzel, M.; Globisch, D.; Carell, T. 5-Hydroxymethylcytosine, the sixth base of the genome. Angew Chem. Int. Ed. Engl. 2011, 50, 6460–6468. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.S.J.; Ortmann, B.M.; Martinelli, A.W.; Houghton, J.W.; Costa, A.S.H.; Burr, S.P.; Antrobus, R.; Frezza, C.; Nathan, J.A. ABHD11 maintains 2-oxoglutarate metabolism by preserving functional lipoylation of the 2-oxoglutarate dehydrogenase complex. Nat. Commun. 2020, 11, 4046. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z.; et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 2013, 45, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 11. [Google Scholar] [CrossRef]
- DiTroia, S.P.; Percharde, M.; Guerquin, M.J.; Wall, E.; Collignon, E.; Ebata, K.T.; Mesh, K.; Mahesula, S.; Agathocleous, M.; Laird, D.J.; et al. Maternal vitamin C regulates reprogramming of DNA methylation and germline development. Nature 2019, 573, 271–275. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Chano, T.; Avnet, S.; Kusuzaki, K.; Bonuccelli, G.; Sonveaux, P.; Rotili, D.; Mai, A.; Baldini, N. Tumour-specific metabolic adaptation to acidosis is coupled to epigenetic stability in osteosarcoma cells. Am. J. Cancer Res. 2016, 6, 859–875. [Google Scholar] [PubMed]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Severson, P.L.; Kulak, M.V.; Futscher, B.W.; Domann, F.E. Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines. Toxicol. Appl. Pharmacol. 2014, 274, 408–416. [Google Scholar] [CrossRef]
- Button, E.L.; Bersten, D.C.; Whitelaw, M.L. HIF has Biff—Crosstalk between HIF1a and the family of bHLH/PAS proteins. Exp. Cell Res. 2017, 356, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, S.; Hudlikar, R.; Wang, L.; Shannar, A.; Peter, R.; Chou, P.J.; Kuo, H.D.; Liu, Z.; Kong, A.N. Redox signaling, mitochondrial metabolism, epigenetics and redox active phytochemicals. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef]
- Katan-Khaykovich, Y.; Struhl, K. Dynamics of global histone acetylation and deacetylation in vivo: Rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002, 16, 743–752. [Google Scholar] [CrossRef]
- Harris, J.; Gouhier, A.; Drouin, J. Mechanisms in endocrinology: Pioneer transcription factors in pituitary development and tumorigenesis. Eur. J. Endocrinol. 2021, 184, R1–R15. [Google Scholar] [CrossRef]
- McBrian, M.A.; Behbahan, I.S.; Ferrari, R.; Su, T.; Huang, T.W.; Li, K.; Hong, C.S.; Christofk, H.R.; Vogelauer, M.; Seligson, D.B.; et al. Histone acetylation regulates intracellular pH. Mol. Cell 2013, 49, 310–321. [Google Scholar] [CrossRef]
- Sutoo, S.; Maeda, T.; Suzuki, A.; Kato, Y. Adaptation to chronic acidic extracellular pH elicits a sustained increase in lung cancer cell invasion and metastasis. Clin. Exp. Metastasis 2020, 37, 133–144. [Google Scholar] [CrossRef]
- Ruch, R.J.; Klaunig, J.E.; Kerckaert, G.A.; Leboeuf, R.A. Modification of gap junctional intercellular communication by changes in extracellular pH in Syrian hamster embryo cells. Carcinogenesis 1990, 11, 909–913. [Google Scholar] [CrossRef]
- Leboeuf, R.A.; Kerckaert, G.A.; Aardema, M.J.; Gibson, D.P. Multistage neoplastic transformation of Syrian hamster embryo cells cultured at pH 6.70. Cancer Res. 1990, 50, 3722–3729. [Google Scholar]
- Yoshida, M.; Kudo, N.; Kosono, S.; Ito, A. Chemical and structural biology of protein lysine deacetylases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 297–321. [Google Scholar] [CrossRef]
- Chrun, E.S.; Modolo, F.; Daniel, F.I. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Res. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S.; Morris, K.V. Transcriptional gene silencing in humans. Nucleic Acids Res. 2016, 44, 6505–6517. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dong, C.; Cui, J.; Wang, Y.; Hong, X. Over-expressed lncRNA HOTAIRM1 promotes tumor growth and invasion through up-regulating HOXA1 and sequestering G9a/EZH2/Dnmts away from the HOXA1 gene in glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2018, 37, 265. [Google Scholar] [CrossRef]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, 2110. [Google Scholar] [CrossRef]
- Ren, W.; Fan, H.; Grimm, S.A.; Guo, Y.; Kim, J.J.; Yin, J.; Li, L.; Petell, C.J.; Tan, X.F.; Zhang, Z.M.; et al. Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc. Natl. Acad. Sci. USA 2020, 117, 18439–18447. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Weinberg, D.N.; Papillon-Cavanagh, S.; Chen, H.; Yue, Y.; Chen, X.; Rajagopalan, K.N.; Horth, C.; McGuire, J.T.; Xu, X.; Nikbakht, H.; et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019, 573, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, X.; Wang, H.; Shao, Q. The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 2018, 10, 2222–2233. [Google Scholar] [PubMed]
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.W.; Yen, R.W.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Brenner, C. KRAS-driven miR-29b expression is required for tumor suppressor gene silencing. Oncotarget 2017, 8, 74755–74766. [Google Scholar] [CrossRef]
- Gazin, C.; Wajapeyee, N.; Gobeil, S.; Virbasius, C.M.; Green, M.R. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature 2007, 449, 1073–1077. [Google Scholar] [CrossRef]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef]
- Gu, T.; Lin, X.; Cullen, S.M.; Luo, M.; Jeong, M.; Estecio, M.; Shen, J.; Hardikar, S.; Sun, D.; Su, J.; et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018, 19, 88. [Google Scholar] [CrossRef]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Fukuda, S.; Umeda, T.; Tanaka, S.; et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019, 9, 1903. [Google Scholar] [CrossRef]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA hypomethylation in epithelial ovarian cancer: Passive demethylation and association with genomic instability. Cancers 2020, 12, 764. [Google Scholar] [CrossRef]
- Oakes, C.C.; Martin-Subero, J.I. Insight into origins, mechanisms, and utility of DNA methylation in B-cell malignancies. Blood 2018, 132, 999–1006. [Google Scholar] [CrossRef]
- Jeong, M.; Goodell, M.A. New answers to old questions from genome-wide maps of DNA methylation in hematopoietic cells. Exp. Hematol. 2014, 42, 609–617. [Google Scholar] [CrossRef]
- Holliday, R.; Grigg, G.W. DNA methylation and mutation. Mutat. Res. 1993, 285, 61–67. [Google Scholar] [CrossRef]
- Ilnytskyy, Y.; Kovalchuk, O. Non-targeted radiation effects-an epigenetic connection. Mutat. Res. 2011, 714, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, M.; Ruan, P.; Teschendorff, A.E.; Wang, S. Detection of epigenetic field defects using a weighted epigenetic distance-based method. Nucleic Acids Res. 2019, 47, e6. [Google Scholar] [CrossRef] [PubMed]
- Pirini, F.; Noazin, S.; Jahuira-Arias, M.H.; Rodriguez-Torres, S.; Friess, L.; Michailidi, C.; Cok, J.; Combe, J.; Vargas, G.; Prado, W.; et al. Early detection of gastric cancer using global, genome-wide and IRF4, ELMO1, CLIP4 and MSC DNA methylation in endoscopic biopsies. Oncotarget 2017, 8, 38501–38516. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, M.; Puig, M.; Carmona, F.J.; Maqueda, M.; Velasquez, A.; Gomez, A.; Labernadie, A.; Lugo, R.; Gabasa, M.; Rigat-Brugarolas, L.G.; et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 2015, 36, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Veland, N.; Hardikar, S.; Zhong, Y.; Gayatri, S.; Dan, J.; Strahl, B.D.; Rothbart, S.B.; Bedford, M.T.; Chen, T. The arginine methyltransferase PRMT6 regulates DNA methylation and contributes to Global DNA hypomethylation in cancer. Cell Rep. 2017, 21, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, C.B.; Nishiyama, A.; Ryan, J.; Nakamura, R.; Yigit, M.; Gluck, I.M.; Trummer, C.; Qin, W.; Bartoschek, M.D.; Traube, F.R.; et al. Recent evolution of a TET-controlled and DPPA3/STELLA-driven pathway of passive DNA demethylation in mammals. Nat. Commun. 2020, 11, 5972. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Brenner, C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014, 9, 1827–1840. [Google Scholar] [CrossRef]
- Gagliardi, M.; Strazzullo, M.; Matarazzo, M.R. DNMT3B functions: Novel insights from human disease. Front. Cell Dev. Biol. 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Mulholland, C.B.; Bultmann, S.; Kori, S.; Endo, A.; Saeki, Y.; Qin, W.; Trummer, C.; Chiba, Y.; Yokoyama, H.; et al. Two distinct modes of DNMT1 recruitment ensure stable maintenance DNA methylation. Nat. Commun. 2020, 11, 1222. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Inflammation-mediated cytosine damage: A mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res. 2007, 67, 5583–5586. [Google Scholar] [CrossRef] [PubMed]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Soares, A.O.; Goff, L.A.; Talasila, A.; Choi, J.A.; Ivenitsky, D.; Karma, S.; Brophy, B.; Devine, S.E.; Meltzer, S.J.; et al. Striking heterogeneity of somatic L1 retrotransposition in single normal and cancerous gastrointestinal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 32215–32222. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Suzuki, K.; Fukui, T.; Takayama, Y.; Kakizawa, N.; Watanabe, F.; Ishikawa, H.; Muto, Y.; Kato, T.; Saito, M.; et al. Overexpression of satellite alpha transcripts leads to chromosomal instability via segregation errors at specific chromosomes. Int. J. Oncol. 2018, 52, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Pettersson, U.; Beard, C.; Jackson-Grusby, L.; Jaenisch, R. DNA hypomethylation leads to elevated mutation rates. Nature 1998, 395, 89–93. [Google Scholar] [CrossRef]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Howard, G.; Eiges, R.; Gaudet, F.; Jaenisch, R.; Eden, A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008, 27, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Jackson-Grusby, L.; Linhart, H.; Meissner, A.; Eden, A.; Lin, H.; Jaenisch, R. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13580–13585. [Google Scholar] [CrossRef]
- Choi, S.H.; Worswick, S.; Byun, H.M.; Shear, T.; Soussa, J.C.; Wolff, E.M.; Douer, D.; Garcia-Manero, G.; Liang, G.; Yang, A.S. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int. J. Cancer 2009, 125, 723–729. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Campan, M.; Long, T.I.; Kim, M.; Woods, C.; Fiala, E.; Ehrlich, M.; Laird, P.W. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005, 33, 6823–6836. [Google Scholar] [CrossRef] [PubMed]
- Tubio, J.M.C.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Van, M.M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De, C.M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 2019, 29, 871–885. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, J.; Noh, K.M.; Choi, W.Y.; Jeon, S.; Oh, G.T.; Kim-Ha, J.; Jin, Y.; Cho, S.W.; Kim, Y.J. Intragenic CpG islands play important roles in bivalent chromatin assembly of developmental genes. Proc. Natl. Acad. Sci. USA 2017, 114, E1885–E1894. [Google Scholar] [CrossRef]
- Guerra-Calderas, L.; González-Barrios, R.; Patiño, C.C.; Alcaraz, N.; Salgado-Albarrán, M.; de León, D.C.; Hernández, C.C.; Sánchez-Pérez, Y.; Maldonado-Martínez, H.A.; De la Rosa-Velazquez, I.A.; et al. CTCF-KDM4A complex correlates with histone modifications that negatively regulate CHD5 gene expression in cancer cell lines. Oncotarget 2018, 9, 17028–17042. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Jeltsch, A.; Broche, J.; Bashtrykov, P. Molecular processes connecting DNA methylation patterns with DNA methyltransferases and histone modifications in mammalian genomes. Genes 2018, 9, 566. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Heberle, E.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Szabo, P.E. Gene body profiles of 5-hydroxymethylcytosine: Potential origin, function and use as a cancer biomarker. Epigenomics 2018, 10, 1029–1032. [Google Scholar] [CrossRef]
- Blagitko-Dorfs, N.; Schlosser, P.; Greve, G.; Pfeifer, D.; Meier, R.; Baude, A.; Brocks, D.; Plass, C.; Lubbert, M. Combination treatment of acute myeloid leukemia cells with DNMT and HDAC inhibitors: Predominant synergistic gene downregulation associated with gene body demethylation. Leukemia 2018, 33, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo, Y.H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Momparler, R.L.; Cote, S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin. Investig. Drugs 2015, 24, 1031–1043. [Google Scholar] [CrossRef]
- Smurova, K.; De Wulf, P. Centromere and pericentromere transcription: Roles and regulation ... in sickness and in health. Front. Genet. 2018, 9, 674. [Google Scholar] [CrossRef]
- Kishikawa, T.; Otsuka, M.; Yoshikawa, T.; Ohno, M.; Yamamoto, K.; Yamamoto, N.; Kotani, A.; Koike, K. Quantitation of circulating satellite RNAs in pancreatic cancer patients. JCI Insight 2016, 1, e86646. [Google Scholar] [CrossRef]
- Desaulniers, D.; Xiao, G.H.; Cummings-Lorbetskie, C.; Stubbert, L.J.; Parfett, C. DNA methylation of repeated elements and cancer-related genes in normal human liver, and in cancer and non-cancer liver cell lines, treated with 5adC or PCB126. Curr. Top. Toxicol. 2016, 12, 47–74. [Google Scholar] [CrossRef]
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development of an IATA for non-genotoxic carcinogenic chemical substances. ALTEX 2016, 33, 359–392. [Google Scholar] [CrossRef] [PubMed]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Parfett, C.L.; Desaulniers, D. A Tox21 approach to altered epigenetic landscapes: Assessing epigenetic toxicity pathways leading to altered gene expression and oncogenic transformation in vitro. Int. J. Mol. Sci. 2017, 18, 1179. [Google Scholar] [CrossRef]
- Paquin, K.L.; Howlett, N.G. Understanding the histone DNA repair code: H4K20me2 makes its mark. Mol. Cancer Res. 2018, 16, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant expression of p16(INK4a) in human cancers—A new biomarker? Cancer Rep. Rev. 2018, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Gonzalez, S.; Lopez-Soto, A. Immunosurveillance of cancer cell stress. Cell Stress 2019, 3, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Xu, B.; Putnam, D.; Thrasher, A.; Li, C.; Yang, J.; Chen, X. MethylationToActivity: A deep-learning framework that reveals promoter activity landscapes from DNA methylomes in individual tumors. Genome Biol. 2021, 22, 24. [Google Scholar] [CrossRef]
- McKenna, E.S.; Roberts, C.W. Epigenetics and cancer without genomic instability. Cell Cycle 2009, 8, 23–26. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stutz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Robertson, K.D. Role of DNA methylation in genome stability. In Genome Stability. From Virus to Human Application; Academic Press: Cambridge, MA, USA, 2016; pp. 409–424. [Google Scholar]
- Jose, C.C.; Wang, Z.; Tanwar, V.S.; Zhang, X.; Zang, C.; Cuddapah, S. Nickel-induced transcriptional changes persist post exposure through epigenetic reprogramming. Epigenet. Chromatin 2019, 12, 75. [Google Scholar] [CrossRef]
- Zhu, Y.; Costa, M. Metals and molecular carcinogenesis. Carcinogenesis 2020, 41, 1161–1172. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, J.; Li, L.; Amato, N.J.; Wang, Z.; Wang, Y. Arsenite targets the zinc finger domains of tet proteins and inhibits tet-mediated oxidation of 5-methylcytosine. Environ. Sci. Technol. 2015, 49, 11923–11931. [Google Scholar] [CrossRef]
- Rea, M.; Gripshover, T.; Fondufe-Mittendorf, Y. Selective inhibition of CTCF binding by iAs directs TET-mediated reprogramming of 5-hydroxymethylation patterns in iAs-transformed cells. Toxicol. Appl. Pharmacol. 2018, 338, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Yin, A.A.; Lu, N.; Etcheverry, A.; Aubry, M.; Barnholtz-Sloan, J.; Zhang, L.H.; Mosser, J.; Zhang, W.; Zhang, X.; Liu, Y.H.; et al. A novel prognostic six-CpG signature in glioblastomas. CNS Neurosci. Ther. 2018, 24, 167–177. [Google Scholar] [CrossRef]
- Nahta, R.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Andrade-Vieira, R.; Bay, S.N.; Brown, D.G.; Calaf, G.M.; Castellino, R.C.; Cohen-Solal, K.A.; et al. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015, 36 (Suppl. S1), S2–S18. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Kondoh, H.; Lleonart, M.E.; Martinez-Leal, J.F.; Mondello, C.; Scovassi, A.I.; Bisson, W.H.; Amedei, A.; Roy, R.; et al. Disruptive chemicals, senescence and immortality. Carcinogenesis 2015, 36 (Suppl. S1), S19–S37. [Google Scholar] [CrossRef]
- Kravchenko, J.; Corsini, E.; Williams, M.A.; Decker, W.; Manjili, M.H.; Otsuki, T.; Singh, N.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; et al. Chemical compounds from anthropogenic environment and immune evasion mechanisms: Potential interactions. Carcinogenesis 2015, 36 (Suppl. S1), S111–S127. [Google Scholar] [CrossRef]
- Henser-Brownhill, T.; Monserrat, J.; Scaffidi, P. Generation of an arrayed CRISPR-Cas9 library targeting epigenetic regulators: From high-content screens to in vivo assays. Epigenetics 2017, 12, 1065–1075. [Google Scholar] [CrossRef]
- Halaburkova, A.; Cahais, V.; Novoloaca, A.; Araujo, M.G.D.S.; Khoueiry, R.; Ghantous, A.; Herceg, Z. Pan-cancer multi-omics analysis and orthogonal experimental assessment of epigenetic driver genes. Genome Res. 2020, 30, 1517–1532. [Google Scholar] [CrossRef]
- Middleton, A.; Cooper, S.; Cull, T.; Stark, R.; Adeleye, Y.B.K.; Clewell, R.; Jennings, P.; Guo, J.; Liu, C.; McMullen, P.; et al. Case studies in cellular stress: Defining adversity/adaptation tipping points. Workshop report. Appl. Vitro Toxicol. 2017, 3, 199–210. [Google Scholar] [CrossRef]
- McMullen, P.D.; Pendse, S.; Adeleye, Y.; Carmichael, P.L.; Andersen, M.E.; Clewell, R.A. Using transcriptomics to evaluate tresholds in genotoxicity dose-response. In Toxicogenomics in Predictive Carcinogenicity; Waters, M.D., Thomas, R.S., Eds.; Royal Society of Chemistry: Cambridge, UK, 2016; pp. 185–208. [Google Scholar]
- Miousse, I.R.; Murphy, L.A.; Lin, H.; Schisler, M.R.; Sun, J.; Chalbot, M.G.; Sura, R.; Johnson, K.; Lebaron, M.J.; Kavouras, I.G.; et al. Dose-response analysis of epigenetic, metabolic, and apical endpoints after short-term exposure to experimental hepatotoxicants. Food Chem. Toxicol. 2017, 109, 690–702. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Alyea, R.A.; Moore, N.P.; Lebaron, M.J.; Gollapudi, B.B.; Rasoulpour, R.J. Is the current product safety assessment paradigm protective for epigenetic mechanisms? J. Pharmacol. Toxicol. Methods 2012, 66, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kurklu, B.; Whitehead, R.H.; Ong, E.K.; Minamoto, T.; Fox, J.G.; Mann, J.R.; Judd, L.M.; Giraud, A.S.; Menheniott, T.R. Lineage-specific RUNX3 hypomethylation marks the preneoplastic immune component of gastric cancer. Oncogene 2015, 34, 2856–2866. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Xiao, G.H.; Leingartner, K.; Chu, I.; Musicki, B.; Tsang, B.K. Comparisons of brain, uterus and liver mRNA expressions of cytochrome P450s, Dnmt-1, and catechol-o-methyltransferase, in prepubertal female Sprague Dawley rats exposed to aryl hydrocarbon receptor agonists. Toxicol. Sci. 2005, 86, 175–184. [Google Scholar] [CrossRef]
- Prins, G.S.; Ye, S.H.; Birch, L.; Zhang, X.; Cheong, A.; Lin, H.; Calderon-Gierszal, E.; Groen, J.; Hu, W.Y.; Ho, S.M.; et al. Prostate cancer risk and DNA methylation signatures in aging rats following developmental BPA exposure: A dose-response analysis. Environ. Health Perspect. 2017, 125, 077007. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Morison, I.M.; Eccles, M.R.; Stockwell, P.A. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. Methods Mol. Biol. 2017, 1537, 249–277. [Google Scholar] [PubMed]
- Lentini, A.; Nestor, C.E. Mapping DNA methylation in mammals: The state of the art. Methods Mol. Biol. 2021, 2198, 37–50. [Google Scholar] [PubMed]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van, N.L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Carmona, J.J.; Accomando, W.P., Jr.; Binder, A.M.; Hutchinson, J.N.; Pantano, L.; Izzi, B.; Just, A.C.; Lin, X.; Schwartz, J.; Vokonas, P.S.; et al. Empirical comparison of reduced representation bisulfite sequencing and Infinium BeadChip reproducibility and coverage of DNA methylation in humans. NPJ Genom. Med. 2017, 2, 13. [Google Scholar] [CrossRef]
- Beck, S. Taking the measure of the methylome. Nat. Biotechnol. 2010, 28, 1026–1028. [Google Scholar] [CrossRef]
- Dreval, K.; Tryndyak, V.; de Conti, A.; Beland, F.A.; Pogribny, I.P. Gene expression and DNA methylation alterations during non-alcoholic steatohepatitis-associated liver carcinogenesis. Front. Genet. 2019, 10, 486. [Google Scholar] [CrossRef]
- Omura, K.; Uehara, T.; Morikawa, Y.; Hayashi, H.; Mitsumori, K.; Minami, K.; Kanki, M.; Yamada, H.; Ono, A.; Urushidani, T. Comprehensive analysis of DNA methylation and gene expression of rat liver in a 2-stage hepatocarcinogenesis model. J. Toxicol. Sci. 2014, 39, 837–848. [Google Scholar] [CrossRef]
- Lempiainen, H.; Muller, A.; Brasa, S.; Teo, S.S.; Roloff, T.C.; Morawiec, L.; Zamurovic, N.; Vicart, A.; Funhoff, E.; Couttet, P.; et al. Phenobarbital mediates an epigenetic switch at the constitutive androstane receptor (CAR) target gene Cyp2b10 in the liver of B6C3F1 mice. PLoS ONE 2011, 6, e18216. [Google Scholar] [CrossRef]
- Tryndyak, V.; Kindrat, I.; Dreval, K.; Churchwell, M.I.; Beland, F.A.; Pogribny, I.P. Effect of aflatoxin B1, benzo[a]pyrene, and methapyrilene on transcriptomic and epigenetic alterations in human liver HepaRG cells. Food Chem. Toxicol. 2018, 121, 214–223. [Google Scholar] [CrossRef]
- Karlic, R.; Chung, H.R.; Lasserre, J.; Vlahovicek, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef]
- Gao, Y.; Widschwendter, M.; Teschendorff, A.E. DNA methylation patterns in normal tissue correlate more strongly with breast cancer status than copy-number variants. EBioMedicine 2018, 31, 243–252. [Google Scholar] [CrossRef]
- Zheng, S.C.; Breeze, C.E.; Beck, S.; Teschendorff, A.E. Identification of differentially methylated cell types in epigenome-wide association studies. Nat. Methods 2018, 15, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Widschwendter, M.; Teschendorff, A.E. Systems-epigenomics inference of transcription factor activity implicates aryl-hydrocarbon-receptor inactivation as a key event in lung cancer development. Genome Biol. 2017, 18, 236. [Google Scholar] [CrossRef] [PubMed]
- Harrill, J.A.; Viant, M.R.; Yauk, C.L.; Sachana, M.; Gant, T.W.; Auerbach, S.S.; Beger, R.D.; Bouhifd, M.; O’Brien, J.; Burgoon, L.; et al. Progress towards an OECD reporting framework for transcriptomics and metabolomics in regulatory toxicology. Regul. Toxicol. Pharmacol. 2021, 125, 105020. [Google Scholar] [CrossRef] [PubMed]
- Noberini, R.; Osti, D.; Miccolo, C.; Richichi, C.; Lupia, M.; Corleone, G.; Hong, S.P.; Colombo, P.; Pollo, B.; Fornasari, L.; et al. Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 2018, 46, 3817–3832. [Google Scholar] [CrossRef] [PubMed]
- Sistare, F.D.; Morton, D.; Alden, C.; Christensen, J.; Keller, D.; Jonghe, S.D.; Storer, R.D.; Reddy, M.V.; Kraynak, A.; Trela, B.; et al. An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: Support for a proposal to modify current regulatory guidelines. Toxicol. Pathol. 2011, 39, 716–744. [Google Scholar] [CrossRef]
- Greally, J.M.; Jacobs, M.N. In vitro and in vivo testing methods of epigenomic endpoints for evaluating endocrine disruptors. ALTEX 2013, 30, 445–471. [Google Scholar] [CrossRef]
- Madrid, A.; Chopra, P.; Alisch, R.S. Species-specific 5 mC and 5 hmC genomic landscapes indicate epigenetic contribution to human brain evolution. Front. Mol. Neurosci. 2018, 11, 39. [Google Scholar] [CrossRef]
- Zeng, J.; Konopka, G.; Hunt, B.G.; Preuss, T.M.; Geschwind, D.; Yi, S.V. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am. J. Hum. Genet. 2012, 91, 455–465. [Google Scholar] [CrossRef]
- Zhang, C.; Hoshida, Y.; Sadler, K.C. Comparative epigenomic profiling of the DNA methylome in mouse and zebrafish uncovers high interspecies divergence. Front. Genet. 2016, 7, 110. [Google Scholar] [CrossRef]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species- and cell type-specific requirements for cellular transformation. Cancer Cell 2004, 6, 171–183. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Yeom, H.; Han, B.I.; Ham, B.J.; Lee, Y.M.; Han, M.R.; Lee, M. Predicting carcinogenic mechanisms of non-genotoxic carcinogens via combined analysis of global DNA methylation and in vitro cell transformation. Int. J. Mol. Sci. 2020, 21, 5387. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, K.; Sasaki, K.; Asada, S.; Tanaka, N.; Umeda, M. An assay method for the prediction of tumor promoting potential of chemicals by the use of Bhas 42 cells. Mutat. Res. 2004, 557, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Fonti, C.; Saumet, A.; Abi-Khalil, A.; Orsetti, B.; Cleroux, E.; Bender, A.; Dumas, M.; Schmitt, E.; Colinge, J.; Jacot, W.; et al. Distinct oncogenes drive different genome and epigenome alterations in human mammary epithelial cells. Int. J. Cancer 2019, 145, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K.; Su, W.; Blagosklonny, M.V.; Costa, M. Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res. 2000, 60, 3375–3378. [Google Scholar] [PubMed]
- Reichard, J.F.; Puga, A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2010, 2, 87–104. [Google Scholar] [CrossRef]
- Zhou, Q.; Xi, S. A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes. Regul. Toxicol. Pharmacol. 2018, 99, 78–88. [Google Scholar] [CrossRef]
- Reichard, J.F.; Schnekenburger, M.; Puga, A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 2007, 352, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, X.; Wang, J.; Liu, Z.; Gaile, D.; Wu, H.; Yu, G.; Mao, G.; Yang, Z.; Di, Z.; et al. Multi-generational impacts of arsenic exposure on genome-wide DNA methylation and the implications for arsenic-induced skin lesions. Environ. Int. 2018, 119, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Bozack, A.K.; Domingo-Relloso, A.; Haack, K.; Gamble, M.V.; Tellez-Plaza, M.; Umans, J.G.; Best, L.G.; Yracheta, J.; Gribble, M.O.; Cardenas, A.; et al. Locus-specific differential DNA methylation and urinary arsenic: An epigenome-wide association study in blood among adults with low-to-moderate arsenic exposure. Environ. Health Perspect. 2020, 128, 67015. [Google Scholar] [CrossRef]
- Paul, S.; Bhattacharjee, P.; Giri, A.K.; Bhattacharjee, P. Arsenic toxicity and epimutagenecity: The new LINEage. Biometals 2017, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.; Eckstein, M.; Eleazer, R.; Smith, C.; Fondufe-Mittendorf, Y.N. Genome-wide DNA methylation reprogramming in response to inorganic arsenic links inhibition of CTCF binding, DNMT expression and cellular transformation. Sci. Rep. 2017, 7, 41474. [Google Scholar] [CrossRef]
- Jiang, J.; Tam, L.M.; Wang, P.; Wang, Y. Arsenite targets the RING finger domain of Rbx1 E3 ubiquitin ligase to inhibit proteasome-mediated degradation of Nrf2. Chem. Res. Toxicol. 2018, 31, 380–387. [Google Scholar] [CrossRef]
- Cardoso, A.P.F.; Al-Eryani, L.; States, J.C. Arsenic-induced carcinogenesis: The impact of miRNA dysregulation. Toxicol. Sci. 2018, 165, 284–290. [Google Scholar] [CrossRef]
- Jiang, R.; Li, Y.; Zhang, A.; Wang, B.; Xu, Y.; Xu, W.; Zhao, Y.; Luo, F.; Liu, Q. The acquisition of cancer stem cell-like properties and neoplastic transformation of human keratinocytes induced by arsenite involves epigenetic silencing of let-7c via Ras/NF-kappaB. Toxicol. Lett. 2014, 227, 91–98. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Q.Y.; Wang, Z.; Zhu, Y.; Kluz, T.; Tan, W.; Li, J.; Wu, F.; Fang, L.; Zhang, X.; et al. Polyadenylation of histone H3.1 mRNA promotes cell transformation by displacing H3.3 from gene regulatory elements. iScience 2020, 23, 101518. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, P.; Paul, S.; Bhattacharjee, P. Understanding the mechanistic insight of arsenic exposure and decoding the histone cipher. Toxicology 2020, 430, 152340. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Sanyal, T.; Bhattacharjee, S.; Bhattacharjee, P. Epigenetic alteration of mismatch repair genes in the population chronically exposed to arsenic in West Bengal, India. Environ. Res. 2018, 163, 289–296. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, A.; Li, C.; Ma, L.; Tang, S.; Wang, Q.; Yang, G.; Li, J. The role of H3K9me2-regulated base excision repair genes in the repair of DNA damage induced by arsenic in HaCaT cells and the effects of Ginkgo biloba extract intervention. Environ. Toxicol. 2021, 36, 850–860. [Google Scholar] [CrossRef]
- Sanyal, T.; Paul, M.; Bhattacharjee, S.; Bhattacharjee, P. Epigenetic alteration of mitochondrial biogenesis regulatory genes in arsenic exposed individuals (with and without skin lesions) and in skin cancer tissues: A case control study. Chemosphere 2020, 258, 127305. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, T.; Bhattacharjee, P.; Bhattacharjee, S.; Bhattacharjee, P. Hypomethylation of mitochondrial D-loop and ND6 with increased mitochondrial DNA copy number in the arsenic-exposed population. Toxicology 2018, 408, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Broday, L.; Peng, W.; Kuo, M.H.; Salnikow, K.; Zoroddu, M.; Costa, M. Nickel compounds are novel inhibitors of histone H4 acetylation. Cancer Res. 2000, 60, 238–241. [Google Scholar] [PubMed]
- Golebiowski, F.; Kasprzak, K.S. Inhibition of core histones acetylation by carcinogenic nickel(II). Mol. Cell. Biochem. 2005, 279, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O’Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef]
- Lee, Y.W.; Klein, C.B.; Kargacin, B.; Salnikow, K.; Kitahara, J.; Dowjat, K.; Zhitkovich, A.; Christie, N.T.; Costa, M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Mol. Cell. Biol. 1995, 15, 2547–2557. [Google Scholar] [CrossRef]
- Chen, H.; Ke, Q.; Kluz, T.; Yan, Y.; Costa, M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol. Cell. Biol. 2006, 26, 3728–3737. [Google Scholar] [CrossRef]
- Chen, H.; Giri, N.C.; Zhang, R.; Yamane, K.; Zhang, Y.; Maroney, M.; Costa, M. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem. 2010, 285, 7374–7383. [Google Scholar] [CrossRef]
- Chen, Q.Y.; DesMarais, T.; Costa, M. Metals and mechanisms of carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 537–554. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutat. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef]
- Murphy, A.; Roy, N.; Sun, H.; Jin, C.; Costa, M. Induction of NUPR1 and AP1 contributes to the carcinogenic potential of nickel. Oncol. Rep. 2021, 45, 41. [Google Scholar] [CrossRef]
- Yin, R.; Mo, J.; Dai, J.; Wang, H. Nickel(II) inhibits tet-mediated 5-methylcytosine oxidation by high affinity displacement of the cofactor iron(II). ACS Chem. Biol. 2017, 12, 1494–1498. [Google Scholar] [CrossRef]
- Karaczyn, A.A.; Golebiowski, F.; Kasprzak, K.S. Ni(II) affects ubiquitination of core histones H2B and H2A. Exp. Cell Res. 2006, 312, 3252–3259. [Google Scholar] [CrossRef]
- Ke, Q.; Ellen, T.P.; Costa, M. Nickel compounds induce histone ubiquitination by inhibiting histone deubiquitinating enzyme activity. Toxicol. Appl. Pharmacol. 2008, 228, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.C.; Xu, B.; Jagannathan, L.; Trac, C.; Mallela, R.K.; Hattori, T.; Lai, D.; Koide, S.; Schones, D.E.; Cuddapah, S. Epigenetic dysregulation by nickel through repressive chromatin domain disruption. Proc. Natl. Acad. Sci. USA 2014, 111, 14631–14636. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.C.; Jagannathan, L.; Tanwar, V.S.; Zhang, X.; Zang, C.; Cuddapah, S. Nickel exposure induces persistent mesenchymal phenotype in human lung epithelial cells through epigenetic activation of ZEB1. Mol. Carcinog. 2018, 57, 794–806. [Google Scholar] [CrossRef]
- Sharapova, T.; Talaty, N.; Buck, W.R.; Fossey, S.; Liguori, M.J.; Van Vleet, T.R. Reduced hepatic global hydroxymethylation in mice treated with non-genotoxic carcinogens is transiently reversible with a methyl supplemented diet. Toxicol. Appl. Pharmacol. 2021, 415, 115439. [Google Scholar] [CrossRef] [PubMed]
- Efimova, O.A.; Koltsova, A.S.; Krapivin, M.I.; Tikhonov, A.V.; Pendina, A.A. Environmental epigenetics and genome flexibility: Focus on 5-hydroxymethylcytosine. Int. J. Mol. Sci. 2020, 21, 3223. [Google Scholar] [CrossRef] [PubMed]
- Pouche, L.; Vitobello, A.; Romer, M.; Glogovac, M.; MacLeod, A.K.; Ellinger-Ziegelbauer, H.; Westphal, M.; Dubost, V.; Stiehl, D.P.; Dumotier, B.; et al. Xenobiotic CAR activators induce Dlk1-Dio3 locus noncoding RNA expression in mouse liver. Toxicol. Sci. 2017, 158, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Couttet, P.; Bolognani, F.; Muller, A.; Dubost, V.; Luisier, R.; Del Rio, E.A.; Vitry, V.; Unterberger, E.B.; Thomson, J.P.; et al. Identification of Dlk1-Dio3 imprinted gene cluster noncoding RNAs as novel candidate biomarkers for liver tumor promotion. Toxicol. Sci. 2013, 131, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I. 2017 Michael Fry award lecture when DNA is actually not a target: Radiation epigenetics as a tool to understand and control cellular response to ionizing radiation. Radiat. Res. 2018, 190, 5–11. [Google Scholar] [CrossRef]
- Wu, C.; Guo, E.; Ming, J.; Sun, W.; Nie, X.; Sun, L.; Peng, S.; Luo, M.; Liu, D.; Zhang, L.; et al. Radiation-induced DNMT3B promotes radioresistance in nasopharyngeal carcinoma through methylation of p53 and p21. Mol. Ther. Oncolyt. 2020, 17, 306–319. [Google Scholar] [CrossRef]
- De Conti, A.; Tryndyak, V.; VonTungeln, L.S.; Churchwell, M.I.; Beland, F.A.; Antunes, A.M.M.; Pogribny, I.P. Genotoxic and epigenotoxic alterations in the lung and liver of mice induced by acrylamide: A 28 day drinking water study. Chem. Res. Toxicol. 2019, 32, 869–877. [Google Scholar] [CrossRef]
- Wu, Q.; Odwin-Dacosta, S.; Cao, S.; Yager, J.D.; Tang, W.Y. Estrogen down regulates COMT transcription via promoter DNA methylation in human breast cancer cells. Toxicol. Appl. Pharmacol. 2019, 367, 12–22. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, S.; Zhang, D. Association of inorganic arsenic exposure with liver cancer mortality: A meta-analysis. Environ. Res. 2014, 135, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Venkatratnam, A.; Marable, C.A.; Keshava, A.M.; Fry, R.C. Relationships among inorganic arsenic, nutritional status CpG methylation and microRNAs: A review of the literature. Epigenet. Insights 2021, 14, 2516865721989719. [Google Scholar] [CrossRef]
- Sun, M.; Tan, J.; Wang, M.; Wen, W.; He, Y. Inorganic arsenic-mediated upregulation of AS3MT promotes proliferation of nonsmall cell lung cancer cells by regulating cell cycle genes. Environ. Toxicol. 2021, 36, 204–212. [Google Scholar] [CrossRef]
- Saintilnord, W.N.; Fondufe-Mittendorf, Y. Arsenic-induced epigenetic changes in cancer development. Semin. Cancer Biol. 2021, 74. [Google Scholar] [CrossRef]
- Stanton, B.A.; Caldwell, K.; Congdon, C.B.; Disney, J.; Donahue, M.; Ferguson, E.; Flemings, E.; Golden, M.; Guerinot, M.L.; Highman, J.; et al. MDI biological laboratory arsenic summit: Approaches to limiting human exposure to arsenic. Curr. Environ. Health Rep. 2015, 2, 329–337. [Google Scholar] [CrossRef]
- Houben, A.J.; D’Onofrio, R.; Kokelj, S.V.; Blais, J.M. Factors affecting elevated arsenic and methyl mercury concentrations in small shield lakes surrounding gold mines near the Yellowknife, NT, (Canada) region. PLoS ONE 2016, 11, e0150960. [Google Scholar] [CrossRef]
- Sanyal, T.; Bhattacharjee, P.; Paul, S.; Bhattacharjee, P. Recent advances in arsenic research: Significance of differential susceptibility and sustainable strategies for mitigation. Front. Public Health 2020, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Chiou, H.Y.; Wang, T.W.; Hsueh, Y.M.; Wang, I.H.; Chen, C.J.; Lee, T.C. Association of blood arsenic levels with increased reactive oxidants and decreased antioxidant capacity in a human population of northeastern Taiwan. Environ. Health Perspect. 2001, 109, 1011–1017. [Google Scholar] [CrossRef]
- Roy, J.S.; Chatterjee, D.; Das, N.; Giri, A.K. Substantial evidences indicate that inorganic arsenic is a genotoxic carcinogen: A review. Toxicol. Res. 2018, 34, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Chowdhury, A.; Arnold, L.L. Inorganic arsenic: A non-genotoxic carcinogen. J. Environ. Sci. 2016, 49, 28–37. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Banerjee, M.; Giri, A.K. Role of genomic instability in arsenic-induced carcinogenicity. A review. Environ. Int. 2013, 53, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Giri, A.K. Epimutagenesis: A prospective mechanism to remediate arsenic-induced toxicity. Environ. Int. 2015, 81, 8–17. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Das, A.; Giri, A.K.; Bhattacharjee, P. Epigenetic regulations in alternative telomere lengthening: Understanding the mechanistic insight in arsenic-induced skin cancer patients. Sci. Total Environ. 2020, 704, 135388. [Google Scholar] [CrossRef]
- Draviam, V.M.; Xie, S.; Sorger, P.K. Chromosome segregation and genomic stability. Curr. Opin. Genet. Dev. 2004, 14, 120–125. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Chatterjee, D.; Singh, K.K.; Giri, A.K. Systems biology approaches to evaluate arsenic toxicity and carcinogenicity: An overview. Int. J. Hyg. Environ. Health 2013, 216, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fowler, B.A. Roles of biomarkers in evaluating interactions among mixtures of lead, cadmium and arsenic. Toxicol. Appl. Pharmacol. 2008, 233, 92–99. [Google Scholar] [CrossRef]
- Cui, X.; Wakai, T.; Shirai, Y.; Yokoyama, N.; Hatakeyama, K.; Hirano, S. Arsenic trioxide inhibits DNA methyltransferase and restores methylation-silenced genes in human liver cancer cells. Hum. Pathol. 2006, 37, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Han, B.R.; Park, W.H. Combination of arsenic trioxide and valproic acid efficiently inhibits growth of lung cancer cells via G2/M-phase arrest and apoptotic cell death. Int. J. Mol. Sci. 2020, 21, 2649. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.T.; Sultan, M.; Vidovic, D.; Dean, C.A.; Cruickshank, B.M.; Lee, K.; Loung, C.Y.; Holloway, R.W.; Hoskin, D.W.; Waisman, D.M.; et al. Retinoic acid and arsenic trioxide induce lasting differentiation and demethylation of target genes in APL cells. Sci. Rep. 2019, 9, 9414. [Google Scholar] [CrossRef]
- Raynal, N.J.; Lee, J.T.; Wang, Y.; Beaudry, A.; Madireddi, P.; Garriga, J.; Malouf, G.G.; Dumont, S.; Dettman, E.J.; Gharibyan, V.; et al. Targeting calcium signaling induces epigenetic reactivation of tumor suppressor genes in cancer. Cancer Res. 2016, 76, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Meehan, R.R. The application of genome-wide 5-hydroxymethylcytosine studies in cancer research. Epigenomics 2017, 9, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, C. Metal carcinogen exposure induces cancer stem cell-like property through epigenetic reprograming: A novel mechanism of metal carcinogenesis. Semin. Cancer Biol. 2019, 57, 95–104. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenet. 2021, 13, 71. [Google Scholar] [CrossRef]
- Torres, C.M.; Biran, A.; Burney, M.J.; Patel, H.; Henser-Brownhill, T.; Cohen, A.S.; Li, Y.; Ben-Hamo, R.; Nye, E.; Spencer-Dene, B.; et al. The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 2016, 353, 1644. [Google Scholar] [CrossRef]
- Howe, C.G.; Gamble, M.V. Influence of arsenic on global levels of histone posttranslational modifications: A review of the literature and challenges in the field. Curr. Environ. Health Rep. 2016, 3, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, J.; Chang, Q.; Beezhold, K.; Lu, Y.; Chen, F. JNK and STAT3 signaling pathways converge on Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells induced by arsenic. Cell Cycle 2013, 12, 112–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, X.; Li, Q.; Arita, A.; Sun, H.; Costa, M. Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol. Appl. Pharmacol. 2009, 236, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.J.; Ren, X.; Chu, F.; Aleshin, M.; Wintz, H.; Burlingame, A.; Smith, M.T.; Vulpe, C.D.; Zhang, L. Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicol. Appl. Pharmacol. 2009, 241, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, D.; Zhao, L.; Yang, Y.; Ding, J.; Dong, L.; Hu, L.; Wang, F.; Zhao, X.; Cai, Y.; et al. Arsenic trioxide reduces global histone H4 acetylation at lysine 16 through direct binding to histone acetyltransferase hMOF in human cells. PLoS ONE 2015, 10, e0141014. [Google Scholar] [CrossRef]
- Herbert, K.J.; Holloway, A.; Cook, A.L.; Chin, S.P.; Snow, E.T. Arsenic exposure disrupts epigenetic regulation of SIRT1 in human keratinocytes. Toxicol. Appl. Pharmacol. 2014, 281, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, J.; Chen, X.; Tsompana, M.; Gaile, D.; Buck, M.; Ren, X. A time-series analysis of altered histone H3 acetylation and gene expression during the course of MMAIII-induced malignant transformation of urinary bladder cells. Carcinogenesis 2017, 38, 378–390. [Google Scholar] [CrossRef]
- Ge, Y.; Zhu, J.; Wang, X.; Zheng, N.; Tu, C.; Qu, J.; Ren, X. Mapping dynamic histone modification patterns during arsenic-induced malignant transformation of human bladder cells. Toxicol. Appl. Pharmacol. 2018, 355, 164–173. [Google Scholar] [CrossRef]
- Ge, Y.; Gong, Z.; Olson, J.R.; Xu, P.; Buck, M.J.; Ren, X. Inhibition of monomethylarsonous acid (MMA(III))-induced cell malignant transformation through restoring dysregulated histone acetylation. Toxicology 2013, 312, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Basu, A.; Mahata, J.; Basu, S.; Sengupta, M.; Das, J.K.; Mukherjee, A.; Sarkar, A.K.; Mondal, L.; Ray, K.; et al. Cytogenetic damage and genetic variants in the individuals susceptible to arsenic-induced cancer through drinking water. Int. J. Cancer 2006, 118, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, P.; Paul, S.; Bhattacharjee, S.; Giri, A.K.; Bhattacharjee, P. Association of H3K79 monomethylation (an epigenetic signature) with arsenic-induced skin lesions. Mutat. Res. 2018, 807, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Borowa-Mazgaj, B.; Steward, C.R.; Beland, F.A.; Pogribny, I.P. Epigenetic effects of low-level sodium arsenite exposure on human liver HepaRG cells. Arch. Toxicol. 2020, 94, 3993–4005. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef]
- Selmin, O.I.; Donovan, M.G.; Skovan, B.; Paine-Murieta, G.D.; Romagnolo, D.F. Arsenic induced BRCA1 CpG promoter methylation is associated with the downregulation of ERalpha and resistance to tamoxifen in MCF7 breast cancer cells and mouse mammary tumor xenografts. Int. J. Oncol. 2019, 54, 869–878. [Google Scholar]
- Lamb, R.; Bonuccelli, G.; Ozsvari, B.; Peiris-Pages, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef]
- Smith, A.H.; Marshall, G.; Liaw, J.; Yuan, Y.; Ferreccio, C.; Steinmaus, C. Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ. Health Perspect. 2012, 120, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, M.; Suzuki, S.; Gi, M.; Kakehashi, A.; Oishi, Y.; Okuno, T.; Yukimatsu, N.; Wanibuchi, H. Dimethylarsinic acid (DMA) enhanced lung carcinogenesis via histone H3K9 modification in a transplacental mouse model. Arch. Toxicol. 2020, 94, 927–937. [Google Scholar] [CrossRef]
- Rojas, D.; Rager, J.E.; Smeester, L.; Bailey, K.A.; Drobna, Z.; Rubio-Andrade, M.; Styblo, M.; Garcia-Vargas, G.; Fry, R.C. Prenatal arsenic exposure and the epigenome: Identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol. Sci. 2015, 143, 97–106. [Google Scholar] [CrossRef]
- Bozack, A.K.; Cardenas, A.; Geldhof, J.; Quamruzzaman, Q.; Rahman, M.; Mostofa, G.; Christiani, D.C.; Kile, M.L. Cord blood DNA methylation of DNMT3A mediates the association between in utero arsenic exposure and birth outcomes: Results from a prospective birth cohort in Bangladesh. Environ. Res. 2020, 183, 109134. [Google Scholar] [CrossRef]
- Bozack, A.K.; Cardenas, A.; Quamruzzaman, Q.; Rahman, M.; Mostofa, G.; Christiani, D.C.; Kile, M.L. DNA methylation in cord blood as mediator of the association between prenatal arsenic exposure and gestational age. Epigenetics 2018, 13, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gunewardena, S.; Yue, C.J.; Klaassen, C.D.; Chorley, B.N.; Corton, J.C. Transplacental arsenic exposure produced 5-methylcytosine methylation changes and aberrant microRNA expressions in livers of male fetal mice. Toxicology 2020, 435, 152409. [Google Scholar] [CrossRef] [PubMed]
- Abuawad, A.; Bozack, A.K.; Saxena, R.; Gamble, M.V. Nutrition, one-carbon metabolism and arsenic methylation. Toxicology 2021, 457, 152803. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, A.L.; Rahman, M.; Persson, L.A.; Vahter, M. The risk of arsenic induced skin lesions in Bangladeshi men and women is affected by arsenic metabolism and the age at first exposure. Toxicol. Appl. Pharmacol. 2008, 230, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Plaza, M.; Tang, W.Y.; Shang, Y.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Ledesma, M.; Leon, M.; Laclaustra, M.; Pollak, J.; et al. Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples. Environ. Health Perspect. 2014, 122, 946–954. [Google Scholar] [CrossRef]
- Janasik, B.; Reszka, E.; Stanislawska, M.; Jablonska, E.; Kuras, R.; Wieczorek, E.; Malachowska, B.; Fendler, W.; Wasowicz, W. Effect of arsenic exposure on NRF2-KEAP1 pathway and epigenetic modification. Biol. Trace Elem. Res. 2018, 185, 11–19. [Google Scholar] [CrossRef]
- Pilsner, J.R.; Liu, X.; Ahsan, H.; Ilievski, V.; Slavkovich, V.; Levy, D.; Factor-Litvak, P.; Graziano, J.H.; Gamble, M.V. Genomic methylation of peripheral blood leukocyte DNA: Influences of arsenic and folate in Bangladeshi adults. Am. J. Clin. Nutr. 2007, 86, 1179–1186. [Google Scholar] [CrossRef]
- Pilsner, J.R.; Liu, X.; Ahsan, H.; Ilievski, V.; Slavkovich, V.; Levy, D.; Factor-Litvak, P.; Graziano, J.H.; Gamble, M.V. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect. 2009, 117, 254–260. [Google Scholar] [CrossRef]
- Arita, A.; Niu, J.; Qu, Q.; Zhao, N.; Ruan, Y.; Nadas, A.; Chervona, Y.; Wu, F.; Sun, H.; Hayes, R.B.; et al. Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Environ. Health Perspect. 2012, 120, 198–203. [Google Scholar] [CrossRef] [PubMed]
- IARC. Nickel and Nickel Compounds (IARC Monographs 100-C); IARC: Lyon, France, 2019; pp. 169–218. Available online: https://monographs.iarc.fr/wp-content/uploads/2018/06/mono100C-10.pdf (accessed on 7 October 2021).
- Trott, D.A.; Cuthbert, A.P.; Overell, R.W.; Russo, I.; Newbold, R.F. Mechanisms involved in the immortalization of mammalian cells by ionizing radiation and chemical carcinogens. Carcinogenesis 1995, 16, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Kluz, T.; Harder, M.E.; Xiong, J.; Costa, M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 2009, 48, 4626–4632. [Google Scholar] [CrossRef]
- Cloos, P.A.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef]
- Chen, H.; Kluz, T.; Zhang, R.; Costa, M. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 2010, 31, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, C.; Zhu, C.; Nie, S.; Qian, X.; Shi, Z.; Shi, M.; Liang, Y.; Ding, X.; Zhang, S.; et al. Hypoxia promotes the metastasis of pancreatic cancer through regulating NOX4/KDM5A-mediated histone methylation modification changes in a HIF1A-independent manner. Clin. Epigenet. 2021, 13, 18. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef]
- Liu, G.; Yan, J.; Wang, X.; Chen, J.; Wang, X.; Dong, Y.; Zhang, S.; Gan, X.; Huang, J.; Chen, X. RPA-mediated recruitment of Bre1 couples histone H2B ubiquitination to DNA replication and repair. Proc. Natl. Acad. Sci. USA 2021, 118, e2017497118. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.J.; Ma, Y.; Dickson, K.A. Histone monoubiquitination in chromatin remodelling: Focus on the histone H2B interactome and cancer. Cancers 2020, 12, 3462. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group. International Agency for Research on Cancer Monographs on the Evaluation of Carcinogenic Risks to Humans. Some Thyrotropic Agents; IARC: Lyon, France, 2001; Volume 79, pp. 1–773. [Google Scholar]
- Lake, B.G. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol. Res. 2018, 7, 697–717. [Google Scholar] [CrossRef]
- Becker, F.F. Morphological classification of mouse liver tumors based on biological characteristics. Cancer Res. 1982, 42, 3918–3923. [Google Scholar] [PubMed]
- Yamada, T.; Ohara, A.; Ozawa, N.; Maeda, K.; Kondo, M.; Okuda, Y.; Abe, J.; Cohen, S.M.; Lake, B.G. Comparison of the hepatic effects of phenobarbital in chimeric mice containing either rat or human hepatocytes with humanized constitutive androstane receptor and pregnane X receptor mice. Toxicol. Sci. 2020, 177, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Ohara, A.; Takahashi, Y.; Kondo, M.; Okuda, Y.; Takeda, S.; Kushida, M.; Kobayashi, K.; Sumida, K.; Yamada, T. Candidate genes responsible for early key events of phenobarbital-promoted mouse hepatocellular tumorigenesis based on differentiation of regulating genes between wild type mice and humanized chimeric mice. Toxicol. Res. 2017, 6, 795–813. [Google Scholar] [CrossRef]
- Negishi, M.; Kobayashi, K.; Sakuma, T.; Sueyoshi, T. Nuclear receptor phosphorylation in xenobiotic signal transduction. J. Biol. Chem. 2020, 295, 15210–15225. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hashimoto, M.; Honkakoski, P.; Negishi, M. Regulation of gene expression by CAR: An update. Arch. Toxicol. 2015, 89, 1045–1055. [Google Scholar] [CrossRef]
- Hori, T.; Saito, K.; Moore, R.; Flake, G.P.; Negishi, M. Nuclear receptor CAR suppresses GADD45B-p38 MAPK signaling to promote phenobarbital-induced proliferation in mouse liver. Mol. Cancer Res. 2018, 16, 1309–1318. [Google Scholar] [CrossRef]
- Phillips, J.M.; Goodman, J.I. Identification of genes that may play critical roles in phenobarbital (PB)-induced liver tumorigenesis due to altered DNA methylation. Toxicol. Sci. 2008, 104, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Bachman, A.N.; Phillips, J.M.; Goodman, J.I. Phenobarbital induces progressive patterns of GC-rich and gene-specific altered DNA methylation in the liver of tumor-prone B6C3F1 mice. Toxicol. Sci. 2006, 91, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Lempiainen, H.; Hackett, J.A.; Nestor, C.E.; Muller, A.; Bolognani, F.; Oakeley, E.J.; Schubeler, D.; Terranova, R.; Reinhardt, D.; et al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome. Genome Biol. 2012, 13, R93. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.P.; Hunter, J.M.; Lempiainen, H.; Muller, A.; Terranova, R.; Moggs, J.G.; Meehan, R.R. Dynamic changes in 5-hydroxymethylation signatures underpin early and late events in drug exposed liver. Nucleic Acids Res. 2013, 41, 5639–5654. [Google Scholar] [CrossRef]
- Globisch, D.; Munzel, M.; Muller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Bruckl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef]
- Bogdanffy, M.S.; Lesniak, J.; Mangipudy, R.; Sistare, F.D.; Colman, K.; Garcia-Tapia, D.; Monticello, T.; Blanset, D. Tg.rasH2 mouse model for assessing carcinogenic potential of pharmaceuticals: Industry survey of current practices. Int. J. Toxicol. 2020, 39, 198–206. [Google Scholar] [CrossRef]
- Misra, P.; Reddy, J.K. Peroxisome proliferator-activated receptor-alpha activation and excess energy burning in hepatocarcinogenesis. Biochimie 2014, 98, 63–74. [Google Scholar] [CrossRef]
- Luisier, R.; Lempiainen, H.; Scherbichler, N.; Braeuning, A.; Geissler, M.; Dubost, V.; Muller, A.; Scheer, N.; Chibout, S.D.; Hara, H.; et al. Phenobarbital induces cell cycle transcriptional responses in mouse liver humanized for constitutive androstane and pregnane x receptors. Toxicol. Sci. 2014, 139, 501–511. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Roy-Chaudhuri, B.; Kim, H.K.; Sayles, L.C.; Zheng, Y.; Chuang, C.H.; Caswell, D.R.; Chu, K.; Zhang, Y.; Winslow, M.M.; et al. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene 2015, 34, 94–103. [Google Scholar] [CrossRef]
- Luk, J.M.; Burchard, J.; Zhang, C.; Liu, A.M.; Wong, K.F.; Shek, F.H.; Lee, N.P.; Fan, S.T.; Poon, R.T.; Ivanovska, I.; et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J. Biol. Chem. 2011, 286, 30706–30713. [Google Scholar] [CrossRef] [PubMed]
- Dudek, K.M.; Suter, L.; Darras, V.M.; Marczylo, E.L.; Gant, T.W. Decreased translation of Dio3 mRNA is associated with drug-induced hepatotoxicity. Biochem. J. 2013, 453, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shen, H.; Xie, H.; Ying, Y.; Jin, K.; Yan, H.; Wang, S.; Xu, M.; Wang, X.; Xu, X.; et al. Dysregulation of ncRNAs located at the DLK1DIO3 imprinted domain: Involvement in urological cancers. Cancer Manag. Res. 2019, 11, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Molina-Pinelo, S.; Salinas, A.; Moreno-Mata, N.; Ferrer, I.; Suarez, R.; Andres-Leon, E.; Rodriguez-Paredes, M.; Gutekunst, J.; Jantus-Lewintre, E.; Camps, C.; et al. Impact of DLK1-DIO3 imprinted cluster hypomethylation in smoker patients with lung cancer. Oncotarget 2018, 9, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, S.; Albo-Castellanos, C.; Urdinguio, R.G.; Canon, S.; Sanchez-Cabo, F.; Martinez-Serrano, A.; Fraga, M.F.; Bernad, A. Deregulation of the imprinted DLK1-DIO3 locus ncRNAs is associated with replicative senescence of human adipose-derived stem cells. PLoS ONE 2018, 13, e0206534. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Kim, H.K.; Chu, K.; Zhang, F.; Xu, J.; Munding, E.M.; Shen, J.; Kay, M.A. miR-122 removal in the liver activates imprinted microRNAs and enables more effective microRNA-mediated gene repression. Nat. Commun. 2018, 9, 5321. [Google Scholar] [CrossRef]
- Peng, Q.; Weng, K.; Li, S.; Xu, R.; Wang, Y.; Wu, Y. A perspective of epigenetic regulation in radiotherapy. Front. Cell Dev. Biol. 2021, 9, 624312. [Google Scholar] [CrossRef]
- Rugo, R.E.; Mutamba, J.T.; Mohan, K.N.; Yee, T.; Chaillet, J.R.; Greenberger, J.S.; Engelward, B.P. Methyltransferases mediate cell memory of a genotoxic insult. Oncogene 2011, 30, 751–756. [Google Scholar] [CrossRef]
- Kalinich, J.F.; Catravas, G.N.; Snyder, S.L. Radioprotective properties of DNA methylation-disrupting agents. Int. J. Radiat. Biol. 1991, 59, 1217–1226. [Google Scholar] [CrossRef]
- Ngalame, N.N.O.; Luz, A.L.; Makia, N.; Tokar, E.J. Arsenic alters exosome quantity and cargo to mediate stem cell recruitment into a cancer stem cell-like phenotype. Toxicol. Sci. 2018, 165, 40–49. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Burgio, E.; Piscitelli, P.; Migliore, L. Ionizing radiation and human health: Reviewing models of exposure and mechanisms of cellular damage. An epigenetic perspective. Int. J. Environ. Res. Public Health 2018, 15, 1971. [Google Scholar] [CrossRef]
- Flunkert, J.; Maierhofer, A.; Dittrich, M.; Muller, T.; Horvath, S.; Nanda, I.; Haaf, T. Genetic and epigenetic changes in clonal descendants of irradiated human fibroblasts. Exp. Cell Res. 2018, 370, 322–332. [Google Scholar] [CrossRef]
- Huang, C.R.; Schneider, A.M.; Lu, Y.; Niranjan, T.; Shen, P.; Robinson, M.A.; Steranka, J.P.; Valle, D.; Civin, C.I.; Wang, T.; et al. Mobile interspersed repeats are major structural variants in the human genome. Cell 2010, 141, 1171–1182. [Google Scholar] [CrossRef]
- Miousse, I.R.; Skinner, C.M.; Sridharan, V.; Seawright, J.W.; Singh, P.; Landes, R.D.; Cheema, A.K.; Hauer-Jensen, M.; Boerma, M.; Koturbash, I. Changes in one-carbon metabolism and DNA methylation in the hearts of mice exposed to space environment-relevant doses of oxygen ions ((16)O). Life Sci. Space Res. 2019, 22, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA methyltransferases in cancer: Biology, paradox, aberrations, and targeted therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef] [PubMed]
- Beland, F.A.; Mellick, P.W.; Olson, G.R.; Mendoza, M.C.; Marques, M.M.; Doerge, D.R. Carcinogenicity of acrylamide in B6C3F(1) mice and F344/N rats from a 2-year drinking water exposure. Food Chem. Toxicol. 2013, 51, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, B.N.; Guhathakurta, S.; Akhtar, A. The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep. 2019, 20, e47630. [Google Scholar] [CrossRef]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef] [PubMed]
- Schalbetter, S.A.; Goloborodko, A.; Fudenberg, G.; Belton, J.M.; Miles, C.; Yu, M.; Dekker, J.; Mirny, L.; Baxter, J. SMC complexes differentially compact mitotic chromosomes according to genomic context. Nat. Cell Biol. 2017, 19, 1071–1080. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A pathway for mitotic chromosome formation. Science 2018, 359, 6135. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Kori, Y.; Lopes, M.; Yuan, Z.F.; Kim, H.J.; Kulej, K.; Janssen, K.A.; Agosto, L.M.; Cunha, J.P.C.D.; Andrews, A.J.; et al. One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res. 2019, 29, 978–987. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Rahhal, R.; Seto, E. Emerging roles of histone modifications and HDACs in RNA splicing. Nucleic Acids Res. 2019, 47, 4911–4926. [Google Scholar] [CrossRef]
- Howe, F.S.; Fischl, H.; Murray, S.C.; Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays 2017, 39, 1–12. [Google Scholar] [CrossRef]
- Cai, X.C.; Kapilashrami, K.; Luo, M. Synthesis and assays of inhibitors of methyltransferases. Methods Enzymol. 2016, 574, 245–308. [Google Scholar]
- Hsiao, K.; Zegzouti, H.; Goueli, S.A. Methyltransferase-Glo: A universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 2016, 8, 321–339. [Google Scholar] [CrossRef]
- Gul, S. Epigenetic assays for chemical biology and drug discovery. Clin. Epigenet. 2017, 9, 41. [Google Scholar] [CrossRef]
- OECD. Guidance Document on Developing And Assessing Adverse Outcome Pathways, 184th ed.; Series on Testing and Assessment; ENV/JM/MONO(2013)6; OECD: Paris, France, 2013; pp. 1–16. [Google Scholar]
- Nyffeler, J.; Willis, C.; Lougee, R.; Richard, A.; Paul-Friedman, K.; Harrill, J.A. Bioactivity screening of environmental chemicals using imaging-based high-throughput phenotypic profiling. Toxicol. Appl. Pharmacol. 2020, 389, 114876. [Google Scholar] [CrossRef] [PubMed]
- Luense, S.; Denner, P.; Fernandez-Montalvan, A.; Hartung, I.; Husemann, M.; Stresemann, C.; Prechtl, S. Quantification of histone H3 Lys27 trimethylation (H3K27me3) by high-throughput microscopy enables cellular large-scale screening for small-molecule EZH2 inhibitors. J. Biomol. Screen. 2015, 20, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Mulji, A.; Haslam, C.; Brown, F.; Randle, R.; Karamshi, B.; Smith, J.; Eagle, R.; Munoz-Muriedas, J.; Taylor, J.; Sheikh, A.; et al. Configuration of a high-content imaging platform for hit identification and pharmacological assessment of JMJD3 demethylase enzyme inhibitors. J. Biomol. Screen. 2012, 17, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Hatch, S.B.; Yapp, C.; Montenegro, R.C.; Savitsky, P.; Gamble, V.; Tumber, A.; Ruda, G.F.; Bavetsias, V.; Fedorov, O.; Atrash, B.; et al. Assessing histone demethylase inhibitors in cells: Lessons learned. Epigenet. Chromatin 2017, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Zhou, G.; Parfett, C.; Cummings-Lorbetskie, C.; Xiao, G.H.; Desaulniers, D. Two-color fluorescent cytosine extension assay for the determination of global DNA methylation. Biotechniques 2017, 62, 157–164. [Google Scholar] [CrossRef]
- Smith, M.T.; Guyton, K.Z.; Kleinstreuer, N.; Borrel, A.; Cardenas, A.; Chiu, W.A.; Felsher, D.W.; Gibbons, C.F.; Goodson, W.H.; Houck, K.A.; et al. The key characteristics of carcinogens: Relationship to the hallmarks of cancer, relevant biomarkers, and assays to measure them. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1887–1903. [Google Scholar] [CrossRef]
- Si, J.; Boumber, Y.A.; Shu, J.; Qin, T.; Ahmed, S.; He, R.; Jelinek, J.; Issa, J.P. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res. 2010, 70, 6968–6977. [Google Scholar] [CrossRef]
- Okochi-Takada, E.; Hattori, N.; Ito, A.; Niwa, T.; Wakabayashi, M.; Kimura, K.; Yoshida, M.; Ushijima, T. Establishment of a high-throughput detection system for DNA demethylating agents. Epigenetics 2018, 13, 147–155. [Google Scholar] [CrossRef]
- Cui, Y.; Hausheer, F.; Beaty, R.; Zahnow, C.; Issa, J.P.; Bunz, F.; Baylin, S.B. A recombinant reporter system for monitoring reactivation of an endogenously DNA hypermethylated gene. Cancer Res. 2014, 74, 3834–3843. [Google Scholar] [CrossRef]
- Hsiao, S.H.; Lee, K.D.; Hsu, C.C.; Tseng, M.J.; Jin, V.X.; Sun, W.S.; Hung, Y.C.; Yeh, K.T.; Yan, P.S.; Lai, Y.Y.; et al. DNA methylation of the Trip10 promoter accelerates mesenchymal stem cell lineage determination. Biochem. Biophys. Res. Commun. 2010, 400, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Jaenisch, R. Monitoring dynamics of DNA methylation at single-cell resolution during development and disease. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 199–206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coronado-Posada, N.; Olivero-Verbel, J. In silico evaluation of pesticides as potential modulators of human DNA methyltransferases. SAR QSAR Environ. Res. 2019, 30, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Anteneh, H.; Hornisch, M.; Wagner, V.; Lu, J.; Radde, N.E.; Bashtrykov, P.; Song, J.; Jeltsch, A. DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation. Nat. Commun. 2020, 11, 3723. [Google Scholar] [CrossRef]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef]
- Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant methylation of LINE-1 transposable elements: A search for cancer biomarkers. Cells 2020, 9, 2017. [Google Scholar] [CrossRef]
- Kakizawa, N.; Suzuki, K.; Abe, I.; Endo, Y.; Tamaki, S.; Ishikawa, H.; Watanabe, F.; Ichida, K.; Saito, M.; Futsuhara, K.; et al. High relative levels of satellite alpha transcripts predict increased risk of bilateral breast cancer and multiple primary cancer in patients with breast cancer and lacking BRCArelated clinical features. Oncol. Rep. 2019, 42, 857–865. [Google Scholar] [CrossRef]
- Bruckmann, N.H.; Pedersen, C.B.; Ditzel, H.J.; Gjerstorff, M.F. Epigenetic reprogramming of pericentromeric satellite DNA in premalignant and malignant lesions. Mol. Cancer Res. 2018, 16, 417–427. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell 2018, 33, 322–336. [Google Scholar] [CrossRef]
- Madakashira, B.P.; Sadler, K.C. DNA methylation, nuclear organization, and cancer. Front. Genet. 2017, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Rybak, P.; Hoang, A.; Bujnowicz, L.; Bernas, T.; Berniak, K.; Zarebski, M.; Darzynkiewicz, Z.; Dobrucki, J. Low level phosphorylation of histone H2AX on serine 139 (gammaH2AX) is not associated with DNA double-strand breaks. Oncotarget 2016, 7, 49574–49587. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, D.T.; Bryce, S.M.; Bemis, J.C.; Kirkland, D.; Dertinger, S.D. gammaH2AX and p53 responses in TK6 cells discriminate promutagens and nongenotoxicants in the presence of rat liver S9. Environ. Mol. Mutagen. 2016, 57, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Schleich, K.; Kase, J.; Dorr, J.R.; Trescher, S.; Bhattacharya, A.; Yu, Y.; Wailes, E.M.; Fan, D.N.Y.; Lohneis, P.; Milanovic, M.; et al. H3K9me3-mediated epigenetic regulation of senescence in mice predicts outcome of lymphoma patients. Nat. Commun. 2020, 11, 3651. [Google Scholar] [CrossRef]
- Paluvai, H.; Di, G.E.; Brancolini, C. The histone code of senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Boix-Chornet, M.; Fraga, M.F.; Villar-Garea, A.; Caballero, R.; Espada, J.; Nunez, A.; Casado, J.; Largo, C.; Casal, J.I.; Cigudosa, J.C.; et al. Release of hypoacetylated and trimethylated histone H4 is an epigenetic marker of early apoptosis. J. Biol. Chem. 2006, 281, 13540–13547. [Google Scholar] [CrossRef]
- Allera, C.; Lazzarini, G.; Patrone, E.; Alberti, I.; Barboro, P.; Sanna, P.; Melchiori, A.; Parodi, S.; Balbi, C. The condensation of chromatin in apoptotic thymocytes shows a specific structural change. J. Biol. Chem. 1997, 272, 10817–10822. [Google Scholar] [CrossRef]
- Pickles, J.C.; Pant, K.; Mcginty, L.A.; Yasaei, H.; Roberts, T.; Scott, A.D.; Newbold, R.F. A mechanistic evaluation of the Syrian hamster embryo cell transformation assay (pH 6.7) and molecular events leading to senescence bypass in SHE cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 802, 50–58. [Google Scholar] [CrossRef]
- Dogan, F.; Forsyth, N.R. Telomerase regulation: A role for epigenetics. Cancers 2021, 13, 1213. [Google Scholar] [CrossRef]
- Baribault, C.; Ehrlich, K.C.; Ponnaluri, V.K.C.; Pradhan, S.; Lacey, M.; Ehrlich, M. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics 2018, 13, 275–289. [Google Scholar] [CrossRef]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Diaz-Lagares, A.; Mendez-Gonzalez, J.; Hervas, D.; Saigi, M.; Pajares, M.J.; Garcia, D.; Crujerias, A.B.; Pio, R.; Montuenga, L.M.; Zulueta, J.; et al. A novel epigenetic signature for early diagnosis in lung cancer. Clin. Cancer Res. 2016, 22, 3361–3371. [Google Scholar] [CrossRef]
- Mari-Alexandre, J.; Diaz-Lagares, A.; Villalba, M.; Juan, O.; Crujeiras, A.B.; Calvo, A.; Sandoval, J. Translating cancer epigenomics into the clinic: Focus on lung cancer. Transl. Res. 2017, 189, 76–92. [Google Scholar] [CrossRef]
- Moran, S.; Martinez-Cardus, A.; Sayols, S.; Musulen, E.; Balana, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Heidecker, B.; Hare, J.M. The use of transcriptomic biomarkers for personalized medicine. Heart Fail Rev. 2007, 12, 1–11. [Google Scholar] [CrossRef]
- Cho, E.; Buick, J.K.; Williams, A.; Chen, R.; Li, H.H.; Corton, J.C.; Fornace, A.J., Jr.; Aubrecht, J.; Yauk, C.L. Assessment of the performance of the TGx-DDI biomarker to detect DNA damage-inducing agents using quantitative RT-PCR in TK6 cells. Environ. Mol. Mutagen. 2019, 60, 122–133. [Google Scholar] [CrossRef]
- Schaap, M.M.; Wackers, P.F.; Zwart, E.P.; Huijskens, I.; Jonker, M.J.; Hendriks, G.; Breit, T.M.; van Steeg, H.; van de Water, B.; Luijten, M. A novel toxicogenomics-based approach to categorize (non-)genotoxic carcinogens. Arch. Toxicol. 2015, 89, 2413–2427. [Google Scholar] [CrossRef]
- Huang, S.H.; Lin, Y.C.; Tung, C.W. Identification of time-invariant biomarkers for non-genotoxic hepatocarcinogen assessment. Int. J. Environ. Res. Public Health 2020, 17, 4298. [Google Scholar] [CrossRef]
- Harrill, J.A.; Everett, L.J.; Haggard, D.E.; Sheffield, T.; Bundy, J.L.; Willis, C.M.; Thomas, R.S.; Shah, I.; Judson, R.S. High-throughput transcriptomics platform for screening environmental chemicals. Toxicol. Sci. 2021, 181, 68–89. [Google Scholar] [CrossRef]
- Cho, E.; Rowan-Carroll, A.; Williams, A.; Corton, J.C.; Li, H.H.; Fornace, A.J., Jr.; Hobbs, C.A.; Yauk, C.L. Development and validation of the TGx-HDACi transcriptomic biomarker to detect histone deacetylase inhibitors in human TK6 cells. Arch. Toxicol. 2021, 95, 1631–1645. [Google Scholar] [CrossRef]
- Furihata, C.; Suzuki, T. Evaluation of 12 mouse marker genes in rat toxicogenomics public data, Open TG-GATEs: Discrimination of genotoxic from non-genotoxic hepatocarcinogens. Mutat. Res. 2019, 838, 9–15. [Google Scholar] [CrossRef]
- Kossler, N.; Matheis, K.A.; Ostenfeldt, N.; Bach, T.D.; Dhalluin, S.; Deschl, U.; Kalkuhl, A. Identification of specific mRNA signatures as fingerprints for carcinogenesis in mice induced by genotoxic and nongenotoxic hepatocarcinogens. Toxicol. Sci. 2015, 143, 277–295. [Google Scholar] [CrossRef]
- Williams, A.; Buick, J.K.; Moffat, I.; Swartz, C.D.; Recio, L.; Hyduke, D.R.; Li, H.H.; Fornace, A.J., Jr.; Aubrecht, J.; Yauk, C.L. A predictive toxicogenomics signature to classify genotoxic versus non-genotoxic chemicals in human TK6 cells. Data Brief 2015, 5, 77–83. [Google Scholar] [CrossRef]
- Ito, Y.; Nakajima, K.; Masubuchi, Y.; Kikuchi, S.; Saito, F.; Akahori, Y.; Jin, M.; Yoshida, T.; Shibutani, M. Expression characteristics of genes hypermethylated and downregulated in rat liver specific to non-genotoxic hepatocarcinogens. Toxicol. Sci. 2019, 169, 122–136. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Guidelines for Carcinogen Risk Assessment; EPA/630/P-03/001F; U.S. Environmental Protection Agency: Washington, DC, USA, 2005; pp. 1–166.
- Kirkland, D.; Kasper, P.; Martus, H.J.; Muller, L.; van Benthem, J.; Madia, F.; Corvi, R. Updated recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 795, 7–30. [Google Scholar] [CrossRef]
- Maegley, K.A.; Krivacic, C.; Bingham, P.; Liu, W.; Brooun, A. Comparison of a high-throughput mass spectrometry method and radioactive filter binding to assay the protein methyltransferase PRMT5. Assay Drug Dev. Technol. 2015, 13, 235–240. [Google Scholar] [CrossRef]
- Qian, K.; Hu, H.; Xu, H.; Zheng, Y.G. Detection of PRMT1 inhibitors with stopped flow fluorescence. Signal Transduct. Target Ther. 2018, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Jayatunga, M.K.; Thompson, S.; McKee, T.C.; Chan, M.C.; Reece, K.M.; Hardy, A.P.; Sekirnik, R.; Seden, P.T.; Cook, K.M.; McMahon, J.B.; et al. Inhibition of the HIF1alpha-p300 interaction by quinone- and indandione-mediated ejection of structural Zn(II). Eur. J. Med. Chem. 2015, 94, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Sekirnik, R.; Rose, N.R.; Thalhammer, A.; Seden, P.T.; Mecinovic, J.; Schofield, C.J. Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II). Chem. Commun. 2009, 42, 6376–6378. [Google Scholar] [CrossRef]
- Lungu, C.; Pinter, S.; Broche, J.; Rathert, P.; Jeltsch, A. Modular fluorescence complementation sensors for live cell detection of epigenetic signals at endogenous genomic sites. Nat. Commun. 2017, 8, 649. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, A.; Bermudez Silva, F.J.; Lopez-Hoyos, M.; Cobaleda, C.; Montoliu, L.; Del, V.M.; Leech, K. Non-animal-derived monoclonal antibodies are not ready to substitute current hybridoma technology. Nat. Methods 2020, 17, 1069–1070. [Google Scholar] [CrossRef]
- Barroso, J.; Halder, M.; Whelan, M. EURL ECVAM Recommendation on Non-Animal-Derived Antibodies; EUR 30185 EN; Publications Office of the European Union: Luxembourg, 2020; pp. 1–74. [Google Scholar] [CrossRef]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol. Cell. Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef]
- Johnson, R.L.; Huang, W.; Jadhav, A.; Austin, C.P.; Inglese, J.; Martinez, E.D. A quantitative high-throughput screen identifies potential epigenetic modulators of gene expression. Anal. Biochem. 2008, 375, 237–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stepanenko, A.A.; Heng, H.H. Transient and stable vector transfection: Pitfalls, off-target effects, artifacts. Mutat. Res. 2017, 773, 91–103. [Google Scholar] [CrossRef]
- Godecke, N.; Zha, L.; Spencer, S.; Behme, S.; Riemer, P.; Rehli, M.; Hauser, H.; Wirth, D. Controlled re-activation of epigenetically silenced Tet promoter-driven transgene expression by targeted demethylation. Nucleic Acids Res. 2017, 45, e147. [Google Scholar] [CrossRef]
- Spencer, S.; Gugliotta, A.; Godecke, N.; Hauser, H.; Wirth, D. Epigenetic modulations rendering cell-to-cell variability and phenotypic metastability. J. Genet. Genom. 2016, 43, 503–511. [Google Scholar] [CrossRef]
- Raynal, N.J.; Da Costa, E.M.; Lee, J.T.; Gharibyan, V.; Ahmed, S.; Zhang, H.; Sato, T.; Malouf, G.G.; Issa, J.J. Repositioning FDA-approved drugs in combination with epigenetic drugs to reprogram colon cancer epigenome. Mol. Cancer Ther. 2017, 16, 397–407. [Google Scholar] [CrossRef]
- Lin, Y.S.; Shaw, A.Y.; Wang, S.G.; Hsu, C.C.; Teng, I.W.; Tseng, M.J.; Huang, T.H.; Chen, C.S.; Leu, Y.W.; Hsiao, S.H. Identification of novel DNA methylation inhibitors via a two-component reporter gene system. J. Biomed. Sci. 2011, 18, 3. [Google Scholar] [CrossRef]
- Teng, I.W.; Hou, P.C.; Lee, K.D.; Chu, P.Y.; Yeh, K.T.; Jin, V.X.; Tseng, M.J.; Tsai, S.J.; Chang, Y.S.; Wu, C.S.; et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res. 2011, 71, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Fraga, M.F.; Esteller, M. Quantfication of global DNA methylation by capillary electrophoresis and mass spectrometry. In DNA Methylation, Methods and Protocols. Springer Protocols. Methods in Molecular Biology; Tost, J., Ed.; Humana Press: New York, NY, USA, 2009; Volume 507, pp. 23–34. [Google Scholar]
- Fraga, M.F.; Uriol, E.; Borja, D.L.; Berdasco, M.; Esteller, M.; Canal, M.J.; Rodriguez, R. High-performance capillary electrophoretic method for the quantification of 5-methyl 2’-deoxycytidine in genomic DNA: Application to plant, animal and human cancer tissues. Electrophoresis 2002, 23, 1677–1681. [Google Scholar] [CrossRef]
- Rocha, M.S.; Castro, R.; Rivera, I.; Kok, R.M.; Smulders, Y.M.; Jakobs, C.; de Almeida, I.T.; Blom, H.J. Global DNA methylation: Comparison of enzymatic- and non-enzymatic-based methods. Clin. Chem. Lab. Med. 2010, 48, 1793–1798. [Google Scholar] [CrossRef] [PubMed]
- Kok, R.M.; Smith, D.E.; Barto, R.; Spijkerman, A.M.; Teerlink, T.; Gellekink, H.J.; Jakobs, C.; Smulders, Y.M. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: Analytical technique, reference values and determinants in healthy subjects. Clin. Chem. Lab. Med. 2007, 45, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Li, X.; Zhang, L.; Liu, D.; Du, W.; Yin, D.; Lyu, N.; Zhao, G.; Guo, C.; Tang, D. Accurate quantification of 5-Methylcytosine, 5-Hydroxymethylcytosine, 5-Formylcytosine, and 5-Carboxylcytosine in genomic DNA from breast cancer by chemical derivatization coupled with ultra performance liquid chromatography- electrospray quadrupole time of flight mass spectrometry analysis. Oncotarget 2017, 8, 91248–91257. [Google Scholar]
- Fernandez, A.F.; Valledor, L.; Vallejo, F.; Canal, M.J.; Fraga, M.F. Quantification of global DNA methylation levels by mass spectrometry. Methods Mol. Biol. 2018, 1708, 49–58. [Google Scholar]
- Shahal, T.; Koren, O.; Shefer, G.; Stern, N.; Ebenstein, Y. Hypersensitive quantification of global 5-hydroxymethylcytosine by chemoenzymatic tagging. Anal. Chim. Acta 2018, 1038, 87–96. [Google Scholar] [CrossRef]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Walker, B.A. A rapid and robust protocol for reduced representation bisulfite sequencing in multiple myeloma. Methods Mol. Biol. 2018, 1792, 179–191. [Google Scholar]
- Karimi, M.; Johansson, S.; Stach, D.; Corcoran, M.; Grander, D.; Schalling, M.; Bakalkin, G.; Lyko, F.; Larsson, C.; Ekstrom, T.J. LUMA (LUminometric Methylation Assay)-a high throughput method to the analysis of genomic DNA methylation. Exp. Cell Res. 2006, 312, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Petronis, A. DNA Methylation Microarrays, Experimental Design and Statistical Analysis; CRC Press: Boca Raton, FL, USA, 2008; pp. 1–256. [Google Scholar]
- Esteller, M. DNA Methylation: Approaches, Methods, and Applications; CRC Press: New York, NY, USA, 2004; pp. 1–223. [Google Scholar]
- Tost, J. DNA Methylation, Methods and Protocols, 2nd ed.; Humana Press: New York, NY, USA, 2009; pp. 1–356. [Google Scholar]
- Noberini, R.; Restellini, C.; Savoia, E.O.; Bonaldi, T. Enrichment of histones from patient samples for mass spectrometry-based analysis of post-translational modifications. Methods 2019, 184, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Chung, T.H.; Sun, X.; Jia, X.Y. Mirror bisulfite sequencing: A method for single-base resolution of hydroxymethylcytosine. Anal. Chem. 2018, 90, 13200–13206. [Google Scholar] [CrossRef] [PubMed]
- Foksinski, M.; Zarakowska, E.; Gackowski, D.; Skonieczna, M.; Gajda, K.; Hudy, D.; Szpila, A.; Bialkowski, K.; Starczak, M.; Labejszo, A.; et al. Profiles of a broad spectrum of epigenetic DNA modifications in normal and malignant human cell lines: Proliferation rate is not the major factor responsible for the 5-hydroxymethyl-2′-deoxycytidine level in cultured cancerous cell lines. PLoS ONE 2017, 12, e0188856. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; He, B.; Xia, B.; Bai, D.; Lu, X.; Cai, J.; Chen, L.; Zhou, A.; Zhu, C.; Meng, H.; et al. Bisulfite-free, nanoscale analysis of 5-hydroxymethylcytosine at single base resolution. J. Am. Chem. Soc. 2018, 140, 13190–13194. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.; Yi, P.; James, S.J. A sensitive new method for rapid detection of abnormal methylation patterns in global DNA and within CpG islands. Biochem. Biophys. Res. Commun. 1999, 262, 624–628. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Karimi, M.; Johansson, S.; Ekstrom, T.J. Using LUMA: A Luminometric-based assay for global DNA-methylation. Epigenetics 2006, 1, 45–48. [Google Scholar]
- Bielefeld-Sevigny, M. AlphaLISA immunoassay platform- the “no-wash” high-throughput alternative to ELISA. Assay Drug Dev. Technol. 2009, 7, 90–92. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Chen, Y.; Liu, J.; Xiao, S.; Lian, F.; Zhang, N.; Ding, H.; Zhang, Y.; Chen, K.; et al. Discovery of potent DOT1L inhibitors by AlphaLISA based high throughput screening assay. Bioorg. Med. Chem. 2018, 26, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Machleidt, T.; Robers, M.B.; Hermanson, S.B.; Dudek, J.M.; Bi, K. TR-FRET cellular assays for interrogating posttranslational modifications of histone H3. J. Biomol. Screen. 2011, 16, 1236–1246. [Google Scholar] [CrossRef]
- Fan, J.; Baeza, J.; Denu, J.M. Investigating histone acetylation stoichiometry and turnover rate. Methods Enzymol. 2016, 574, 125–148. [Google Scholar]
- Simon, R.P.; Robaa, D.; Alhalabi, Z.; Sippl, W.; Jung, M. KATching-up on small molecule modulators of lysine acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic lesions causing genetic lesions in human cancer: Promoter hypermethylation of DNA repair genes. Eur. J. Cancer 2000, 36, 2294–2300. [Google Scholar] [CrossRef]
- Vos, S.; Van Diest, P.J.; Moelans, C.B. A systematic review on the frequency of BRCA promoter methylation in breast and ovarian carcinomas of BRCA germline mutation carriers: Mutually exclusive, or not? Crit. Rev. Oncol. Hematol. 2018, 127, 29–41. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Vuong, L.D.; Truong, V.L.; Ta, T.V.; Nguyen, N.T.; Nguyen, H.P.; Chu, H.H. Genetic and epigenetic alterations of the EGFR and mutually independent association with BRCA1, MGMT, and RASSF1A methylations in Vietnamese lung adenocarcinomas. Pathol. Res. Pract. 2019, 215, 885–892. [Google Scholar] [CrossRef]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, G.; Yin, J.; Li, L.; Tan, Y.; Wei, H.; Liu, B.; Deng, L.; Tang, J.; Chen, Y.; et al. GSTP1 and cancer: Expression, methylation, polymorphisms and signaling (Review). Int. J. Oncol. 2020, 56, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, S.; Sekine, Y.; Shineha, R.; Nishihira, T.; Sato, K. Elevation of the placental glutathione S-transferase form (GST-pi) in tumor tissues and the levels in sera of patients with cancer. Cancer Res. 1989, 49, 5225–5229. [Google Scholar] [PubMed]
- Satoh, K.; Kitahara, A.; Soma, Y.; Inaba, Y.; Hatayama, I.; Sato, K. Purification, induction, and distribution of placental glutathione transferase: A new marker enzyme for preneoplastic cells in the rat chemical hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3964–3968. [Google Scholar] [CrossRef]
- Jain, S.; Chen, S.; Chang, K.C.; Lin, Y.J.; Hu, C.T.; Boldbaatar, B.; Hamilton, J.P.; Lin, S.Y.; Chang, T.T.; Chen, S.H.; et al. Impact of the location of CpG methylation within the GSTP1 gene on its specificity as a DNA marker for hepatocellular carcinoma. PLoS ONE 2012, 7, e35789. [Google Scholar] [CrossRef]
- Tian, M.; Peng, S.; Martin, F.L.; Zhang, J.; Liu, L.; Wang, Z.; Dong, S.; Shen, H. Perfluorooctanoic acid induces gene promoter hypermethylation of glutathione-S-transferase Pi in human liver L02 cells. Toxicology 2012, 296, 48–55. [Google Scholar] [CrossRef]
- Alnaes, G.I.; Ronneberg, J.A.; Kristensen, V.N.; Tost, J. Heterogeneous DNA methylation patterns in the GSTP1 promoter lead to discordant results between assay technologies and impede its implementation as epigenetic biomarkers in breast cancer. Genes 2015, 6, 878–900. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Jiao, Y.; Feng, Y.; Wang, X. Regulation of tumor suppressor gene CDKN2A and encoded p16-INK4a protein by covalent modifications. Biochemistry 2018, 83, 1289–1298. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Fontana, R.; Ranieri, M.; La, M.G.; Vivo, M. Dual role of the alternative reading frame ARF protein in cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant expression of p14(ARF) in human cancers: A new biomarker? Tumor Microenviron. 2018, 1, 37–44. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, L.; Delgado, M.D.; Leon, J. MYC oncogene contributions to release of cell cycle brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef]
- Krimpenfort, P.; IJpenberg, A.; Song, J.Y.; van der Valk, M.; Nawijn, M.; Zevenhoven, J.; Berns, A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007, 448, 943–946. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef]
- Kong, Y.; Hsieh, C.H.; Alonso, L.C. ANRIL: A lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front. Endocrinol. 2018, 9, 405. [Google Scholar] [CrossRef]
- Herman, J.G.; Jen, J.; Merlo, A.; Baylin, S.B. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res. 1996, 56, 722–727. [Google Scholar]
- Hinshelwood, R.A.; Melki, J.R.; Huschtscha, L.I.; Paul, C.; Song, J.Z.; Stirzaker, C.; Reddel, R.R.; Clark, S.J. Aberrant de novo methylation of the p16INK4A CpG island is initiated post gene silencing in association with chromatin remodelling and mimics nucleosome positioning. Hum. Mol. Genet. 2009, 18, 3098–3109. [Google Scholar] [CrossRef]
- Xiao, Z.; He, Y.; Liu, C.; Xiang, L.; Yi, J.; Wang, M.; Shen, T.; Shen, L.; Xue, Y.; Shi, H.; et al. Targeting P16INK4A in uterine serous carcinoma through inhibition of histone demethylation. Oncol. Rep. 2019, 41, 2667–2678. [Google Scholar] [CrossRef]
- Kim, K.Y.; Lewis, J.S., Jr.; Chen, Z. Current status of clinical testing for human papillomavirus in oropharyngeal squamous cell carcinoma. J. Pathol. Clin. Res. 2018, 4, 213–226. [Google Scholar] [CrossRef]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Ramsey, M.R.; Balasubramanian, P.; Castrillon, D.H.; Depinho, R.A. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene 2004, 23, 379–385. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Ohtani, N. Deciphering the mechanism for induction of senescence-associated secretory phenotype (SASP) and its role in aging and cancer development. J. Biochem. 2019, 166, 289–295. [Google Scholar] [CrossRef]
- Wheeler, A.R. Directive to Prioritize Efforts to Reduce Animal Testing, September 10, 2019; Memorandum from the US-EPA Administrator to EPA Management; EPA: Washington, DC, USA, 2019.
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2016, 17, 95–103. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Tomasi, T.B.; Magner, W.J.; Khan, A.N. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol. Immunother. 2006, 55, 1159–1184. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Dicato, M.; Diederich, M.F. Anticancer potential of naturally occurring immunoepigenetic modulators: A promising avenue? Cancer 2019, 125, 1612–1628. [Google Scholar] [CrossRef]
- Vijayan, S.; Sidiq, T.; Yousuf, S.; van den Elsen, P.J.; Kobayashi, K.S. Class I transactivator, NLRC5: A central player in the MHC class I pathway and cancer immune surveillance. Immunogenetics 2019, 71, 273–282. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Axelrod, M.L.; Cook, R.S.; Johnson, D.B.; Balko, J.M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 2019, 25, 2392–2402. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.S.; Poulton, K.V.; Poles, A. Factors affecting HLA expression: A review. Int. J. Immunogenet. 2019, 46, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Saigi, M.; Alburquerque-Bejar, J.J.; Sanchez-Cespedes, M. Determinants of immunological evasion and immunocheckpoint inhibition response in non-small cell lung cancer: The genetic front. Oncogene 2019, 38, 5921–5932. [Google Scholar] [CrossRef]
- Yoshihama, S.; Vijayan, S.; Sidiq, T.; Kobayashi, K.S. NLRC5/CITA: A key player in cancer immune surveillance. Trends Cancer 2017, 3, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef]
- Ramsuran, V.; Kulkarni, S.; O’hUigin, C.; Yuki, Y.; Augusto, D.G.; Gao, X.; Carrington, M. Epigenetic regulation of differential HLA-A allelic expression levels. Hum. Mol. Genet. 2015, 24, 4268–4275. [Google Scholar] [CrossRef]
- Siebenkas, C.; Chiappinelli, K.B.; Guzzetta, A.A.; Sharma, A.; Jeschke, J.; Vatapalli, R.; Baylin, S.B.; Ahuja, N. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PLoS ONE 2017, 12, e0179501. [Google Scholar] [CrossRef]
- Swets, M.; Seneby, L.; Boot, A.; van Wezel, T.; Gelderblom, H.; van de Velde, C.J.; van den Elsen, P.J.; Kuppen, P.J. Promoter methylation and mRNA expression of HLA-G in relation to HLA-G protein expression in colorectal cancer. Hum. Immunol. 2016, 77, 764–772. [Google Scholar] [CrossRef]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef]
- Garziera, M.; Scarabel, L.; Toffoli, G. Hypoxic modulation of HLA-G expression through the metabolic sensor HIF-1 in human cancer cells. J. Immunol. Res. 2017, 2017, 4587520. [Google Scholar] [CrossRef]
- Yaghi, L.; Poras, I.; Simoes, R.T.; Donadi, E.A.; Tost, J.; Daunay, A.; de Almeida, B.S.; Carosella, E.D.; Moreau, P. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget 2016, 7, 63690–63707. [Google Scholar] [CrossRef]
- Wijdeven, R.H.; van Luijn, M.M.; Wierenga-Wolf, A.F.; Akkermans, J.J.; van den Elsen, P.J.; Hintzen, R.Q.; Neefjes, J. Chemical and genetic control of IFNgamma-induced MHCII expression. EMBO Rep. 2018, 19, e45553. [Google Scholar] [CrossRef]
- Toor, S.M.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Abu Nada, M.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2019, 10, 2936. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3, e96793. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked global DNA hypomethylation is associated with constitutive PD-L1 expression in melanoma. iScience 2018, 4, 312–325. [Google Scholar] [CrossRef]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Darvin, P.; Sasidharan Nair, V.; Elkord, E. PD-L1 expression in human breast cancer stem cells is epigenetically regulated through posttranslational histone modifications. J. Oncol. 2019, 2019, 3958908. [Google Scholar] [CrossRef]
- Nair, V.S.; Toor, S.M.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenet. 2018, 10, 104. [Google Scholar] [CrossRef]
- Nair, V.S.; El Salhat, H.; Taha, R.Z.; John, A.; Ali, B.R.; Elkord, E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin. Epigenet. 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef]
- Xue, G.; Cui, Z.J.; Zhou, X.H.; Zhu, Y.X.; Chen, Y.; Liang, F.J.; Tang, D.N.; Huang, B.Y.; Zhang, H.Y.; Hu, Z.H.; et al. DNA methylation biomarkers predict objective responses to PD-1/PD-L1 inhibition blockade. Front. Genet. 2019, 10, 724. [Google Scholar] [CrossRef]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Sanchez-Del-Campo, L.; Marti-Diaz, R.; Montenegro, M.F.; Gonzalez-Guerrero, R.; Hernandez-Caselles, T.; Martinez-Barba, E.; Pinero-Madrona, A.; Cabezas-Herrera, J.; Goding, C.R.; Rodriguez-Lopez, J.N. MITF induces escape from innate immunity in melanoma. J. Exp. Clin. Cancer Res. 2021, 40, 117. [Google Scholar] [CrossRef]
- Ferrari de, A.L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Bhat, J.; Dubin, S.; Dananberg, A.; Quabius, E.S.; Fritsch, J.; Dowds, C.M.; Saxena, A.; Chitadze, G.; Lettau, M.; Kabelitz, D. Histone deacetylase inhibitor modulates NKG2D receptor expression and memory phenotype of human gamma/delta T cells upon interaction with tumor cells. Front. Immunol. 2019, 10, 569. [Google Scholar] [CrossRef]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef]
- Lian, Y.; Meng, L.; Ding, P.; Sang, M. Epigenetic regulation of MAGE family in human cancer progression-DNA methylation, histone modification, and non-coding RNAs. Clin. Epigenet. 2018, 10, 115. [Google Scholar] [CrossRef]
- Al-Khadairi, G.; Decock, J. Cancer testis antigens and immunotherapy: Where do we stand in the targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef]
- Gibbs, Z.A.; Whitehurst, A.W. Emerging contributions of cancer/testis antigens to neoplastic behaviors. Trends Cancer 2018, 4, 701–712. [Google Scholar] [CrossRef]
- Fukuyama, T.; Yamazaki, T.; Fujita, T.; Uematsu, T.; Ichiki, Y.; Kaneko, H.; Suzuki, T.; Kobayashi, N. Helicobacter pylori, a carcinogen, induces the expression of melanoma antigen-encoding gene (Mage)-A3, a cancer/testis antigen. Tumour Biol. 2012, 33, 1881–1887. [Google Scholar] [CrossRef]
- Ono, T.; Sato, S.; Kimura, N.; Tanaka, M.; Shibuya, A.; Old, L.J.; Nakayama, E. Serological analysis of BALB/C methylcholanthrene sarcoma Meth A by SEREX: Identification of a cancer/testis antigen. Int. J. Cancer 2000, 88, 845–851. [Google Scholar] [CrossRef]
- Jin, S.; Cao, S.; Grigorev, A.; Li, J.; Meng, Q.; Wang, C.; Feng, M.; Hu, J.; Jiang, F.; Yu, Y. Establishment of cancer/testis antigen profiling based on clinicopathological characteristics in resected pathological stage III non-small cell lung cancer. Cancer Manag. Res. 2018, 10, 2031–2046. [Google Scholar] [CrossRef]
- Kim, Y.D.; Park, H.R.; Song, M.H.; Shin, D.H.; Lee, C.H.; Lee, M.K.; Lee, S.Y. Pattern of cancer/testis antigen expression in lung cancer patients. Int. J. Mol. Med. 2012, 29, 656–662. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Liu, Q.L.; Luo, M.; Huang, C.; Chen, H.N.; Zhou, Z.G. Epigenetic regulation of epithelial to mesenchymal transition in the cancer metastatic cascade: Implications for cancer therapy. Front. Oncol. 2021, 11, 657546. [Google Scholar] [CrossRef]
- Liu, C.; Fennell, L.J.; Bettington, M.L.; Walker, N.I.; Dwine, J.; Leggett, B.A.; Whitehall, V.L.J. DNA methylation changes that precede onset of dysplasia in advanced sessile serrated adenomas. Clin. Epigenet. 2019, 11, 90. [Google Scholar] [CrossRef]
- Bragelmann, J.; Barahona, P.C.; Marcelain, K.; Roessler, S.; Goeppert, B.; Gallegos, I.; Colombo, A.; Sanhueza, V.; Morales, E.; Rivera, M.T.; et al. Epigenome-wide analysis of methylation changes in the sequence of gallstone disease, dysplasia, and gallbladder cancer. Hepatology 2021, 73, 2293–2310. [Google Scholar] [CrossRef]
- Hernandez-Meza, G.; von Felden, J.; Gonzalez-Kozlova, E.E.; Garcia-Lezana, T.; Peix, J.; Portela, A.; Craig, A.J.; Sayols, S.; Schwartz, M.; Losic, B.; et al. DNA methylation profiling of human hepatocarcinogenesis. Hepatology 2020, 74, 183. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Kono, T.J.Y.; Knutson, C.G.; Parry, N.M.; Seiler, C.L.; Fox, J.G.; Tannenbaum, S.R.; Tretyakova, N.Y. Multi-omics characterization of inflammatory bowel disease-induced hyperplasia/dysplasia in the Rag2(−/−)/Il10(−/−) mouse model. Int. J. Mol. Sci. 2020, 22, 364. [Google Scholar] [CrossRef] [PubMed]
- Gougousis, S.; Mouchtaropoulou, E.; Besli, I.; Vrochidis, P.; Skoumpas, I.; Constantinidis, I. HPV-related oropharyngeal cancer and biomarkers based on epigenetics and microbiome profile. Front. Cell Dev. Biol. 2020, 8, 625330. [Google Scholar] [CrossRef]
- Walker, C.; Burggren, W. Remodeling the epigenome and (epi)cytoskeleton: A new paradigm for co-regulation by methylation. J. Exp. Biol. 2020, 223, jeb220632. [Google Scholar] [CrossRef]
- Karki, M.; Jangid, R.K.; Anish, R.; Seervai, R.N.H.; Bertocchio, J.P.; Hotta, T.; Msaouel, P.; Jung, S.Y.; Grimm, S.L.; Coarfa, C.; et al. A cytoskeletal function for PBRM1 reading methylated microtubules. Sci. Adv. 2021, 7, eabf2866. [Google Scholar] [CrossRef]
- Westcott, J.M.; Prechtl, A.M.; Maine, E.A.; Dang, T.T.; Esparza, M.A.; Sun, H.; Zhou, Y.; Xie, Y.; Pearson, G.W. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J. Clin. Investig. 2015, 125, 1927–1943. [Google Scholar] [CrossRef]
- Rorth, P. Fellow travellers: Emergent properties of collective cell migration. EMBO Rep. 2012, 13, 984–991. [Google Scholar] [CrossRef]
- Summerbell, E.R.; Mouw, J.K.; Bell, J.S.K.; Knippler, C.M.; Pedro, B.; Arnst, J.L.; Khatib, T.O.; Commander, R.; Barwick, B.G.; Konen, J.; et al. Epigenetically heterogeneous tumor cells direct collective invasion through filopodia-driven fibronectin micropatterning. Sci. Adv. 2020, 6, eaaz6197. [Google Scholar] [CrossRef]
- Grochowski, C.M.; Loomes, K.M.; Spinner, N.B. Jagged1 (JAG1): Structure, expression, and disease associations. Gene 2016, 576, 381–384. [Google Scholar] [CrossRef]
- Yuan, S.; Natesan, R.; Sanchez-Rivera, F.J.; Li, J.; Bhanu, N.V.; Yamazoe, T.; Lin, J.H.; Merrell, A.J.; Sela, Y.; Thomas, S.K.; et al. Global regulation of the histone mark H3K36me2 underlies epithelial plasticity and metastatic progression. Cancer Discov. 2020, 10, 854–871. [Google Scholar] [CrossRef]
- Schneider, J.; Wood, A.; Lee, J.S.; Schuster, R.; Dueker, J.; Maguire, C.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Shilatifard, A. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol. Cell 2005, 19, 849–856. [Google Scholar] [CrossRef]
- Wu, J.; Chai, H.; Xu, X.; Yu, J.; Gu, Y. Histone methyltransferase SETD1A interacts with HIF1alpha to enhance glycolysis and promote cancer progression in gastric cancer. Mol. Oncol. 2020, 14, 1397–1409. [Google Scholar] [CrossRef]
- Wu, J.; Chai, H.; Shan, H.; Pan, C.; Xu, X.; Dong, W.; Yu, J.; Gu, Y. Histone methyltransferase SETD1A induces epithelial-mesenchymal transition to promote invasion and metastasis through epigenetic reprogramming of snail in gastric cancer. Front. Cell Dev. Biol. 2021, 9, 657888. [Google Scholar] [CrossRef]
- Diaz-Lopez, A.; Diaz-Martin, J.; Moreno-Bueno, G.; Cuevas, E.P.; Santos, V.; Olmeda, D.; Portillo, F.; Palacios, J.; Cano, A. Zeb1 and Snail1 engage miR-200f transcriptional and epigenetic regulation during EMT. Int. J. Cancer 2015, 136, E62–E73. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Fan, X.; Jin, S.; Li, Y.; Khadaroo, P.A.; Dai, Y.; He, L.; Zhou, D.; Lin, H. Genetic and epigenetic regulation of E-cadherin signaling in human hepatocellular carcinoma. Cancer Manag. Res. 2019, 11, 8947–8963. [Google Scholar] [CrossRef]
- De Santis, G.; Miotti, S.; Mazzi, M.; Canevari, S.; Tomassetti, A. E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene 2009, 28, 1206–1217. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Nardone, V.; Lucarelli, A.P.; Dalle, V.A.; Fanelli, R.; Tomassetti, A.; Belvisi, L.; Civera, M.; Parisini, E. Crystal structure of human E-cadherin-EC1EC2 in complex with a peptidomimetic competitive inhibitor of cadherin homophilic interaction. J. Med. Chem. 2016, 59, 5089–5094. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.J.; Kim, J.H.; Lee, H.S.; Jang, J.J.; Kang, G.H. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am. J. Pathol. 2003, 163, 1371–1378. [Google Scholar] [CrossRef]
- Dumont, N.; Wilson, M.B.; Crawford, Y.G.; Reynolds, P.A.; Sigaroudinia, M.; Tlsty, T.D. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 14867–14872. [Google Scholar] [CrossRef]
- Tsujiuchi, T.; Shimizu, K.; Itsuzaki, Y.; Onishi, M.; Sugata, E.; Fujii, H.; Honoki, K. CpG site hypermethylation of E-cadherin and Connexin26 genes in hepatocellular carcinomas induced by a choline-deficient L-Amino Acid-defined diet in rats. Mol. Carcinog. 2007, 46, 269–274. [Google Scholar] [CrossRef]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Stroik, S.; Hendrickson, E.A. Telomere fusions and translocations: A bridge too far? Curr. Opin. Genet. Dev. 2020, 60, 85–91. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Herate, C.; Sabatier, L. Telomere instability initiates and then boosts carcinogenesis by the butterfly effect. Curr. Opin. Genet. Dev. 2020, 60, 92–98. [Google Scholar] [CrossRef]
- Cesare, A.J.; Karlseder, J. A three-state model of telomere control over human proliferative boundaries. Curr. Opin. Cell Biol. 2012, 24, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Kolquist, K.A.; Ellisen, L.W.; Counter, C.M.; Meyerson, M.; Tan, L.K.; Weinberg, R.A.; Haber, D.A.; Gerald, W.L. Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat. Genet. 1998, 19, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Komosa, M.; Nunes, N.M.; Tabori, U. DNA methylation of the TERT promoter and its impact on human cancer. Curr. Opin. Genet. Dev. 2020, 60, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, S.; Selig, S. Human subtelomeric DNA methylation: Regulation and roles in telomere function. Curr. Opin. Genet. Dev. 2020, 60, 9–16. [Google Scholar] [CrossRef]
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 3760. [Google Scholar] [CrossRef] [PubMed]
- Feretzaki, M.; Pospisilova, M.; Valador, F.R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Getz, G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef]
- Kaur, S.; Bronson, S.M.; Pal-Nath, D.; Miller, T.W.; Soto-Pantoja, D.R.; Roberts, D.D. Functions of thrombospondin-1 in the tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 4570. [Google Scholar] [CrossRef] [PubMed]
- Perez-Janices, N.; Blanco-Luquin, I.; Tunon, M.T.; Barba-Ramos, E.; Ibanez, B.; Zazpe-Cenoz, I.; Martinez-Aguillo, M.; Hernandez, B.; Martinez-Lopez, E.; Fernandez, A.F.; et al. EPB41L3, TSP-1 and RASSF2 as new clinically relevant prognostic biomarkers in diffuse gliomas. Oncotarget 2015, 6, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Watnick, R.S.; Rodriguez, R.K.; Wang, S.; Blois, A.L.; Rangarajan, A.; Ince, T.; Weinberg, R.A. Thrombospondin-1 repression is mediated via distinct mechanisms in fibroblasts and epithelial cells. Oncogene 2015, 34, 2823–2835. [Google Scholar] [CrossRef]
- Goncalves, C.S.; Le, B.E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef]
- Smith, J.; Zyoud, A.; Allegrucci, C. A case of identity: HOX genes in normal and cancer stem cells. Cancers 2019, 11, 512. [Google Scholar] [CrossRef]
- Le Boiteux, E.; Court, F.; Guichet, P.O.; Vaurs-Barriere, C.; Vaillant, I.; Chautard, E.; Verrelle, P.; Costa, B.M.; Karayan-Tapon, L.; Fogli, A.; et al. Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage. Mol. Oncol. 2021, 15, 1995–2010. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von, D.A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Li, H.H.; Chen, R.; Hyduke, D.R.; Williams, A.; Frotschl, R.; Ellinger-Ziegelbauer, H.; O’Lone, R.; Yauk, C.L.; Aubrecht, J.; Fornace, A.J., Jr. Development and validation of a high-throughput transcriptomic biomarker to address 21st century genetic toxicology needs. Proc. Natl. Acad. Sci. USA 2017, 114, E10881–E10889. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Chappell, G.A.; Rager, J.E. Epigenetics in chemical-induced genotoxic carcinogenesis. Curr. Opin. Toxicol. 2017, 6, 10–17. [Google Scholar] [CrossRef]
- Mascolo, M.G.; Perdichizzi, S.; Vaccari, M.; Rotondo, F.; Zanzi, C.; Grilli, S.; Paparella, M.; Jacobs, M.N.; Colacci, A. The transformics assay: First steps for the development of an integrated approach to investigate the malignant cell transformation in vitro. Carcinogenesis 2018, 39, 955–967. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C.; Tcheremenskaia, O. Nongenotoxic carcinogenicity of chemicals: Mechanisms of action and early recognition through a new set of structural alerts. Chem. Rev. 2013, 113, 2940–2957. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.I.; Kay, S.J.E.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D.; Bateman, A.; et al. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [PubMed]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015, 43, D168–D173. [Google Scholar] [CrossRef]
- House, J.S.; Grimm, F.A.; Jima, D.D.; Zhou, Y.H.; Rusyn, I.; Wright, F.A. A pipeline for high-throughput concentration response modeling of gene expression for toxicogenomics. Front. Genet. 2017, 8, 168. [Google Scholar] [CrossRef]
- Larman, H.B.; Scott, E.R.; Wogan, M.; Oliveira, G.; Torkamani, A.; Schultz, P.G. Sensitive, multiplex and direct quantification of RNA sequences using a modified RASL assay. Nucleic Acids Res. 2014, 42, 9146–9157. [Google Scholar] [CrossRef]
- Grimm, F.A.; Iwata, Y.; Sirenko, O.; Chappell, G.A.; Wright, F.A.; Reif, D.M.; Braisted, J.; Gerhold, D.L.; Yeakley, J.M.; Shepard, P.; et al. A chemical-biological similarity-based grouping of complex substances as a prototype approach for evaluating chemical alternatives. Green Chem. 2016, 18, 4407–4419. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Asmus, N.; Papale, L.A.; Madrid, A.; Alisch, R.S. Simultaneous targeted methylation sequencing (sTM-Seq). Curr. Protoc. Hum. Genet. 2019, 101, e81. [Google Scholar] [CrossRef]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; Gaykalova, D.A.; et al. Epigenetic regulation of gene expression in cancer: Techniques, resources and analysis. Brief Funct. Genom. 2018, 17, 49–63. [Google Scholar] [CrossRef]
- Wiehle, L.; Thorn, G.J.; Raddatz, G.; Clarkson, C.T.; Rippe, K.; Lyko, F.; Breiling, A.; Teif, V.B. DNA (de)methylation in embryonic stem cells controls CTCF-dependent chromatin boundaries. Genome Res. 2019, 29, 750–761. [Google Scholar] [CrossRef]
- Canzio, D.; Nwakeze, C.L.; Horta, A.; Rajkumar, S.M.; Coffey, E.L.; Duffy, E.E.; Duffie, R.; Monahan, K.; O’Keeffe, S.; Simon, M.D.; et al. Antisense lncRNA transcription mediates DNA demethylation to drive stochastic protocadherin alpha promoter choice. Cell 2019, 177, 639–653. [Google Scholar] [CrossRef]
- Mohni, K.N.; Wessel, S.R.; Zhao, R.; Wojciechowski, A.C.; Luzwick, J.W.; Layden, H.; Eichman, B.F.; Thompson, P.S.; Mehta, K.P.M.; Cortez, D. HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 2019, 176, 144–153. [Google Scholar] [CrossRef]
- Mahmood, N.; Rabbani, S.A. DNA methylation readers and cancer: Mechanistic and therapeutic applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- Mellen, M.; Ayata, P.; Heintz, N. 5-hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc. Natl. Acad. Sci. USA 2017, 114, E7812–E7821. [Google Scholar] [CrossRef]
- Booth, M.J.; Ost, T.W.; Beraldi, D.; Bell, N.M.; Branco, M.R.; Reik, W.; Balasubramanian, S. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc. 2013, 8, 1841–1851. [Google Scholar] [CrossRef]
- Liu, Y.; Siejka-Zielinska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Bockler, B.; Song, C.X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Yu, M.; Hon, G.C.; Szulwach, K.E.; Song, C.X.; Zhang, L.; Kim, A.; Li, X.; Dai, Q.; Shen, Y.; Park, B.; et al. Base-resolution analysis of 5-hydroxymethylcytosine in the Mammalian genome. Cell 2012, 149, 1368–1380. [Google Scholar] [CrossRef]
- Tierling, S.; Schmitt, B.; Walter, J. Comprehensive evaluation of commercial bisulfite-based DNA methylation kits and development of an alternative protocol with improved conversion performance. Genet. Epigenet. 2018, 10, 1179237X18766097. [Google Scholar] [CrossRef]
- Paparella, M.; Colacci, A.; Jacobs, M.N. Uncertainties of testing methods: What do we (want to) know about carcinogenicity? ALTEX 2017, 34, 235–252. [Google Scholar] [CrossRef]
- Cohen, S.M. The relevance of experimental carcinogenicity studies to human safety. Curr. Opin. Toxicol. 2017, 3, 6–11. [Google Scholar] [CrossRef]
- Goodman, J.I. Goodbye to the bioassay. Toxicol. Res. 2018, 7, 558–564. [Google Scholar] [CrossRef]
- Sarigiannis, D.A.; Karakitsios, S.; Dominguez-Romero, E.; Papadaki, K.; Brochot, C.; Kumar, V.; Schumacher, M.; Sy, M.; Mielke, H.; Greiner, M.; et al. Physiology-based toxicokinetic modelling in the frame of the European Human Biomonitoring Initiative. Environ. Res. 2019, 172, 216–230. [Google Scholar] [CrossRef]
- OECD. Guidance Document on the Characterisation, Validation and Reporting of PBK Models for Regulatory Purposes; Series on Testing and Assessment, no. 331; Environment Directorate; Organisation for Economic Cooperation and Development: Paris, France, 2021; pp. 1–96. [Google Scholar]
- Cheong, A.; Zhang, X.; Cheung, Y.Y.; Tang, W.Y.; Chen, J.; Ye, S.H.; Medvedovic, M.; Leung, Y.K.; Prins, G.S.; Ho, S.M. DNA methylome changes by estradiol benzoate and bisphenol A links early-life environmental exposures to prostate cancer risk. Epigenetics 2016, 11, 674–689. [Google Scholar] [CrossRef]
- Aravind, L.; Abhiman, S.; Iyer, L.M. Natural history of the eukaryotic chromatin protein methylation system. Prog. Mol. Biol. Transl. Sci. 2011, 101, 105–176. [Google Scholar]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar]
- Angrish, M.M.; Allard, P.; McCullough, S.D.; Druwe, I.L.; Helbling, C.L.; Hines, E.; Chorley, B.N. Epigenetic applications in adverse outcome pathways and environmental risk evaluation. Environ. Health Perspect. 2018, 126, 045001. [Google Scholar] [CrossRef]
- Wirbisky-Hershberger, S.E.; Sanchez, O.F.; Horzmann, K.A.; Thanki, D.; Yuan, C.; Freeman, J.L. Atrazine exposure decreases the activity of DNMTs, global DNA methylation levels, and dnmt expression. Food Chem. Toxicol. 2017, 109, 727–734. [Google Scholar] [CrossRef]
- Sugai, T.; Yoshida, M.; Eizuka, M.; Uesugii, N.; Habano, W.; Otsuka, K.; Sasaki, A.; Yamamoto, E.; Matsumoto, T.; Suzuki, H. Analysis of the DNA methylation level of cancer-related genes in colorectal cancer and the surrounding normal mucosa. Clin. Epigenet. 2017, 9, 55. [Google Scholar] [CrossRef]
- Lim, P.L.; Tan, W.; Latchoumycandane, C.; Mok, W.C.; Khoo, Y.M.; Lee, H.S.; Sattabongkot, J.; Beerheide, W.; Lim, S.G.; Tan, T.M.; et al. Molecular and functional characterization of drug-metabolizing enzymes and transporter expression in the novel spontaneously immortalized human hepatocyte line HC-04. Toxicol. Vitro 2007, 21, 1390–1401. [Google Scholar] [CrossRef]
- Kleensang, A.; Vantangoli, M.M.; Odwin-Dacosta, S.; Andersen, M.E.; Boekelheide, K.; Bouhifd, M.; Fornace, A.J., Jr.; Li, H.H.; Livi, C.B.; Madnick, S.; et al. Genetic variability in a frozen batch of MCF-7 cells invisible in routine authentication affecting cell function. Sci. Rep. 2016, 6, 28994. [Google Scholar] [CrossRef]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Nery, E.D.; Ebner, D.; Montoya, M.C.; Ostling, P.; Pietiainen, V.; Price, L.S.; et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822–32840. [Google Scholar] [CrossRef]
- Schock, B.C.; Koostra, J.; Kwack, S.; Hackman, R.M.; Van Der Vliet, A.; Cross, C.E. Ascorbic acid in nasal and tracheobronchial airway lining fluids. Free Radic. Biol. Med. 2004, 37, 1393–1401. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef]
- Jiang, J.; Wolters, J.E.; van Breda, S.G.; Kleinjans, J.C.; de Kok, T.M. Development of novel tools for the in vitro investigation of drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1523–1537. [Google Scholar] [CrossRef]
- Li, M.; Li, X.; Zhuang, Y.; Flemington, E.K.; Lin, Z.; Shan, B. Induction of a novel isoform of the lncRNA HOTAIR in Claudin-low breast cancer cells attached to extracellular matrix. Mol. Oncol. 2017, 11, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Wolters, J.E.J.; van Breda, S.G.J.; Caiment, F.; Claessen, S.M.; de Kok, T.M.C.M.; Kleinjans, J.C.S. Nuclear and mitochondrial DNA methylation patterns induced by valproic acid in human hepatocytes. Chem. Res. Toxicol. 2017, 30, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Wolters, J.E.J.; van Breda, S.G.J.; Grossmann, J.; Fortes, C.; Caiment, F.; Kleinjans, J.C.S. Integrated ‘omics analysis reveals new drug-induced mitochondrial perturbations in human hepatocytes. Toxicol. Lett. 2018, 289, 1–13. [Google Scholar] [CrossRef] [PubMed]








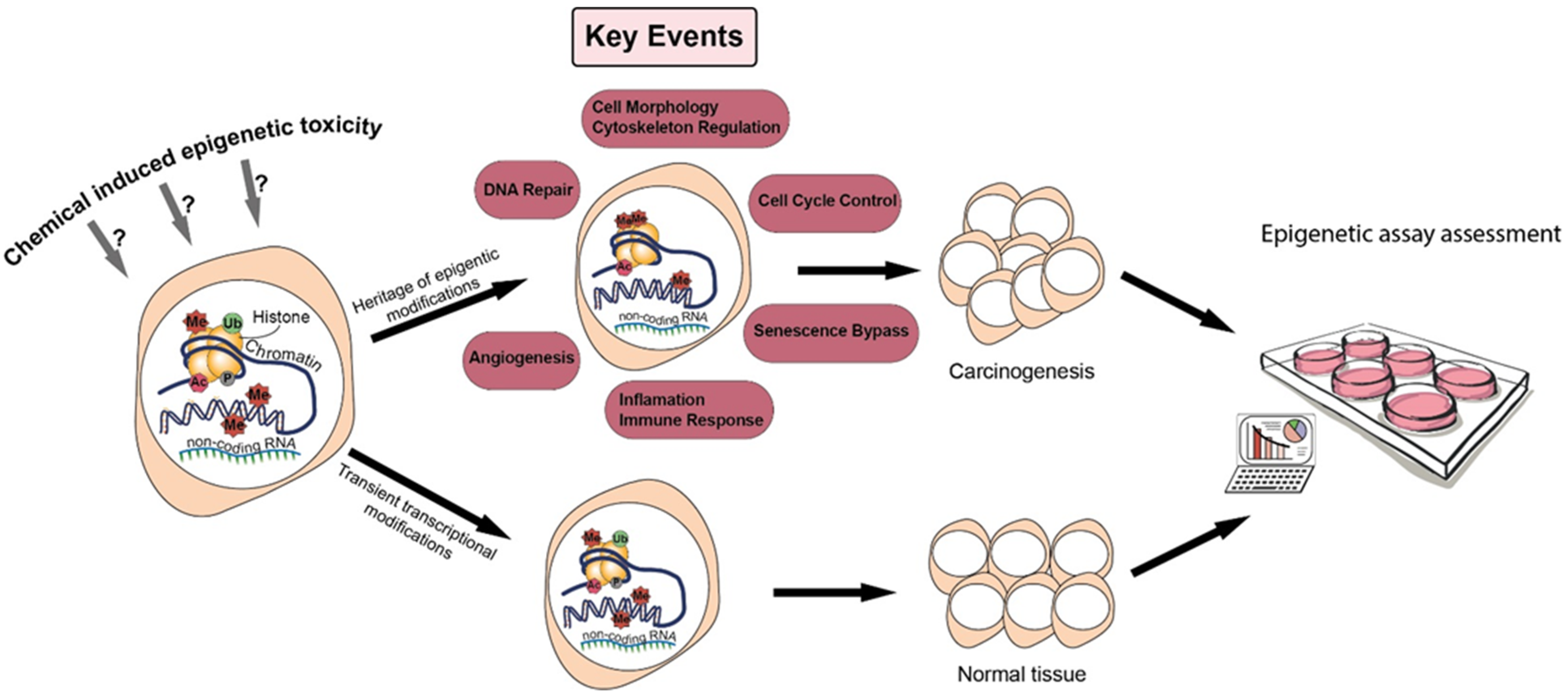{kind=link}
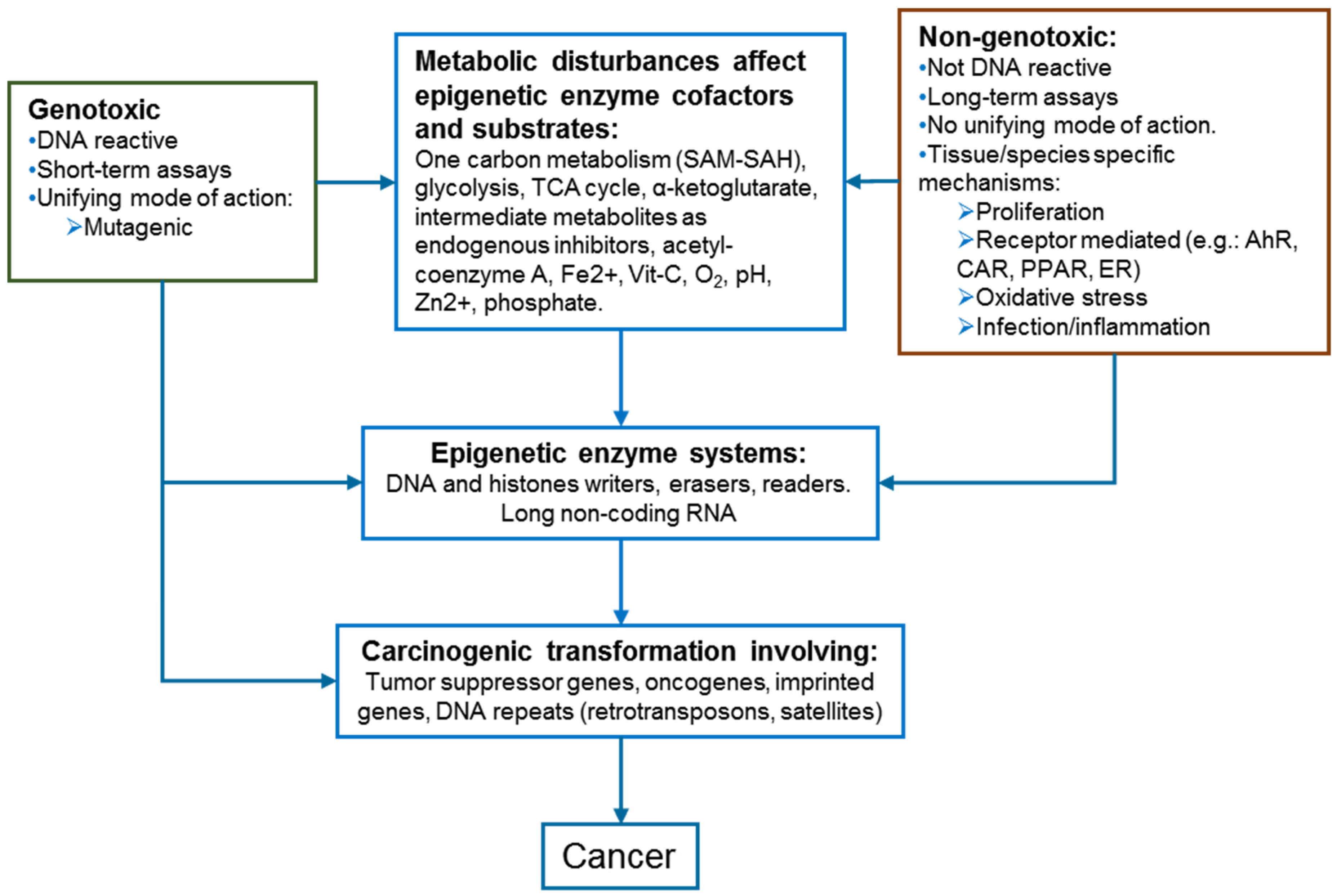{kind=link}
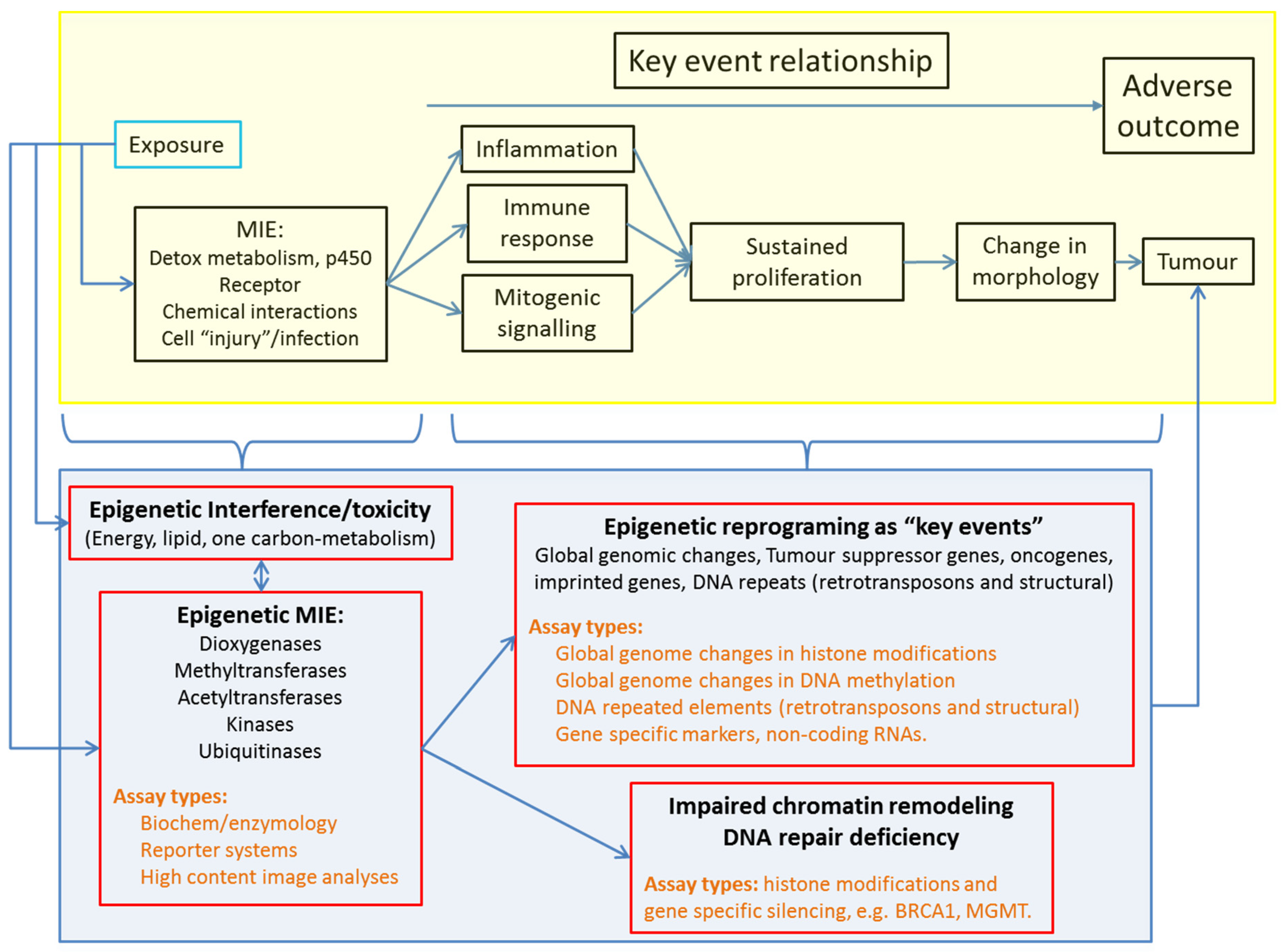{kind=link}
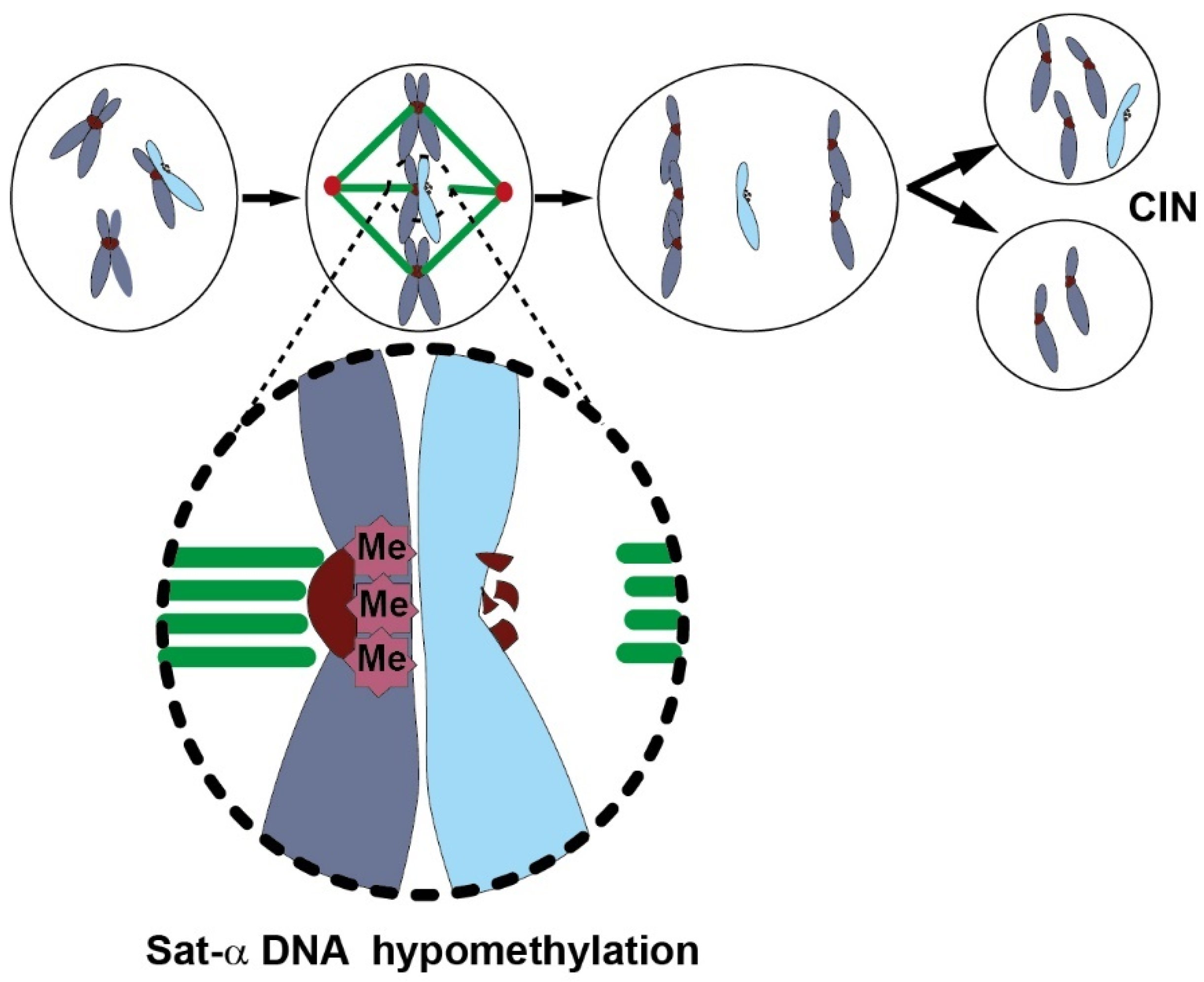{kind=link}
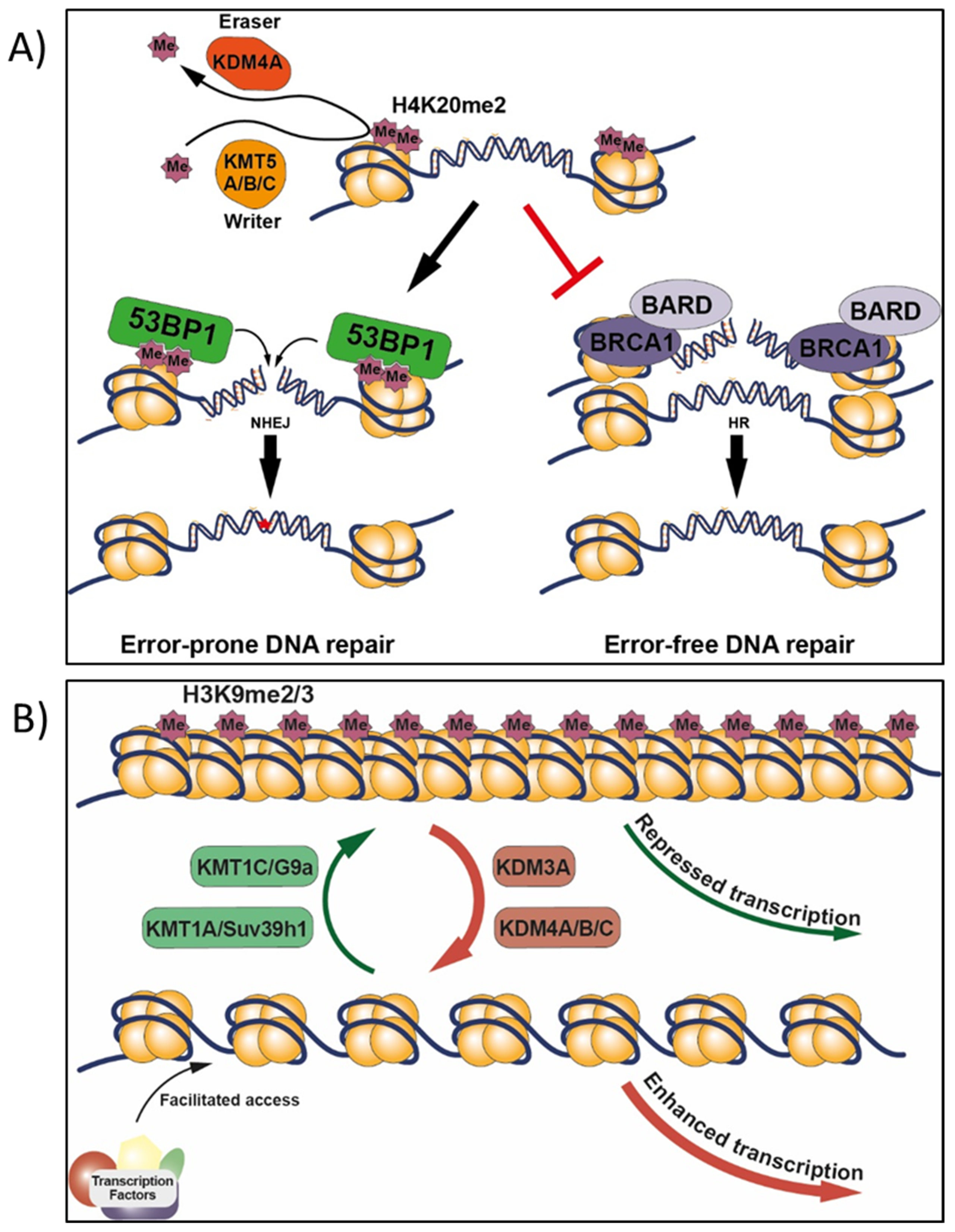{kind=link}
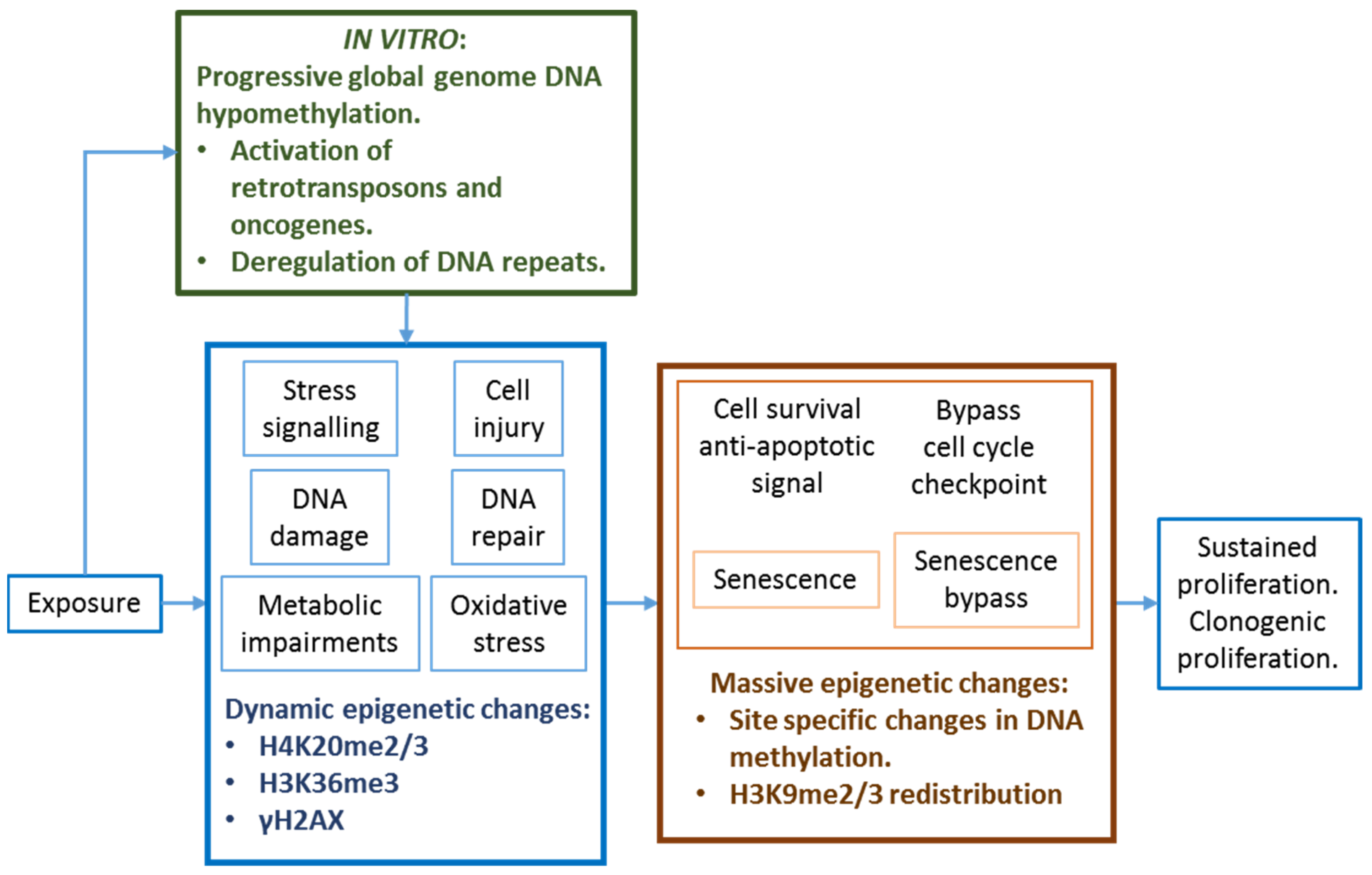{kind=link}
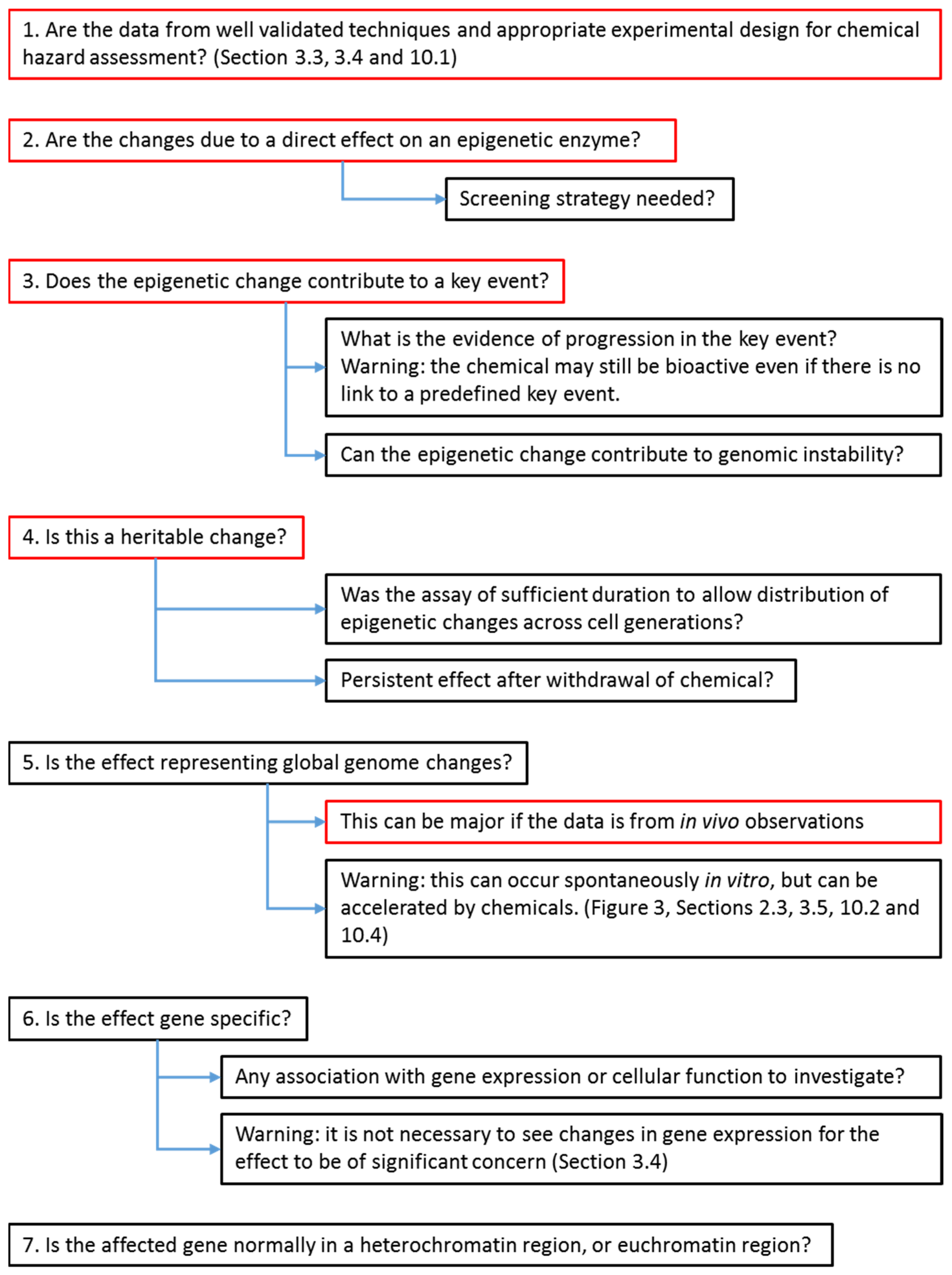{kind=link}
| (1) Sufficient evidence to be considered as ca ontributor to a carcinogenic MIE or KE. | |
| (2) Assay robustness and reproducibility across users and laboratories. | |
| (3) Dose/concentration–response: | |
| |
| (4) Protocol: | |
| |
| Arsenicals | References |
|---|---|
| (1) Oxidative stress and arsenic metabolism that limit the availability of SAM for DNMT and other MT. | [233,234] |
| (2) Reduction in DNMT mRNA expression and interference with DNMT activities. | [235] |
| (3) Global genome DNA hypomethylation and site-specific changes in DNAm including retrotransposons. | [236,237,238] |
| (4) Reduction in CTCF binding to the DNA. CTCF act as a repressor, insulator, or as a transcription factor. | [239] |
| (5) Binding of sulfhydryl groups of cysteine residues and displacement of zinc ions from the zinc finger DNA binding domains of numerous proteins (TET, histone acetyltransferase, etc.). | [240] |
| (6) Interference with TET enzyme expression and activities | [189,190] |
| (7) Change in DNAm inducing changes in ncRNAs (miRNAs and LncRNAs). | [241,242] |
| (8) Imbalance in expression of histone variants. | [243] |
| (9) Histone post-transcriptional modifications | [244] |
| (10) DNA repair mechanisms | [245,246] |
| (11) Mitochondrial biogenesis and mitochondrial DNA copy number. | [247,248] |
| Nickel | |
| (1) Reduction in core histones acetylation. | [249,250] |
| (2) Redistribution of the silencing mark H3K9me2 and 5mC (p16 silencing). | [251,252,253] |
| (3) Reduction in the histone methyltransferase activities of G9a (targeting H3K9 dimethylation) and Suv39h1 (targeting H3K9 trimethylation). | [254] |
| (4) Inhibition of the lysine demethylases; KDM3A/JMJD1A acting on H3K9me1 and me2, while KDM4A-D/JMJD2A-D on H3K9me2 and me3.Nickel ions inactivate 2-oxoglutarate-dependent dioxygenases by replacing the cation Fe2+ at the catalytic sites (effects of hypoxia and oxidative stress). | [255,256,257,258] |
| (5) Inhibition of other dioxygenases, including TET1, HIF prolyl hydroxylases and the DNA repair enzyme ABH2. | [71,255,259] |
| (6) Deregulation of the ubiquitination/deubiquitination machinery. | [260,261] |
| (7) Interference with the Zn2+ finger protein CTCF. | [262,263] |
| (8) Long-term effects on gene expression associated with abundance of H3K4me3 and H3K27me3 in promoters. | [188,263] |
| Phenobarbital | |
| (1) Induction of the stress-response protein GADD45, as mediator of DNA demethylation. | [90,91,264] |
| (2) 5hmC precedes global DNA hypomethylation, and global loss of 5hmC as an early marker of hepatocarcinogenesis and genome flexibility. | [264,265] |
| (3) DNAm-dependent expression of miRNAs and LncRNAs from the imprinted Dlk1-Dio3 locus. | [266,267] |
| (4) Reduction in hepatic expression of epigenetic system components, and L1 ORF1 hypomethylation in carcinogenic target tissue only (in the liver but not in the kidney). | [201] |
| Irradiation | |
| (1) Persistent GGDHo and in distant non-target cells. | [137,268] |
| (2) DNA hypomethylation of more recent L1 retrotransposons | [137,268] |
| (3) Over expression of DNMT3B contributing to p53 and p21 silencing by DNA methylation. | [269] |
| Other chemicals (acrylamide, 2-acetylaminofluorene, 1,3-butadiene, furan, and methapyrilene) | |
| Carcinogen target tissues show decreases in abundance of H4K20 methylation and of its corresponding histone methyltransferase family (KMT5A/B/C), compared to non-target tissues. | [270] |
| Estradiol-17β | |
| DNA hypermethylation of the distal promoter of catechol-o-methyltransferase that reduces the expression of this protective enzyme against the formation of the mutagenic catecholestrogens. | [271] |
| 1. Screening assays for chemical interference with enzyme activities: |
Dioxygenases, methyltransferases, acetyltransferases, kinases, ubiquitinases.
|
| 2. Absolute global genome changes: |
Robust measures of multiple epigenetic marks within the same sample by LC–MS/MS, CE–MS.
|
| 3. Index of global genome changes in DNAm: |
|
| 4. Markers of specific key events (tissue/species specific): |
Detoxification and DNA repair pathway:
|
| 5. Next-generation sequencing epigenetic methodologies: |
Transcriptomic analyses of epigenetic driver genes.
|
| Screening | Potential to Predict Key Events and to Provide a Comprehensive Analysis | |
|---|---|---|
| In vitro Short-term enzymology |
| Limited under short-term experiment |
| In vitro Heritable epigenetic memory/reprograming (Long-term > 3 weeks) |
| NGS-based methodologies for DNA or HPTM followed by validation, pathway analyses, and demonstration of affected pathway |
| In vivo Heritable epigenetic memory/reprograming (Long-term > 3 weeks) |
| NGS-based methodologies for DNA or HPTM followed by validation, pathway analyses, and demonstration of affected pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desaulniers, D.; Vasseur, P.; Jacobs, A.; Aguila, M.C.; Ertych, N.; Jacobs, M.N. Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications. Int. J. Mol. Sci. 2021, 22, 10969. https://doi.org/10.3390/ijms222010969
Desaulniers D, Vasseur P, Jacobs A, Aguila MC, Ertych N, Jacobs MN. Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications. International Journal of Molecular Sciences. 2021; 22(20):10969. https://doi.org/10.3390/ijms222010969
Chicago/Turabian StyleDesaulniers, Daniel, Paule Vasseur, Abigail Jacobs, M. Cecilia Aguila, Norman Ertych, and Miriam N. Jacobs. 2021. "Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications" International Journal of Molecular Sciences 22, no. 20: 10969. https://doi.org/10.3390/ijms222010969
APA StyleDesaulniers, D., Vasseur, P., Jacobs, A., Aguila, M. C., Ertych, N., & Jacobs, M. N. (2021). Integration of Epigenetic Mechanisms into Non-Genotoxic Carcinogenicity Hazard Assessment: Focus on DNA Methylation and Histone Modifications. International Journal of Molecular Sciences, 22(20), 10969. https://doi.org/10.3390/ijms222010969