
Insight into the Binding and Hydrolytic Preferences of hNudt16 Based on Nucleotide Diphosphate Substrates

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
2.1. Hydrolytic Activity of hNudt16 on Dinucleotides Containing Diphosphate Bridge
2.2. Assessment of the Compliance of the Properties of hNudt16 Mutants with the Wild-Type Protein by Circular Dichroism (CD), Differential Scanning Fluorimetry (DSF)
2.2.1. Comparison of the Secondary Structure of hNudt16 and Its Mutants by CD
2.2.2. Analysis of Protein Thermal Stability by DSF
2.3. hNudt16 Stability in the Presence of Different Ligands and Its Substrate Affinity Studies
2.3.1. hNudt16-Ligand Complex Thermal Stability Screen by DSF and NanoDSF
2.3.2. Characterization of hNudt16-Ligand Complexes by Microscale Thermophoresis (MST)
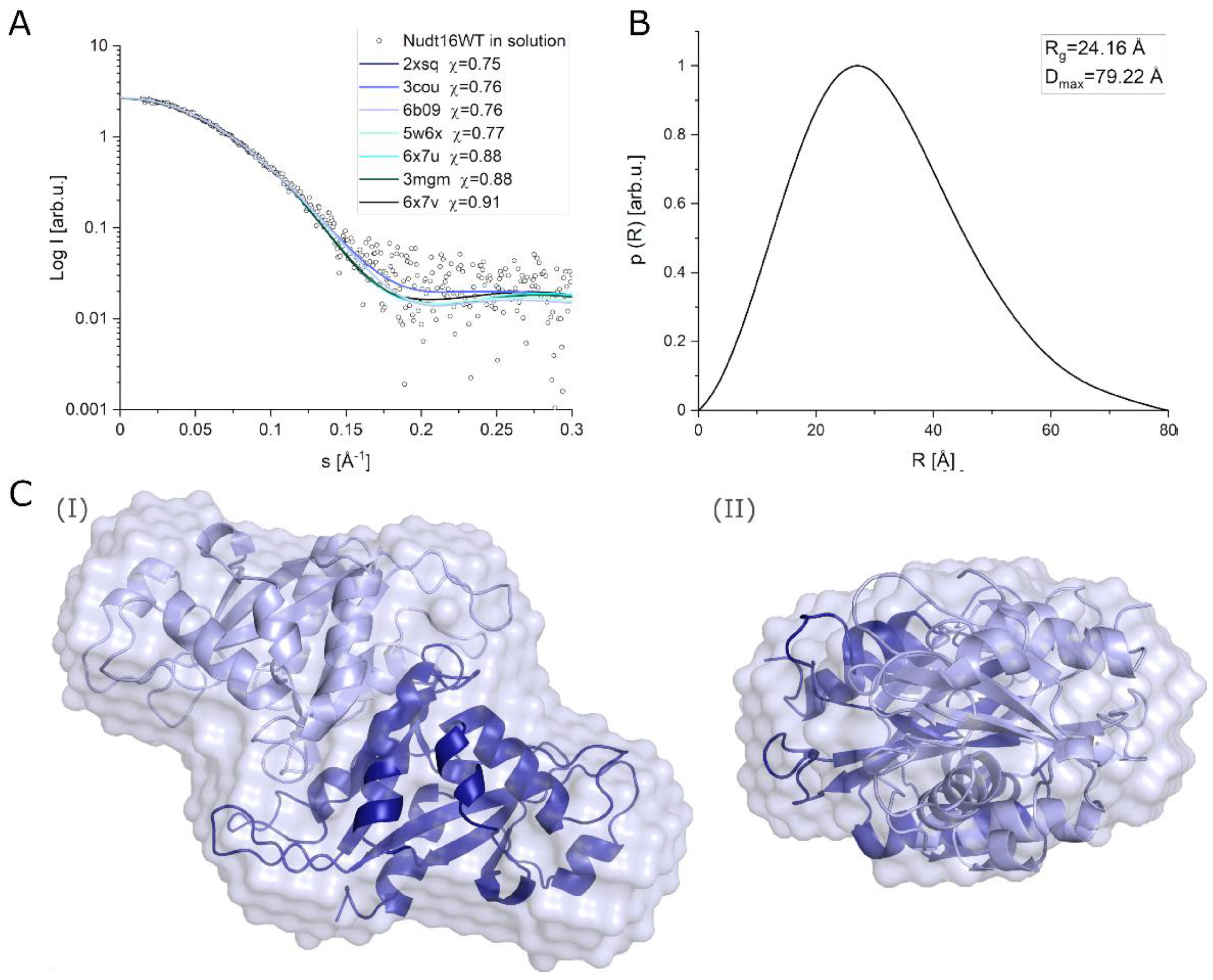
2.3.3. Determination of the Structure of hNudt16 in Solution by SAXS
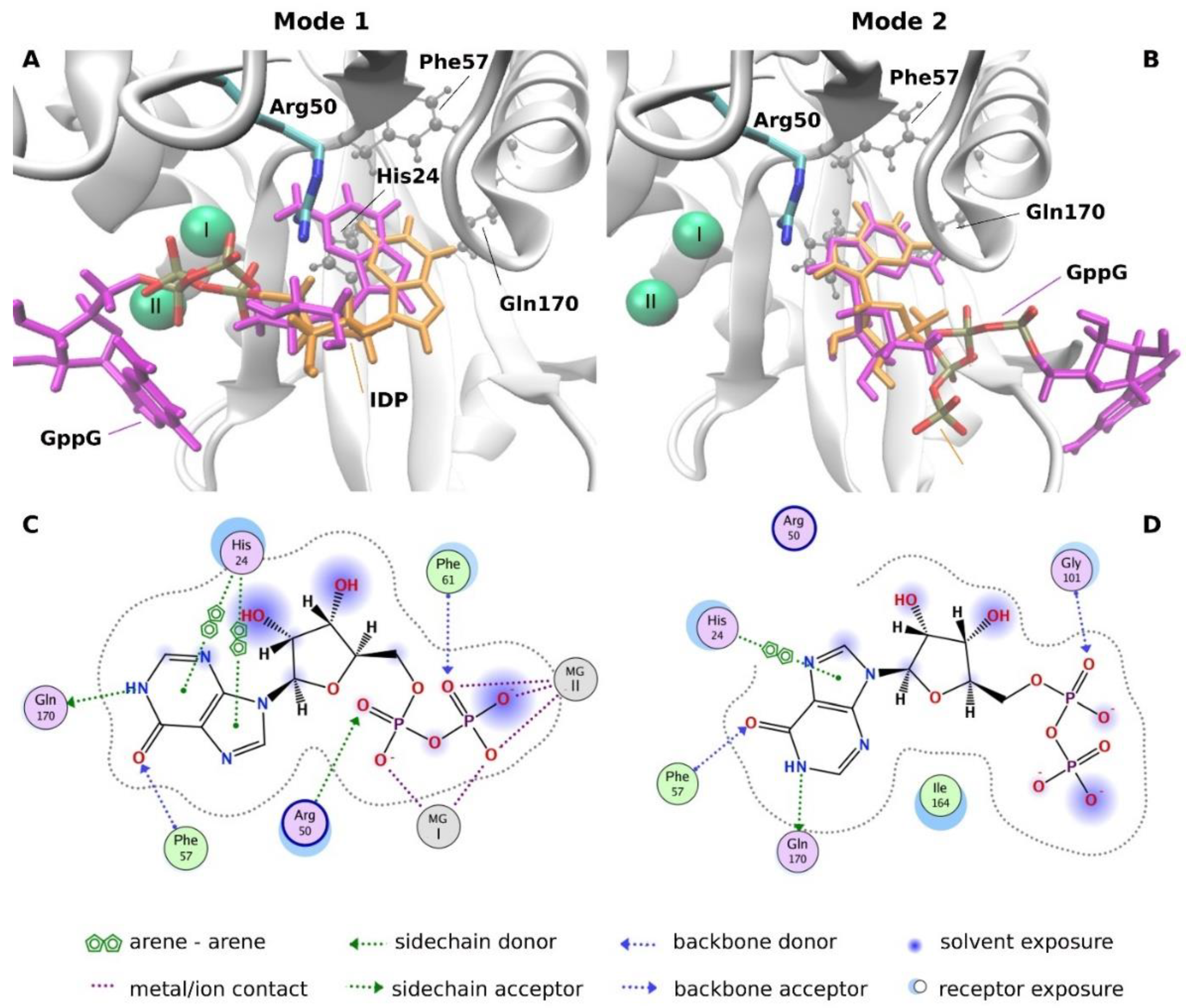
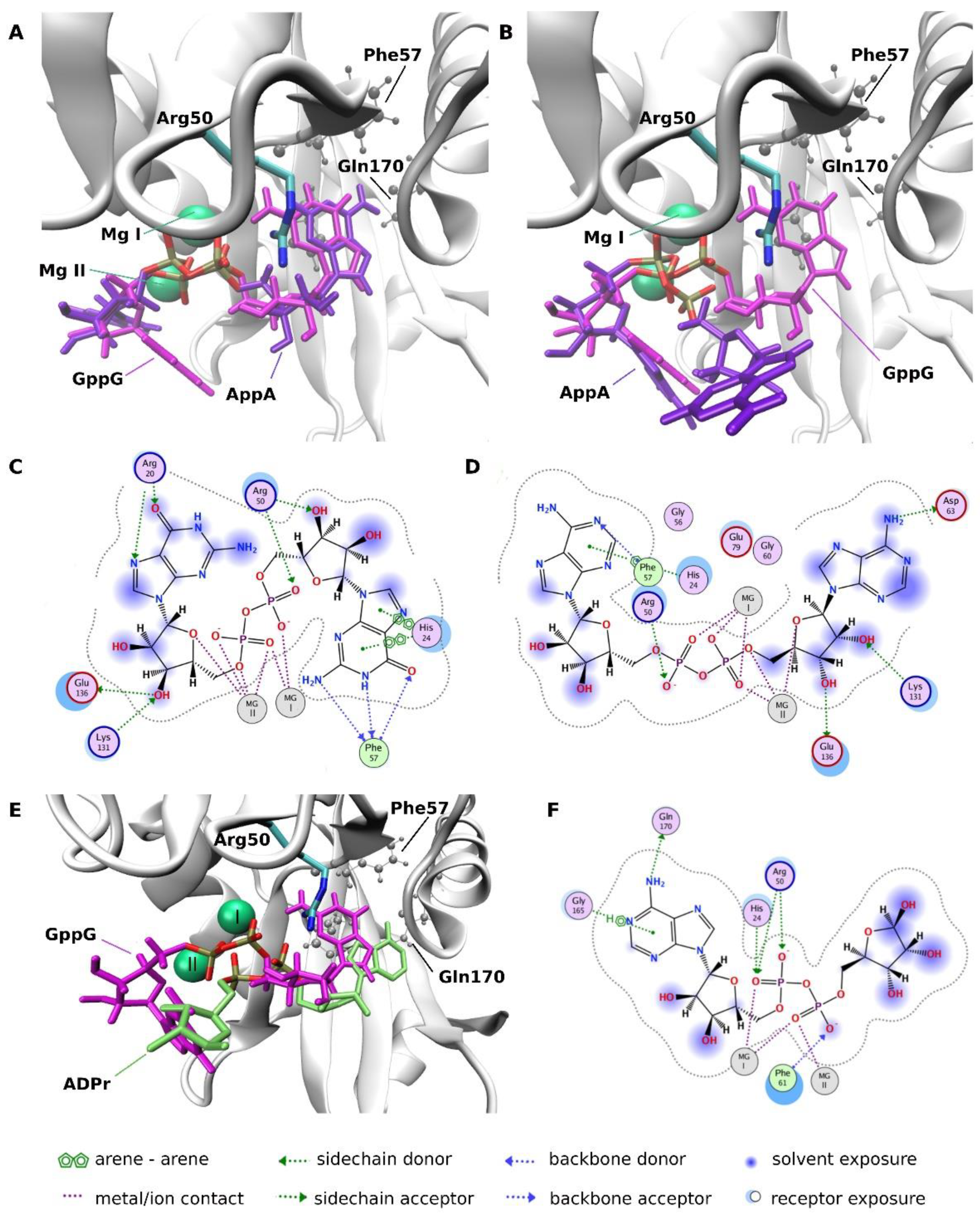
2.3.4. Molecular Docking Analysis
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of hNudt16
4.2. Site-Directed Mutagenesis of hNudt16
4.3. Chemical Synthesis of Cap Analogs
4.4. Circular Dichroism (CD)
4.5. Small Angle X-ray Scattering (SAXS)
4.6. Differential Scanning Fluorimetry (DSF)
4.6.1. Protein Thermal Stability Screen
4.6.2. hNudt16-Ligand Thermal Stability Screen
4.7. Microscale Thermophoresis (MST)
4.8. Enzymatic Assays
4.9. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NUDIX | Nucleoside diphosphate linked to another moiety X |
| GDP | Guanosine diphosphate |
| GMP | Guanosine monophosphate |
| m7GDP | 7-methylguanosine diphosphate |
| m32,2,7GDP | 2,2,7-trimethylguanosine |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced NAD+ |
| FAD | Flavin adenine dinucleotide |
| dpCoA | Dephospho-coenzyme A |
| IDP | Inosine diphosphate |
| IMP | Inosine monophosphate |
| ITP | Inosine triphosphate |
| CDP | Cytidine diphosphate |
| ADPr | Adenosine diphosphate ribose |
| CD | Circular dichroism |
| SAXS | Small-angle X-ray scattering |
| DSF | Differential Scanning Fluorimetry |
| nanoDSF | Low-volume Differential Scanning Fluorimetry |
| TCEP | (Tris(2-carboxyethyl)phosphine) |
| MST | Microscale Thermophoresis |
| HPLC | High Performance Liquid Chromatography |
| PDB | Protein Data Bank |
| dNTP | Deoxynucleotide triphosphate |
References
- Bessman, M.J.; Frick, D.; O’Handley, S.F. The MutT proteins or “nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J. Biol. Chem. 1996, 271, 25059–25062. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.; Xia, Z.; Azurmendi, H.; Saraswat, V.; Legler, P.; Massiah, M.; Gabelli, S.; Bianchet, M.; Kang, L.-W.; Amzel, L.M. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dunn, C.A.; Jones, C.R.; D’Souza, G.; Bessman, M.J. The 26 Nudix hydrolases of Bacillus cereus, a close relative of Bacillus anthracis. J. Biol. Chem. 2004, 279, 24861–24865. [Google Scholar] [CrossRef]
- Zheng, Q.-C.; Li, Z.-S.; Sun, M.; Zhang, Y.; Sun, C.-C. Homology modeling and substrate binding study of Nudix hydrolase Ndx1 from Thermos thermophilus HB. Biochem. Biophys. Res. Commun. 2005, 333, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Peculis, B.A.; Reynolds, K.; Cleland, M. Metal determines efficiency and substrate specificity of the nuclear NUDIX decapping proteins X29 and H29K (Nudt16). J. Biol. Chem. 2007, 282, 24792–24805. [Google Scholar] [CrossRef]
- Abeygunawardana, C.; Weber, D.J.; Gittis, A.G.; Frick, D.; Lin, J.; Miller, A.-F.; Bessman, M.J.; Mildvan, A.S. Solution structure of the MutT enzyme, a nucleoside Triphosphate Pyrophosphohydrolase. Biochemistry 1995, 34, 14997–15005. [Google Scholar] [CrossRef]
- Ghosh, T.; Peterson, B.; Tomasevic, N.; Peculis, B. Xenopus U8 snoRNA binding protein is a conserved nuclear decapping enzyme. Mol. Cell 2004, 13, 817–828. [Google Scholar] [CrossRef]
- Taylor, M.J.; Peculis, B.A. Evolutionary conservation supports ancient origin for Nudt16, a nuclear-localized, RNA-binding, RNA-decapping enzyme. Nucleic Acids Res. 2008, 36, 6021–6034. [Google Scholar] [CrossRef][Green Version]
- Song, M.-G.; Li, Y.; Kiledjian, M. Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell 2010, 40, 423–432. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, J.; Li, Y.; Li, Z.; Zhang, N.; Xu, X.; Wang, T.; Guan, Z.; Gao, G.F.; Yan, J. hNUDT16: A universal decapping enzyme for small nucleolar RNA and cytoplasmic mRNA. Protein Cell 2011, 2, 64–73. [Google Scholar] [CrossRef]
- Li, Y.; Song, M.; Kiledjian, M. Differential utilization of decapping enzymes in mammalian mRNA decay pathways. RNA 2011, 17, 419–428. [Google Scholar] [CrossRef]
- Grzela, R.; Nasilowska, K.; Lukaszewicz, M.; Tyras, M.; Stepinski, J.; Jankowska-Anyszka, M.; Bojarska, E.; Darzynkiewicz, E. Hydrolytic activity of human Nudt16 enzyme on dinucleotide cap analogs and short capped oligonucleotides. RNA 2018, 24, 633–642. [Google Scholar] [CrossRef]
- Sharma, S.; Grudzien-Nogalska, E.; Hamilton, K.; Jiao, X.; Yang, J.; Tong, L.; Kiledjian, M. Mammalian Nudix proteins cleave nucleotide metabolite caps on RNAs. Nucleic Acids Res. 2020, 48, 6788–6798. [Google Scholar] [CrossRef]
- Kulikova, V.A.; Nikiforov, A.A. Role of NUDIX hydrolases in NAD and ADP-Ribose metabolism in mammals. Biochemistry 2020, 85, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, N.; Iyama, T.; Tsuchimoto, D.; Sakumi, K.; Ohno, M.; Behmanesh, M.; Nakabeppu, Y. NUDT16 and ITPA play a dual protective role in maintaining chromosome stability and cell growth by eliminating dIDP/IDP and dITP/ITP from nucleotide pools in mammals. Nucleic Acids Res. 2010, 38, 2891–2903. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Abolhassani, N.; Tsuchimoto, D.; Nonaka, M.; Nakabeppu, Y. NUDT16 is a (deoxy)inosine diphosphatase, and its deficiency induces accumulation of single-strand breaks in nuclear DNA and growth arrest. Nucleic Acids Res. 2010, 38, 4834–4843. [Google Scholar] [CrossRef] [PubMed]
- Trésaugues, L.; Lundbäck, T.; Welin, M.; Flodin, S.; Nyman, T.; Silvander, C.; Gräslund, S.; Nordlund, P. Structural basis for the specificity of human NUDT16 and its regulation by inosine monophosphate. PLoS ONE 2015, 10, e0131507. [Google Scholar] [CrossRef]
- Palazzo, L.; Daniels, C.; Nettleship, J.; Rahman, N.; McPherson, R.L.; Ong, S.; Kato, K.; Nureki, O.; Leung, A.K.L.; Ahel, I. ENPP 1 processes protein ADP -ribosylation in vitro. FEBS J. 2016, 283, 3371–3388. [Google Scholar] [CrossRef]
- Thirawatananond, P.; McPherson, R.L.; Malhi, J.; Nathan, S.; Lambrecht, M.J.; Brichacek, M.; Hergenrother, P.J.; Leung, A.K.L.; Gabelli, S.B. Structural analyses of NudT16–ADP-ribose complexes direct rational design of mutants with improved processing of poly(ADP-ribosyl)ated proteins. Sci. Rep. 2019, 9, 5940. [Google Scholar] [CrossRef]
- Palazzo, L.; Thomas, B.; Jemth, A.-S.; Colby, T.; Leidecker, O.; Feijs, K.L.; Zaja, R.; Loseva, O.; Puigvert, J.C.; Matic, I.; et al. Processing of protein ADP-ribosylation by Nudix hydrolases. Biochem. J. 2015, 468, 293–301. [Google Scholar] [CrossRef]
- Daniels, C.M.; Ong, S.-E.; Leung, A.K.L. ADP-Ribosylated peptide enrichment and site identification: The phosphodiesterase-based method. Methods Mol. Biol. 2017, 1608, 79–93. [Google Scholar] [CrossRef]
- Munnur, D.; Bartlett, E.; Mikolčević, P.; Kirby, I.T.; Rack, J.; Mikoč, A.; Cohen, M.S.; Ahel, I. Reversible ADP-ribosylation of RNA. Nucleic Acids Res. 2019, 47, 5658–5669. [Google Scholar] [CrossRef]
- Zhang, F.; Lou, L.; Peng, B.; Song, X.; Reizes, O.; Almasan, A.; Gong, Z. Nudix hydrolase NUDT16 regulates 53BP1 protein by reversing 53BP1 ADP-ribosylation. Cancer Res. 2020, 80, 999–1010. [Google Scholar] [CrossRef]
- Carreras-Puigvert, J.; Zitnik, M.; Jemth, A.-S.; Carter, M.; Unterlass, J.E.; Hallström, B.; Loseva, O.; Karem, Z.; Montaño, J.M.C.; Lindskog, C.; et al. A comprehensive structural, biochemical and biological profiling of the human NUDIX hydrolase family. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Scarsdale, J.N.; Peculis, B.A.; Wright, H.T. Crystal structures of U8 snoRNA decapping nudix hydrolase, X29, and Its metal and cap complexes. Structure 2006, 14, 331–343. [Google Scholar] [CrossRef]
- Jankowski, J.; Seibt, B.; Henning, L.; Zidek, W.; Schlüter, H.; Jankowski, V. Identification of dinucleoside polyphosphates in adrenal glands. Biochem. Biophys. Res. Commun. 2003, 304, 365–370. [Google Scholar] [CrossRef]
- Jankowski, J.; Hagemann, J.; Tepel, M.; van der Giet, M.; Stephan, N.; Henning, L.; Gouni-Berthold, H.; Sachinidis, A.; Zidek, W.; Schlüter, H. Dinucleotides as growth-promoting extracellular mediators. Presence of dinucleoside diphosphates Ap2A, Ap2G, and Gp2G in releasable granules of platelet. J. Biol. Chem. 2001, 276, 8904–8909. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, V.; van der Giet, M.; Mischak, H.; Morgan, M.; Zidek, W.; Jankowski, J. Dinucleoside polyphosphates: Strong endogenous agonists of the purinergic system. Br. J. Pharmacol. 2009, 157, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Di Liberto, V.; Mudò, G.; Garozzo, R.; Frinchi, M.; Fernandez-Dueñas, V.; Di Iorio, P.; Ciccarelli, R.; Caciagli, F.; Condorelli, D.F.; Ciruela, F.; et al. The guanine-based purinergic system: The tale of an orphan neuromodulation. Front. Pharmacol. 2016, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Frick, D.N. Fluorescent probe displacement assays reveal unique nucleic acid binding properties of human nudix enzymes. Anal. Biochem. 2020, 595, 113622. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Gao, K.; Oerlemans, R.; Groves, M.R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12, 85–104. [Google Scholar] [CrossRef]
- Cimmperman, P.; Baranauskienė, L.; Jachimovičiūtė, S.; Jachno, J.; Torresan, J.; Michailovienė, V.; Matulienė, J.; Sereikaitė, J.; Bumelis, V.; Matulis, D. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 2008, 95, 3222–3231. [Google Scholar] [CrossRef]
- Winiewska, M.; Bugajska, E.M.N.; Poznański, J. ITC-derived binding affinity may be biased due to titrant (nano)-aggregation. Binding of halogenated benzotriazoles to the catalytic domain of human protein kinase CK. PLoS ONE 2017, 12, e0173260. [Google Scholar] [CrossRef]
- Svergun, D. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef]
- Michel, M.; Homan, E.J.; Wiita, E.; Pedersen, K.; Almlöf, I.; Gustavsson, A.-L.; Lundbäck, T.; Helleday, T.; Berglund, U.W. In silico druggability assessment of the NUDIX Hydrolase protein family as a workflow for target prioritization. Front. Chem. 2020, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Wojtczak, B.A.; Warminski, M.; Kowalska, J.; Lukaszewicz, M.; Honcharenko, M.; Smith, C.I.E.; Strömberg, R.; Darzynkiewicz, E.; Jemielity, J. Clickable trimethylguanosine cap analogs modified within the triphosphate bridge: Synthesis, conjugation to RNA and susceptibility to degradation. RSC Adv. 2016, 6, 8317–8328. [Google Scholar] [CrossRef]
- Steogon;pinski, J.; Bretner, M.; Jankowska, M.; Felczak, K.; Stolarski, R.; Wieczorek, Z.; Caipostalcode, A.-L.; Rhoads, R.E.; Temeriusz, A.; Haber, D.; et al. Synthesis and properties of P1, P2-, P1, P3- and P1, P4-dinucleoside Di-, Tri- and tetraphosphate mRNA 5’-cap analogues. Nucleosides Nucleotides Nucleic Acids 1995, 14, 717–721. [Google Scholar] [CrossRef]
- Stepinski, J.; Jemielity, J.; Lewdorowicz, M.; Jankowska-Anyszka, M.; Darzynkiewicz, E. Catalytic efficiency of divalent metal salts in dinucleoside 5′,5′-triphosphate bond formation. Collect. Czechoslov. Chem. Commun. 2002, 5, 154–158. [Google Scholar] [CrossRef]
- Niedzwiecka, A.; Stepinski, J.; Antosiewicz, J.M.; Darzynkiewicz, E.; Stolarski, R. Biophysical approach to studies of cap–eIF4E interaction by synthetic cap analogs. In Methods in Enzymology; Lorsch, J., Ed.; Translation Initiation: Reconstituted Systems and Biophysical Methods; Academic Press: Cambridge, UK, 2007; Volume 430, pp. 209–245. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and differentiation of data by simplified least squares procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Vivó-Truyols, G.; Schoenmakers, P. Automatic selection of optimal Savitzky-Golay smoothing. Anal. Chem. 2006, 78, 4598–4608. [Google Scholar] [CrossRef] [PubMed]
- Girardot, R.; Viguier, G.; Pérez, J.; Ounsy, M. FOXTROT: A JAVA-based application to reduce and analyse SAXS and WAXS piles of 2D data at synchrotron SOLEIL. In Proceedings of the Synchrotron Soleil, canSAS-VIII, Tokai, Japan, 14–16 April 2015. [Google Scholar]
- Konarev, P.; Volkov, V.V.; Sokolova, A.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Semenyuk, A.V.; Svergun, D.I. GNOM—A program package for small-angle scattering data processing. J. Appl. Crystallogr. 1991, 24, 537–540. [Google Scholar] [CrossRef]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Czapinska, H.; Winiewska-Szajewska, M.; Szymaniec-Rutkowska, A.; Piasecka, A.; Bochtler, M.; Poznański, J. Halogen atoms in the protein-ligand system. structural and thermodynamic studies of the binding of bromobenzotriazoles by the catalytic subunit of human protein kinase CK2. J. Phys. Chem. B 2021, 125, 2491–2503. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dinucleotide | Hydrolysis Products | Hydrolysis Rate | |
|---|---|---|---|
| (µM/min/µg) | (µM/min/[E]0) | ||
| GppG | GMP | 227.27 ± 18.25 | 5454.5 ± 436.3 |
| m7GppG | m7GMP + GMP | 30.11 ± 2.81 | 722.6 ± 68.6 |
| AppA | AMP | 1.70 ± 0.12 | 40.8 ± 2.9 |
| dpCoA | AMP + dpCop | 0.75 ± 0.05 | 18.0 ± 1.3 |
| NADH | AMP + NMNH | 0.78 ± 0.06 | 18.7 ± 1.5 |
| NAD | NMN + AMP | 0.14 ± 0.02 | 3.4 ± 0.4 |
| ADPr | nd | nd | |
| m7GDP | nd | nd | |
| nanoDSF (Dye-Free) | DSF (with SYPRO Orange) | |||
|---|---|---|---|---|
| Ligand | Tm (°C) | ∆Tm (°C) | Tm (°C) | ∆Tm (°C) |
| apo | 65.1 ± 0.1 | - | 63.8 ± 0.1 | - |
| GppG | 83.1 ± 0.1 | 18.0 ± 0.1 | 81.2 ± 0.1 | 17.5 ± 0.1 |
| IDP | 82.9 ± 0.1 | 17.8 ± 0.1 | 81.5 ± 0.1 | 17.8 ± 0.1 |
| m7GppG | 80.2 ± 0.1 | 15.2 ± 0.1 | 78.2 ± 0.1 | 14.4 ± 0.1 |
| GDP | 78.6 ± 0.1 | 13.5 ± 0.1 | 77.0 ± 0.1 | 13.3 ± 0.1 |
| GpppG | 76.0 ± 0.1 | 10.9 ± 0.1 | 74.8 ± 0.1 | 11.0 ± 0.1 |
| IMP | 75.6 ± 0.1 | 10.5 ± 0.1 | 74.1 ± 0.1 | 10.4 ± 0.1 |
| m7GpppG | 73.5 ± 0.1 | 8.4 ± 0.1 | 72.3 ± 0.1 | 8.5 ± 0.1 |
| GMP | 69.1 ± 0.1 | 4.0 ± 0.1 | 68.0 ± 0.1 | 4.3 ± 0.1 |
| dpCoA | 68.1 ± 0.1 | 3.1 ± 0.1 | 67.0 ± 0.1 | 3.3 ± 0.1 |
| NADH | 67.4 ± 0.1 | 2.4 ± 0.1 | 64.9 ± 0.1 | 1.1 ± 0.1 |
| AppA | 66.8 ± 0.1 | 1.7 ± 0.1 | 65.2 ± 0.1 | 1.5 ± 0.1 |
| CDP | 66.8 ± 0.1 | 1.66 ± 0.1 | 65.6 ± 0.1 | 1.81 ± 0.1 |
| m7GDP | 66.7 ± 0.1 | 1.6 ± 0.1 | 64.4 ± 0.1 | 0.7 ± 0.1 |
| NAD | 66.1 ± 0.1 | 1.1 ± 0.1 | 63.7 ± 0.1 | 0.0 ± 0.1 |
| ADPr | 66.0 ± 0.1 | 0.9 ± 0.1 | 63.6 ± 0.1 | −0.1 ± 0.1 |
| Ligand | Kd1 (nM) | Kd2 (µM) | ΔGdiss1 (kJ/mol) (for Kd1) | ΔGdiss2 (kJ/mol) (for Kd2) |
|---|---|---|---|---|
| IDP | 42 ± 11 | 1.2 ± 0.2 | 42.00 | 33.80 |
| GppG | 73 ± 23 | 1.6 ± 0.4 | 40.62 | 33.01 |
| m7GppG | 220 ± 87 | 30.2 ± 2.5 | 37.92 | 25.75 |
| GDP | 258 ± 45 | 45.6 ± 2.4 | 37.53 | 24.73 |
| m7GpppG | (1.7 ± 0.6) × 103 | 230 ± 57 | 33.58 | 19.32 |
| IMP | (4.9 ± 1.8) × 103 | 41 ± 10 | 30.24 | 24.98 |
| dpCoA | (43 ± 11) × 103 | ≥15,019 | 24.90 | ≤10.38 |
| AppA | (833 ± 164) × 103 | nd | 17.54 | nd |
| ADPr | (1826 ± 497) × 103 | ≥19,900 | 15.60 | ≤9.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrabąszczewska, M.; Winiewska-Szajewska, M.; Ostrowska, N.; Bojarska, E.; Stępiński, J.; Mancewicz, Ł.; Łukaszewicz, M.; Trylska, J.; Taube, M.; Kozak, M.; et al. Insight into the Binding and Hydrolytic Preferences of hNudt16 Based on Nucleotide Diphosphate Substrates. Int. J. Mol. Sci. 2021, 22, 10929. https://doi.org/10.3390/ijms222010929
Chrabąszczewska M, Winiewska-Szajewska M, Ostrowska N, Bojarska E, Stępiński J, Mancewicz Ł, Łukaszewicz M, Trylska J, Taube M, Kozak M, et al. Insight into the Binding and Hydrolytic Preferences of hNudt16 Based on Nucleotide Diphosphate Substrates. International Journal of Molecular Sciences. 2021; 22(20):10929. https://doi.org/10.3390/ijms222010929
Chicago/Turabian StyleChrabąszczewska, Magdalena, Maria Winiewska-Szajewska, Natalia Ostrowska, Elżbieta Bojarska, Janusz Stępiński, Łukasz Mancewicz, Maciej Łukaszewicz, Joanna Trylska, Michał Taube, Maciej Kozak, and et al. 2021. "Insight into the Binding and Hydrolytic Preferences of hNudt16 Based on Nucleotide Diphosphate Substrates" International Journal of Molecular Sciences 22, no. 20: 10929. https://doi.org/10.3390/ijms222010929
APA StyleChrabąszczewska, M., Winiewska-Szajewska, M., Ostrowska, N., Bojarska, E., Stępiński, J., Mancewicz, Ł., Łukaszewicz, M., Trylska, J., Taube, M., Kozak, M., Darżynkiewicz, E., & Grzela, R. (2021). Insight into the Binding and Hydrolytic Preferences of hNudt16 Based on Nucleotide Diphosphate Substrates. International Journal of Molecular Sciences, 22(20), 10929. https://doi.org/10.3390/ijms222010929