
Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis


Abstract
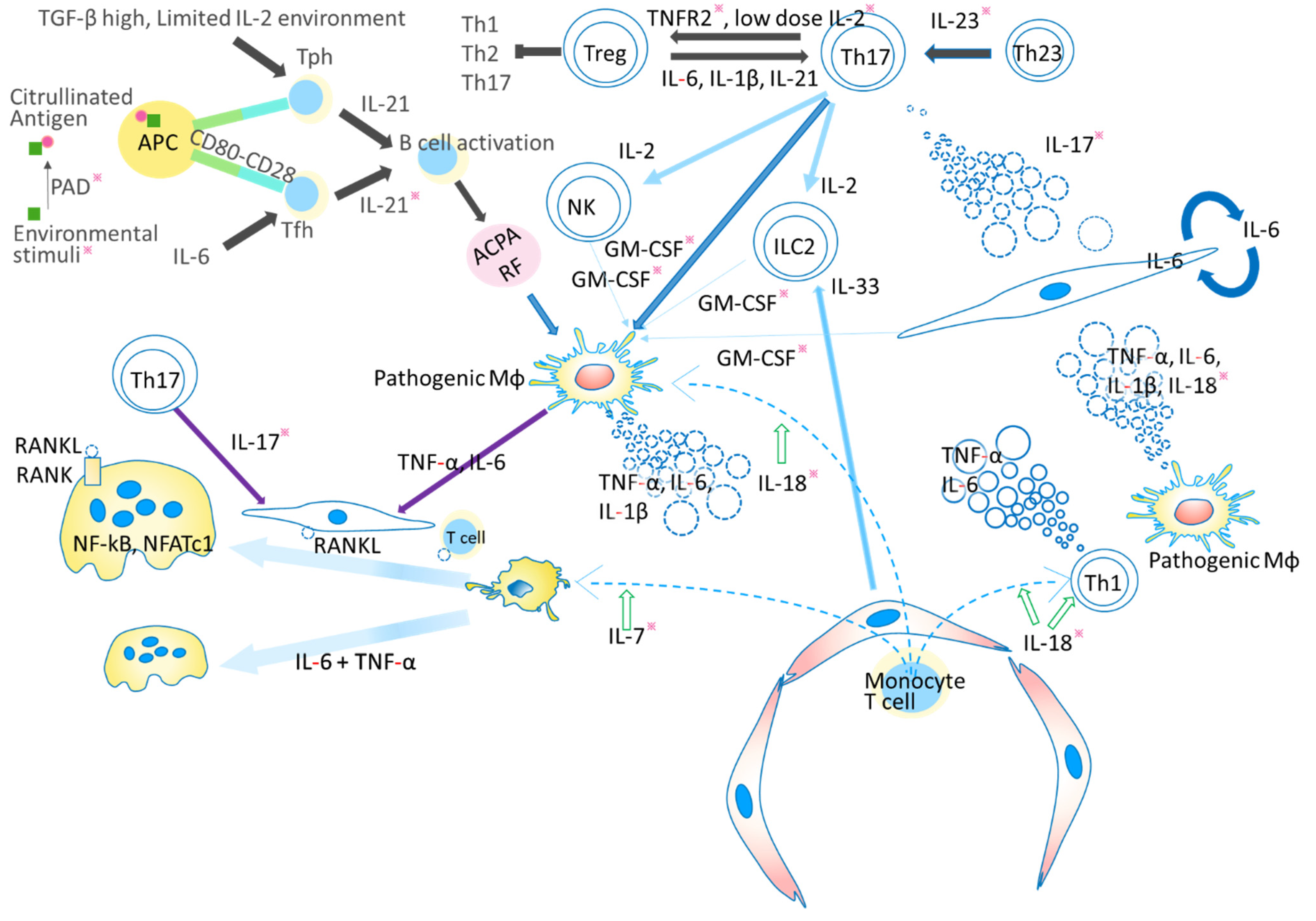
1. Introduction
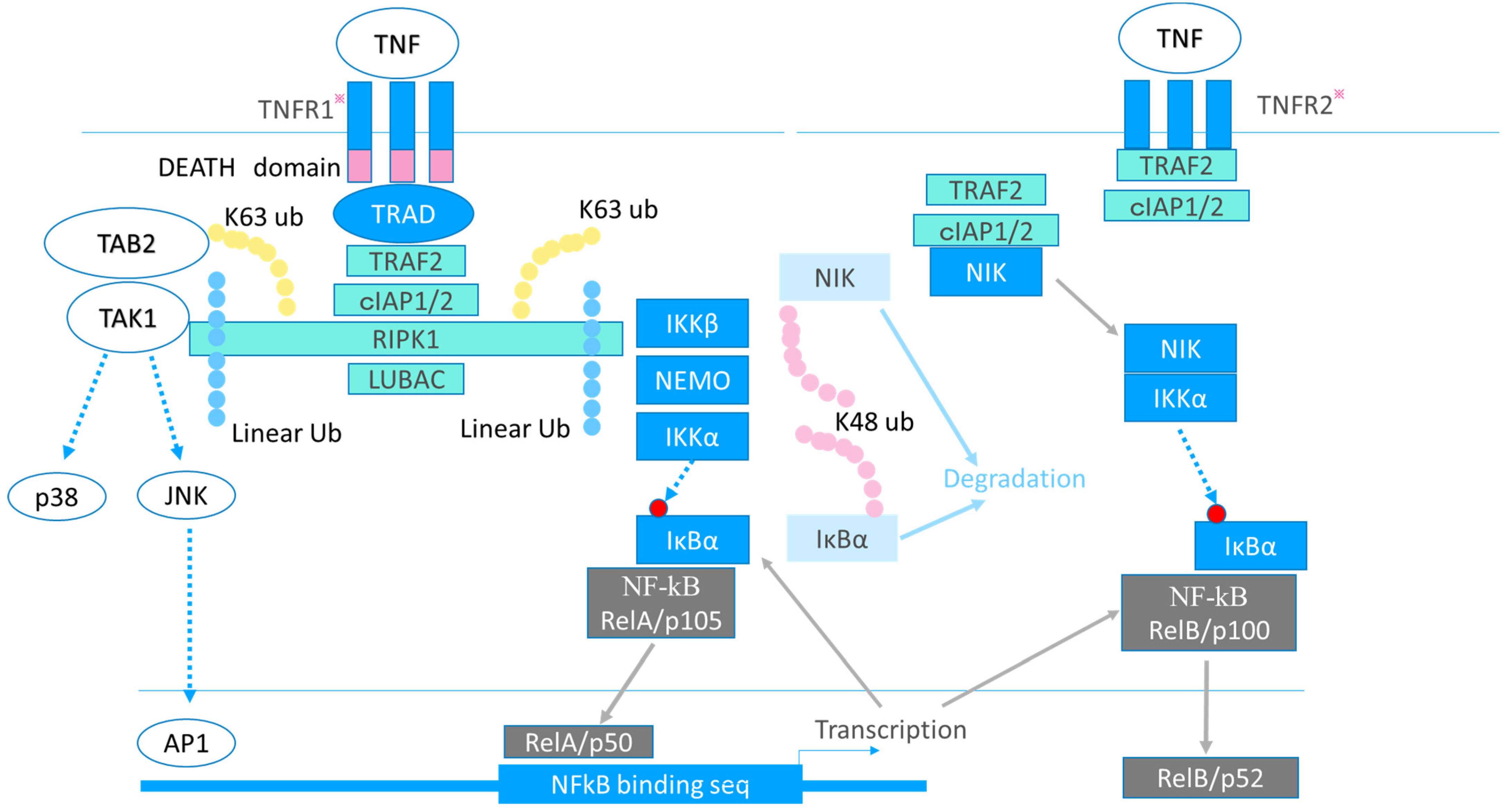
2. TNF and RA
2.1. ACPA Stimulate Macrophages to Produce TNF
2.2. Multidirectional Function of TNF in RA Pathogenesis
2.3. TNFR1 and TNFR2
2.4. TNF in Epigenetics
2.5. Evaluation of Anti-TNF Agents and Challenges of the Future
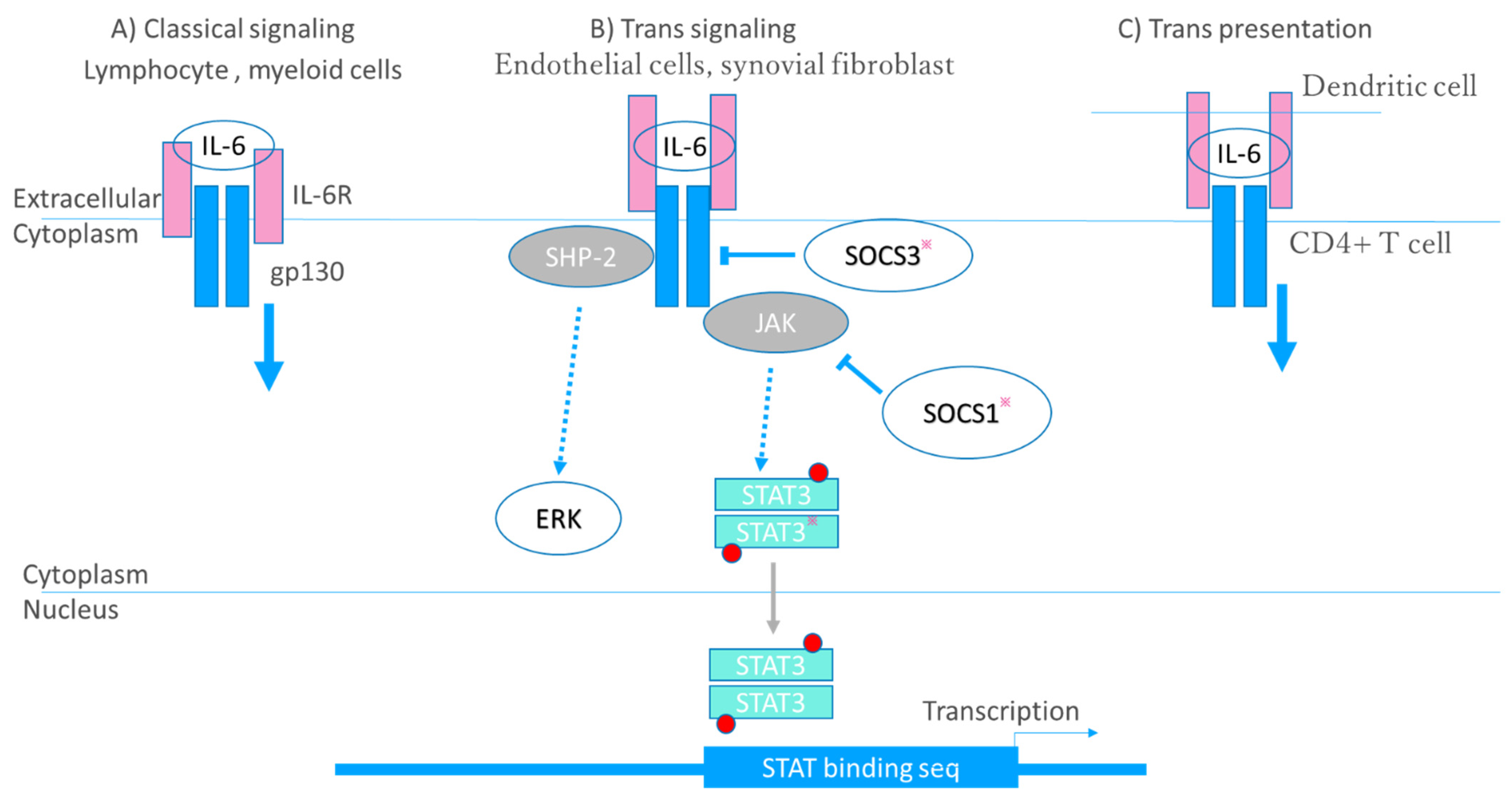
3. IL-6 and RA
3.1. Coordinated Interaction of TNF, IL-17, and IL-6 in RA Pathogenesis
3.2. Receptor–Ligand Interaction
3.3. Multidirectional Function of IL-6 in RA Pathogenesis
3.4. Evaluation of Anti-IL-6 Agents
3.5. RA-Related Comorbidities and Cytokine-Targeted Therapies
4. IL-23/IL-17 and RANKL Elicit Bone Resorption by Driving the Function of Osteoclasts
4.1. IL-23 and IL-17
4.2. RANKL Regulation by Cytokines
4.3. Evaluation of Anti-IL-17 Agents
4.4. Clinical Evaluation of Anti-IL-23 Agents
4.5. IL-7 and IL-21
5. bDMARDs, JAK/STAT Inhibitors and Potential Molecular Targets for Treatment of Bone Resorption in RA
5.1. bDMARDs and Bone Metabolism
5.2. JAK/STAT Inhibitors and Bone Metabolism
5.3. Reactive Oxygen Species and Bone Metabolism
6. GM-CSF and the Pathogenesis of RA
7. IL-1β and the Pathogenesis of RA
8. IL-18 and the Pathogenesis of RA
9. IL-33 and the Pathogenesis of RA
10. IL-2 and RA
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Kuroda, T.; Tanabe, N.; Kobayashi, D.; Sato, H.; Wada, Y.; Murakami, S.; Saeki, T.; Nakano, M.; Narita, I. Treatment with biologic agents improves the prognosis of patients with rheumatoid arthritis and amyloidosis. J. Rheumatol. 2012, 39, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Malmström, V.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Smoking, citrullination and genetic varia-bility in the immunopathogenesis of rheumatoid arthritis. Semin. Immunol. 2011, 23, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, G.A.; Visser, H.; de Jong, B.A.; van den Hoogen, F.H.; Hazes, J.M.; Breedveld, F.C.; van Venrooijet, W.J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 43, 155–163. [Google Scholar] [CrossRef]
- James, E.A.; Moustakas, A.K.; Papadopoulos, G.K.; Bondinas, G.; Buckner, J.H.; Kwok, W.K. HLA–DR1001 presents “altered-self” peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets. Arthritis Rheum. 2010, 62, 2909–2918. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor through Fc receptor IIa engagement by rheumatoid arthritis–specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. 2018, 58, 678–688. [Google Scholar] [CrossRef]
- Ciobanu, D.A.; Poenariu, I.S.; Crînguș, L.I.; Vreju, F.A.; Turcu-Stiolica, A.; Tica, A.A.; Padureanu, V.; Dumitrascu, R.M.; Banicioiu-Covei, S.; Dinescu, S.C.; et al. JAK/STAT pathway in pathology of rheumatoid arthritis. Exp. Ther. Med. 2020, 20, 498–3503. [Google Scholar] [CrossRef]
- Alam, J.; Jantan, I.; Bukhari, S.N.A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 2017, 92, 615–633. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J.D. Cytokines in rheumatoid arthritis—Shaping the immunological landscape. Nat. Rev. Rheumatol. 2016, 12, 63–68. [Google Scholar] [CrossRef]
- Thornton, S.; Boivin, G.P.; Kim, K.N.; Finkelman, F.D.; Hirsch, R. Heterogeneous effects of IL-2 on collagen-induced arthritis. J. Immunol. 2000, 165, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Iwasaki, T.; Kitano, S.; Satake, A.; Nomura, S.; Furukawa, T.; Matsui, K.; Sano, H. IL-2-Anti-IL-2 Mono-clonal antibody immune complexes inhibit collagen induced arthritis by augmenting regulatory T cell functions. J. Immunol. 2018, 201, 1899–1906. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Hong, W.; Guo, Y.; Zhang, P.; Fang, Y.; Wang, X.; Chen, X.; Lu, S.; Wei, W. Ontogeny of synovial macrophages and the roles of synovial macrophages from different origins in arthritis. Front. Immunol. 2019, 10, 1146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef]
- England, B.R.; Thiele, G.M.; Mikuls, T.R. Anticitrullinated protein antibodies: Origin and role in the pathogenesis of rheumatoid arthritis. Curr. Opin. Rheumatol. 2017, 29, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.E.; Kongpachith, S.; Lingampalli, N.; Adamska, J.Z.; Cannon, B.J.; Mao, R.; Blum, L.K.; Robinson, W.H. Affinity maturation drives epitope spreading and generation of proinflammatory anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2018, 70, 1946–1958. [Google Scholar] [CrossRef]
- Finsterbusch, M.; Voisin, M.B.; Beyrau, M.; Williams, T.J.; Nourshargh, S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J. Exp. Med. 2014, 30, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef]
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9, a031245. [Google Scholar] [CrossRef] [PubMed]
- Notley, C.A.; Inglis, J.J.; Alzabin, S.; McCann, F.E.; McNamee, K.E.; Williams, R.O. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J. Exp. Med. 2008, 205, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.; Drutskaya, M.; Schlienz, D.; Gorshkova, E.; Kurz, K.; Morawietz, L.; Nedospasov, S. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann. Rheum. Dis. 2020, 79, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Proske, M.; Duffey, M.; Stangl, H.; Martinez, G.F.; Peters, N.; Kraske, A.; Straub, R.H.; Bethea, J.R.; Kontermann, R.E.; et al. Selective activation of tumor necrosis factor receptor II induces antiinflammatory responses and alleviates experimental arthritis. Arthritis Rheumatol. 2018, 70, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Lamontain, V.; Schmid, T.; Weber-Steffens, D.; Zeller, D.; Jenei-Lanzl, Z.; Wajant, H.; Straub, R.H.; Männel, D.N. Stimulation of TNF receptor type 2 expands regulatory T cells and ameliorates established collagen-induced arthritis in mice. Cell. Mol. Immunol. 2019, 16, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Mori, L.; Iselin, S.; De Libero, G.; Lesslauer, W. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1-treated and TNFR1-deficient mice. J. Immunol. 1996, 157, 3178–3182. [Google Scholar]
- Tseng, W.Y.; Huang, Y.S.; Clanchy, F.; McNamee, K.; Perocheau, D.; Ogbechi, J.; Luo, S.F.; Feldmann, M.; McCann, F.E.; Williams, R.O. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc. Natl. Acad. Sci. USA 2019, 116, 21666–21672. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Sakaguchi, S. Foxp3⁺ T(reg) cells in humoral immunity. Int. Immunol. 2014, 26, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Beilhack, A. Targeting regulatory T cells by addressing tumor necrosis factor and its receptors in allogeneic hematopoietic cell transplantation and cancer. Front. Immunol. 2019, 10, 2040. [Google Scholar] [CrossRef]
- Santinon, F.; Batignes, M.; Mebrek, M.L.; Biton, J.; Clavel, G.; Hervé, R.; Lemeiter, D.; Breckler, M.; Busato, F.; Tost, J.; et al. Involvement of tumor necrosis factor receptor type II in FoxP3 stability and as a marker of Treg cells Specifically expanded by anti-tumor necrosis factor treatments in rheumatoid arthritis. Arthritis Rheumatol. 2020, 72, 576–587. [Google Scholar] [CrossRef]
- Nie, H.; Zheng, Y.; Li, R.; Guo, T.B.; He, D.; Fang, L.; Liu, X.; Xiao, L.; Chen, X.; Wan, B.; et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 2013, 19, 322–328. [Google Scholar] [CrossRef]
- Lubrano di Ricco, M.; Ronin, E.; Collares, D.; Divoux, J.; Grégoire, S.; Wajant, H.; Gomes, T.; Grin-berg-Bleyer, Y.; Baud, V.; Marodon, G.; et al. Tumor necrosis factor receptor family costimulation in-creases regulatory T-cell activation and function via NF-κB. Eur. J. Immunol. 2020, 50, 972–985. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Liao, Y.; Teh, P.; Pascutti, M.F.; Oja, A.E.; Garnham, A.L.; Gloury, R.; Tempany, J.C.; Sidwell, T.; Cuadrado, E.; et al. The TNF Receptor Superfamily-NF-kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep. 2017, 20, 2906–2920. [Google Scholar] [CrossRef]
- Tang, W.; Lu, Y.; Tian, Q.Y.; Zhang, Y.; Guo, F.J.; Liu, G.Y.; Syed, N.M.; Lai, Y.; Lin, E.A.; Kong, L.; et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 2011, 332, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hu, W.; Yi, Y.S.; Hettinghouse, A.; Sun, G.; Bi, Y.; He, W.; Zhang, L.; Gao, G.; Liu, J.; et al. TNFR2/14-3-3ε signaling complex instructs macrophage plasticity in inflammation and autoimmunity. J. Clin. Investig. 2021, 16, 131.e144016. [Google Scholar]
- Karouzakis, E.; Raza, K.; Kolling, C.; Buckley, C.D.; Gay, S.; Filer, A.; Ospelt, C. Analysis of early changes in DNA methylation in synovial fibroblasts of RA patients before diagnosis. Sci. Rep. 2018, 8, 7370. [Google Scholar] [CrossRef]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Boyle, D.L.; Firestein, G.S. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J. Immunol. 2013, 190, 1297–1303. [Google Scholar] [CrossRef]
- Angiolilli, C.; Grabiec, A.M.; Ferguson, B.S.; Ospelt, C.; Malvar-Fernandez, B.; van Es, I.E.; van Baarsen, L.G.; Gay, S.; McKinsey, T.A.; Tak, P.P.; et al. Inflammatory cytokines epigenetically regulate rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing HDAC5 expression. Ann. Rheum. Dis. 2016, 75, 430–438. [Google Scholar] [CrossRef]
- Loh, C.; Park, S.H.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-induced inflammatory genes escape repres-sion in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Gowhari-Shabgah, A.; Shariati-Sarabi, Z.; Tavakkol-Afshari, J.; Ghoryani, M.; Mohammadi, M. Possible anti-inflammatory effects of mesenchymal stem cells transplantation via changes in CXCL8 levels in patients with refractory rheumatoid arthritis. Int. J. Mol. Cell. Med. 2019, 8, 191–199. [Google Scholar] [PubMed]
- Lee, J.H.; Kim, B.; Jin, W.J.; Kim, H.H.; Ha, H.; Lee, Z.H. Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: Relevance for arthritis. Arthritis Res. Ther. 2017, 19, 163. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.M.; Lundell, A.C.; Andersson, K.; Nordström, I.; Theander, E.; Rudin, A. Blood chemokine profile in untreated early rheumatoid arthritis: CXCL10 as a disease activity marker. Arthritis Res. Ther. 2017, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Ota, M.; Sumitomo, S.; Ishigaki, K.; Suzuki, A.; Sakata, T.; Tsuchida, Y.; Inui, H.; Hirose, J.; Kochi, Y.; et al. Parsing multiomics landscape of activated synovial fibroblasts highlights drug targets linked to genetic risk of rheumatoid arthritis. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Kobayashi, D.; Ito, S.; Takai, C.; Hasegawa, E.; Nomura, Y.; Otani, H.; Abe, A.; Ishikawa, H.; Murasawa, A.; Narita, I.; et al. Efficacy and safety of infliximab: A comparison with other biological disease-modifying anti-rheumatic drugs. Mod. Rheumatol. 2018, 28, 599–605. [Google Scholar] [CrossRef]
- Maneiro, J.R.; Souto, A.; Gomez-Reino, J.J. Risks of malignancies related to tofacitinib and biological drugs in rheumatoid arthritis: Systematic review, meta-analysis, and network meta-analysis. Semin. Arthritis Rheum. 2017, 47, 149–156. [Google Scholar] [CrossRef]
- Singh, J.A.; Hossain, A.; Tanjong Ghogomu, E.; Kotb, A.; Christensen, R.; Mudano, A.S.; Maxwell, L.J.; Shah, N.P.; Tugwell, P.; Wells, G.A. Biologics or tofacitinib for rheumatoid arthritis in incomplete responders to methotrexate or other traditional disease-modifying anti-rheumatic drugs: A systematic re-view and network meta-analysis. Cochrane Database Syst. Rev. 2016, 5, CD012183. [Google Scholar] [CrossRef]
- Singh, J.A.; Hossain, A.; Tanjong Ghogomu, E.; Mudano, A.S.; Tugwell, P.; Wells, G.A. Biologic or to-facitinib monotherapy for rheumatoid arthritis in people with traditional disease-modifying an-ti-rheumatic drug (DMARD) failure: A Cochrane Systematic Review and network meta-analysis (NMA). Cochrane Database Syst. Rev. 2016, 11, CD012437. [Google Scholar]
- Singh, J.A.; Hossain, A.; Tanjong Ghogomu, E.; Mudano, A.S.; Maxwell, L.J.; Buchbinder, R.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E.; Tugwell, P.; Wells, G.A. Biologics or tofacitinib for people with rheumatoid arthritis unsuccessfully treated with biologics: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2017, 3, CD012591. [Google Scholar] [CrossRef]
- Singh, J.A.; Hossain, A.; Mudano, A.S.; Tanjong Ghogomu, E.; Suarez-Almazor, M.E.; Buchbinder, R.; Maxwell, L.J.; Tugwell, P.; Wells, G.A. Biologics or tofacitinib for people with rheumatoid arthritis naive to methotrexate: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2017, 5, CD012657. [Google Scholar] [CrossRef] [PubMed]
- Janke, K.; Biester, K.; Krause, D.; Richter, B.; Schürmann, C.; Hirsch, K.; Hörn, H.; Kerekes, M.F.; Kohlepp, P.; Wieseler, B. Comparative effectiveness of biological medicines in rheumatoid arthritis: Systematic review and network meta-analysis including aggregate results from reanalysed individual patient data. BMJ 2020, 370, m2288. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; De Simone, C.; Genovese, G.; Berti, E.; Cugno, M.; Marzano, A.V. Paradoxical skin reactions to biologics in patients with rheumatologic disorders. Front. Pharmacol. 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.; Stoop, A.A.; Williams, R.O. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Lee, A.; Qiao, Y.; Grigoriev, G.; Chen, J.; Park-Min, K.H.; Park, S.H.; Ivashkiv, L.B.; Kalliolias, G.D. Tumor necrosis fac-tor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013, 65, 928–938. [Google Scholar] [CrossRef]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef]
- Slowikowski, K.; Nguyen, H.N.; Noss, E.H.; Simmons, D.P.; Mizoguchi, F.; Watts, G.F.M.; Gurish, M.F.; Brenner, M.B.; Raychaudhuri, S. CUX1 and IkappaBzeta (NFKBIZ) mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts. Proc. Natl. Acad. Sci. USA 2020, 117, 5532–5541. [Google Scholar] [CrossRef]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef]
- Annemann, M.; Plaza-Sirvent, C.; Schuster, M.; Katsoulis-Dimitriou, K.; Kliche, S.; Schraven, B.; Schmitz, I. Atypical IkappaB proteins in immune cell differentiation and function. Immunol. Lett. 2016, 171, 26–35. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Chen, Z.; Larjo, A.; Kanduri, K.; Nousiainen, K.; Äijo, T.; Ricaño-Ponce, I.; Hrdlickova, B.; Tuomela, S.; Laajala, E.; et al. Genome-wide analysis of STAT3-mediated transcription during early human Th17 cell differentiation. Cell Rep. 2017, 19, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell biology: A decade of discovery and diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Eto, D.; Yang, J.A.; Lao, C.; Crotty, S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J. Immunol. 2013, 190, 3049–3053. [Google Scholar] [CrossRef]
- Schmitt, N.; Liu, Y.; Bentebibel, S.E.; Munagala, I.; Bourdery, L.; Venuprasad, K.; Banchereau, J.; Ueno, H. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat. Immunol. 2014, 15, 856–865. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H.; Song, J.; Sugimoto, M.; Hagihara, K.; Kishimoto, T.; Yoshizaki, K.; Nishimoto, N. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1521–1529. [Google Scholar] [CrossRef]
- Sun, A.; Benet, L.Z. Late-stage failures of monoclonal antibody drugs: A retrospective case study analysis. Pharmacology 2020, 105, 145–163. [Google Scholar] [CrossRef]
- Haugeberg, G.; Uhlig, T.; Falch, J.A.; Halse, J.I.; Kvien, T.K. Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: Results from 394 patients in the Oslo County Rheumatoid Arthritis register. Arthritis Rheum. 2000, 43, 522–530. [Google Scholar] [CrossRef]
- Luque Ramos, A.; Redeker, I.; Hoffmann, F.; Callhoff, J.; Zink, A.; Albrecht, K. Comorbidities in patients with rheumatoid arthritis and their association with patient-reported outcomes: Results of claims data linked to questionnaire survey. J. Rheumatol. 2019, 46, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Wijbrandts, C.A.; Klaasen, R.; Dijkgraaf, M.G.; Gerlag, D.M.; van Eck-Smit, B.L.; Tak, P.P. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: Arrest of bone loss. Ann. Rheum. Dis. 2009, 68, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Hoff, M.; Kvien, T.K.; Kälvesten, J.; Elden, A.; Kavanaugh, A.; Haugeberg, G. Adalimumab reduces hand bone loss in rheumatoid arthritis independent of clinical response: Subanalysis of the PREMIER study. BMC Musculoskelet. Disord. 2011, 12, 54. [Google Scholar] [CrossRef]
- Haugeberg, G.; Helgetveit, K.B.; Førre, Ø.; Garen, T.; Sommerseth, H.; Prøven, A. Generalized bone loss in early rheumatoid arthritis patients followed for ten years in the biologic treatment era. BMC Musculoskelet. Disord. 2014, 15, 289. [Google Scholar] [CrossRef]
- Zerbini, C.A.F.; Clark, P.; Mendez-Sanchez, L.; Pereira, R.M.R.; Messina, O.D.; Uña, C.R.; Adachi, J.D.; Lems, W.F.; Cooper, C.; Lane, N.E.; et al. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos. Int. 2017, 28, 429–446. [Google Scholar] [CrossRef]
- Kume, K.; Amano, K.; Yamada, S.; Kanazawa, T.; Ohta, H.; Hatta, K.; Amano, K.; Kuwaba, N. The effect of tocilizumab on bone mineral density in patients with methotrexate-resistant active rheumatoid arthritis. Rheumatology 2014, 53, 900–903. [Google Scholar] [CrossRef]
- Chen, Y.M.; Chen, H.H.; Huang, W.N.; Liao, T.L.; Chen, J.P.; Chao, W.C.; Lin, C.T.; Hung, W.T.; Hsieh, C.W.; Hsieh, T.Y.; et al. Tocilizumab potentially prevents bone loss in patients with anticitrullinated protein antibody-positive rheumatoid arthritis. PLoS ONE 2017, 12, e0188454. [Google Scholar] [CrossRef]
- Gabay, C.; Burmester, G.R.; Strand, V.; Msihid, J.; Zilberstein, M.; Kimura, T.; van Hoogstraten, H.; Boklage, S.H.; Sadeh, J.; Graham, N.M.H.; et al. Sarilumab and adalimumab differential effects on bone remodeling and cardiovascular risk biomarkers, and predictions of treatment outcomes. Arthritis Res. Ther. 2020, 22, 70. [Google Scholar] [CrossRef]
- Santos-Moreno, P.; Burgos-Angulo, G.; Martinez-Ceballos, M.A.; Pizano, A.; Echeverri, D.; Bautista-Niño, P.K.; Roks, A.J.M.; Rojas-Villarraga, A. Inflammaging as a link between autoimmunity and cardiovascular disease: The case of rheumatoid arthritis. RMD Open 2021, 7, e001470. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Gravos, A.; Georgiopoulos, G.; Terentes-Printzios, D.; Ioakeimidis, N.; Vassilopoulos, D.; Stamatelopoulos, K.; Tousoulis, D. The effect of TNF-a antagonists on aortic stiffness and wave reflections: A meta-analysis. Clin. Rheumatol. 2018, 37, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Fumery, M.; Singh, A.G.; Singh, N.; Prokop, L.J.; Dulai, P.S.; Sandborn, W.J.; Curtis, J.R. Comparative risk of cardiovascular events with biologic and synthetic disease-modifying antirheumatic drugs in patients with rheumatoid arthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2020, 72, 541–576. [Google Scholar] [CrossRef] [PubMed]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; Barbera, L.L.; Ciccia, F.; Guggino, G. Role of IL-23/IL-17 pathway in rheumatic diseases: An overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef]
- Tang, C.; Chen, S.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Bunte, K.; Beikler, T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Teng, W.M.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector functions of CD4+ T cells at the site of local autoimmune inflammation-lessons from rheumatoid arthritis. Front. Immnol. 2019, 10, 353. [Google Scholar] [CrossRef]
- Paulissen, S.M.; van Hamburg, J.P.; Dankers, W.; Lubberts, E. The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine 2015, 74, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Penatti, A.; Facciotti, F.; De Matteis, R.; Larghi, P.; Paroni, M.; Murgo, A.; De Lucia, O.; Pagani, M.; Pierannunzii, L.; Truzzi, M.; et al. Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 103. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wu, L.; Li, X. IL-17 family: Cytokines, receptors and signaling. Cytokine 2013, 64, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Vandooren, B.; Cantaert, T.; Noordenbos, T.; Tak, P.P.; Baeten, D. The abundant synovial expression of the RANK/RANKL/Osteoprotegerin system in peripheral spondylarthritis is partially disconnected from inflammation. Arthritis Rheum. 2008, 58, 718–729. [Google Scholar] [CrossRef]
- Aureal, M.; Machuca-Gayet, I.; Coury, F. Rheumatoid arthritis in the view of osteoimmunology. Biomolecules 2020, 11, 48. [Google Scholar] [CrossRef]
- Takayanagi, H.; Iizuka, H.; Juji, T.; Nakagawa, T.; Yamamoto, A.; Miyazaki, T.; Koshihara, Y.; Oda, H.; Nakamura, K.; Tanaka, S. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 259–269. [Google Scholar] [CrossRef]
- Hashizume, M.; Hayakawa, H.; Mihara, M. IL-6 trans-signaling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology 2008, 47, 1635–1640. [Google Scholar] [CrossRef]
- Ganesan, R.; Rasool, M. Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol. Immunol. 2017, 91, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ke, D.; Fu, X.; Xue, Y.; Wu, H.; Zhang, Y.; Chen, X.; Hou, J. IL-17A regulates the autophagy activity of osteoclast precursors through RANKL-JNK1signaling during osteoclastogenesis in vitro. Biochem. Biophys. Res. Commun. 2018, 497, 890–896. [Google Scholar] [CrossRef]
- O’Brien, W.; Fissel, B.M.; Maeda, Y.; Yan, J.; Ge, X.; Gravallese, E.M.; Aliprantis, A.O.; Charles, J.F. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 2016, 68, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Li, J.; Xing, L.; Yao, Z. Bone remodeling and the role of TRAF3 in osteoclastic bone resorption. Front. Immunol. 2018, 9, 2263. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Sato, K.; Miyazaki, T.; Kitaura, H.; Kayama, H.; Miyoshi, F.; Araki, Y.; Akiyama, Y.; Takeda, K.; Mimura, T. Combination of tumor necrosis factor alpha and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis Rheumatol. 2014, 66, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Sato, K.; Miyazaki, T.; Aizaki, Y.; Tanaka, S.; Sekikawa, M.; Kozu, N.; Kadono, Y.; Oda, H.; Mimura, T. Characterization and function of tumor necrosis factor and interleukin-6-induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol. 2021, 73, 1145–1154. [Google Scholar] [CrossRef]
- Blauvelt, A.; Chiricozzi, A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef]
- Genovese, M.C.; Van den Bocsh, F.; Roberson, S.A.; Bojin, S.; Biagini, I.M.; Ryan, P.; Sloan-Lancaster, J. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010, 62, 929–939. [Google Scholar] [CrossRef]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Mazurov, V.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013, 72, 863–869. [Google Scholar] [CrossRef]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; Ikawa, T.; et al. One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: Phase II, dose-finding, doubleblind, randomized, placebo-controlled study. J. Rheumatol. 2014, 41, 414421. [Google Scholar] [CrossRef]
- Martin, D.A.; Churchill, M.; Flores-Suarez, L.F.; Cardiel, M.H.; Wallace, D.; Martin, R.; Phillips, K.; Kaine, J.L.; Dong, H.; Salinger, D.; et al. A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R164. [Google Scholar] [CrossRef]
- Genovese, M.C.; Greenwald, M.; Cho, C.-S.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.Y.; et al. A Phase II randomized study of subcutaneous ixekizumab, an anti–interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis fac-tor inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef]
- Kunwar, S.; Dahal, K.; Sharma, S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: A systematic literature review and meta-analysis of randomized controlled trials. Rheumatol. Int. 2016, 36, 1065–1075. [Google Scholar] [CrossRef]
- Wu, D.; Hou, S.Y.; Zhao, S.; Hou, L.X.; Jiao, T.; Xu, N.N.; Zhang, N. Meta-analysis of IL-17 inhibitors in two populations of rheumatoid arthritis patients: Biologic-naïve or tumor necrosis factor inhibitors inadequate responders. Clin. Rheumatol. 2019, 38, 2747–2756. [Google Scholar] [CrossRef]
- Huang, Y.; Fan, Y.; Liu, Y.; Xie, W.; Zhang, Z. Efficacy and safety of secukinumab in active rheumaqtoid arthritis with an inadequate response to tumor necrosis factor inhibitors: A meta-analysis of Phase III randomized controlled trials. Clin. Rheumatol. 2019, 38, 2765–2776. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.; Taylor, P.C.; McInnes, I.B.; Schett, G.; Landewé, R.; Baeten, D.; Ionescu, L.; Strimenopoulou, F.; Watling, M.I.L.; Shaw, S. Efficacy and safety of bimekizumab as add-on therapy for rheumatoid arthritis in patients with inadequate response to certolizumab pegol: A proof-of-concept study. Ann. Rheum. Dis. 2019, 78, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H.; Kotake, S. IL-23 and Th17 disease in inflamma-tory arthritis. J. Clin. Med. 2017, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef]
- Bikker, A.; Hack, C.E.; Lafeber, F.P.J.G.; van Roon, J.A.G. Interleukin-7: A key mediator in T-cell driven autoimmunity, inflammation, and tissue destruction. Curr. Pharm. Des. 2012, 18, 2347–2356. [Google Scholar] [CrossRef]
- Burska, A.N.; Neilan, J.; Chisman, R.E.; Pitaksalee, R.; Hodgett, R.; Marzo-Ortega, H.; Conaghan, P.G.; West, R.; Emery, P.; Ponchel, F. Serum IL-7 as diagnostic biomarker for rheumatoid arthritis, validation with EULAR 2010 classification criteria. Clin. Exp. Rheumatol. 2018, 36, 115–120. [Google Scholar]
- Leung, S.; Liu, X.; Fang, L.; Chen, X.; Guo, T.; Zhang, J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell. Mol. Immunol. 2010, 7, 182–189. [Google Scholar] [CrossRef]
- Long, D.; Chen, Y.; Wu, H.; Zhao, M.; Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun. 2019, 99, 1–14. [Google Scholar] [CrossRef]
- Dinesh, P.; Rasool, M.J. Multifaceted role of IL-21 in rheumatoid arthritis: Current understanding and future perspectives. J. Cell Physiol. 2018, 233, 3918–3928. [Google Scholar] [CrossRef]
- Hua, F.; Comer, G.M.; Stockert, L.; Jin, B.; Nowak, J.; Pleasic-Williams, S.; Wunderlich, D.; Cheng, J.; Beebe, J.S. Anti-IL21 receptor monoclonal antibody (ATR-107): Safety, pharmacokinetics, and pharmacodynamic evaluation in healthy volunteers: A phase I, first-in-human study. J. Clin. Pharmacol. 2014, 54, 14–22. [Google Scholar] [CrossRef]
- Ignatenko, S.; Skrumsager, B.K.; Mouritzen, U. Safety, PK, and PD of recombinant anti-interleukin-21 monoclonal an-tibody in a first-in-human trial. Int. J. Clin. Pharmacol. Ther. 2016, 54, 243–252. [Google Scholar] [CrossRef]
- Ellis, J.; van Maurik, A.; Fortunato, L.; Gisbert, S.; Chen, K.; Schwartz, A.; McHugh, S.; Want, A.; Franco, S.S.; Oliveira, J.; et al. Anti-IL-7 receptor α monoclonal antibody (GSK2618960) in healthy subjects—A randomized, double-blind, placebo-controlled study. Br. J. Clin. Pharmacol. 2019, 85, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; af Klint, E.; Ernestam, S.; Catrina, S.B.; Makrygiannakis, D.; Botusan, I.R.; Klareskog, L.; Ulfgren, A.K. Anti-tumor necrosis factor therapy increases synovial osteoprotegerin expression in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Vis, M.; Havaardsholm, E.A.; Haugenberg, G.; Uhlig, T.; Voskuyl, A.E.; van de Stadt, R.J.; Dijkmans, B.A.; Woolf, A.D.; Kvien, T.K.; Lems, W.F. Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann. Rheum. Dis 2006, 65, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Takita, C.; Fujikawa, Y.; Itonaga, I.; Taira, H.; Kawashima, M.; Torisu, T. Infliximab acts directly on human osteoclast precursors and enhances osteoclast formation induced by receptor activator of nuclear factor kappaB ligand in vitro. Mod. Rheumatol. 2005, 15, 97–103. [Google Scholar] [CrossRef]
- Lorenzo, J. Cytokines and bone: Osteoimmunology. Bone Regul. Osteoporos. Ther. 2020, 262, 177–230. [Google Scholar]
- Palmqvist, P.; Persson, E.; Conaway, H.; Lemer, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulates the expression of receptor activator of NF-kB ligand, osteoprotegerin, and receptor activator of NF-kB in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef] [PubMed]
- Finzel, S.; Kraus, S.; Figueiredo, C.P.; Regensburger, A.; Kocijian, R.; Rech, J.; Schett, G. Comparison of the effects of tocilizumab monotherapy and adalimumab in combination with methotrexate on bone erosion repair in rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Matsuo, S.; Takai, H.; Uchiyama, Y.; Mihara, M.; Suzuki, M. Early effects of tocilizumab on bone and bone marrow lesions in a collagen-induced arthritis monkey model. Exp. Mol. Pathol. 2008, 84, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Bozec, A.; Luo, Y.; Engdahl, C.; Figueiredo, C.; Bang, H.; Schett, G. Abatacept blocks anti-citrullinated protein antibody and rheumatoid factor mediated cytokine production in human macrophages in IDO-dependent manner. Arthritis Res. Ther. 2018, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, L.J.; Singh, J. Abatacept for rheumatoid arthritis: A Cochrane systematic review. J. Rheumatol. 2010, 37, 234–245. [Google Scholar] [CrossRef]
- Axmann, R.; Herman, S.; Zaiss, M.; Franz, S.; Polzer, K.; Zwerina, J.; Hermann, M.; Smolen, J.; Schett, G. CTLA-4 directly inhibits osteoclast formation. Ann. Rheum. Dis. 2008, 67, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Bozec, A.; Zaiss, M.M.; Kagwiria, R.; Voll, R.; Rauh, M.; Chen, Z.; Mueller-Schmucker, S.; Kroczek, R.A.; Heinzerling, L.; Moser, M.; et al. T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci. Transl. Med. 2014, 6, 235ra60. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kajiya, H.; Omata, Y.; Matsumoto, T.; Sato, Y.; Kobayashi, T.; Nakamura, S.; Kaneko, Y.; Nakamura, S.; Koyama, T.; et al. CTLA4-Ig directly inhibits osteoclastogenesis by interfering with intracellular calcium oscillations in bone marrow macrophages. J. Bone Miner. Res. 2019, 34, 1744–1752. [Google Scholar] [CrossRef]
- Sims, N.A. The JAK1/STAT3/SOCS3 axis in bone development, physiology, and pathology. Exp. Mol. Med. 2020, 52, 1185–1197. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Winkler, I.G.; Pettit, A.R.; Raggatt, L.J.; Jacobsen, R.N.; Forristal, C.E.; Barbier, V.; Nowlan, B.; Cisterne, A.; Bendall, L.J.; Sims, N.A.; et al. Hematopoietic stem cell mobilizing agents G-CSF, cyclophosphamide or AMD3100 have distinct mechanism of action on bone marrow HSC niches and bone formation. Leukemia 2012, 26, 1594–1601. [Google Scholar] [CrossRef]
- LeBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Cascão, R.; Finnilä, M.A.J.; Lopes, I.P.; da Glória, V.G.; Saarakkala, S.; Zioupos, P.; Canhão, H.; Fonseca, J.E. Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology 2018, 57, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Xu, L.L.; Jian, L.L.; Yu, R.H.; Zhao, J.X.; Sun, L.; Du, G.H.; Liu, X.Y. Stattic inhibits RANKL-mediated osteoclastogenesis by suppressing activation of STAT3 and NF-κB pathways. Int. Immunopharmacol. 2018, 58, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef]
- Wright, H.L.; Lyon, M.; Chapman, E.A.; Moots, R.J.; Edwards, S.W. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front. Immunol. 2021, 11, 584116. [Google Scholar] [CrossRef]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106, 852–859. [Google Scholar] [CrossRef]
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 2015, 33, 359–370. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, K.W.; Kim, B.M.; Lee, K.A.; Lee, S.H. N-acetyl-l-cysteine controls osteoclastogenesis through regulating Th17 differentiation and RANKL in rheumatoid arthritis. Korean J. Intern. Med. 2019, 34, 210–219. [Google Scholar] [CrossRef]
- Kanai, T.; Kondo, N.; Okada, M.; Sano, H.; Okumura, G.; Kijima, Y.; Ogose, A.; Kawashima, H.; Endo, N. The JNK pathway represents a novel target in the treatment of rheumatoid arthritis through the suppression of MMP-3. J. Orthop. Surg. Res. 2020, 15, 87. [Google Scholar] [CrossRef]
- Haworth, C.; Brennan, F.M.; Chantry, D.; Turner, M.; Maini, R.N.; Feldmann, M. Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: Regulation by tumor necrosis factor-alpha. Eur. J. Immunol. 1991, 21, 2575–2579. [Google Scholar] [CrossRef]
- Alvaro-Gracia, J.M.; Zvaifler, N.J.; Firestein, G.S. Cytokines in chronic inflammatory arthritis. IV. Granulocyte/macrophage colony-stimulating factor-mediated induction of class II MHC antigen on human monocytes: A possible role in rheumatoid arthritis. J. Exp. Med. 1989, 170, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Tanabe, N.; Sakatsume, M.; Nozawa, S.; Mutsuka, T.; Ishikawa, H.; Tohyama, C.T.; Nakazono, K.; Murasawa, A.; Nakano, M.; et al. Interleukin-2 levels are elevated in the bone marrow serum of patients with mutilans-type rheumatoid arthritis. Clin. Rheumatol. 2002, 21, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Watanabe, H.; Kondoh, G.; Tanaka, A.; Yasuda, K.; Kopf, M.; Potocnik, A.J.; Stockinger, B.; Sakaguchi, N.; Sakaguch, S. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity 2018, 48, 1220–1232.e5. [Google Scholar]
- Avci, A.B.; Feist, E.; Burmester, G.R. Targeting GM-CSF in rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. S98), 39–44. [Google Scholar]
- Mulherin, D.; Fitzgerld, O.; Breshinhan, B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hercus, T.R.; Thomas, D.; Guthridge, M.A.; Ekert, P.G.; King-Scott, J.; Parker, M.W.; Lopez, A.F. The granulocyte-macrophage colony-stimulating factor receptor: Linking its structure to cell signaling and its role in disease. Blood 2009, 114, 1289–1298. [Google Scholar] [CrossRef]
- Sato, N.; Sakamaki, K.; Terada, N.; Arai, K.; Miyajima, A. Signal transduction by the high-affinity GM-CSF receptor: Two distinct cytoplasmic regions of the common beta subunit responsible for different signaling. EMBO J. 1993, 12, 4181–4189. [Google Scholar] [CrossRef]
- Burmester, G.R.; Feist, E.; Sleeman, M.A.; Wang, B.; White, B.; Magrini, F. Mavrilimumab, a human monoclonal anti-body targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann. Rheum. Dis. 2011, 70, 1542–1549. [Google Scholar] [CrossRef]
- Burmester, G.R.; Weinblatt, M.E.; McInnes, I.B.; Porter, D.; Barbarash, O.; Vatutin, M.; Szombati, I.; Esfandiari, E.; Sleeman, M.A.; Kane, C.D.; et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Takuchi, T.; Tanaka, Y.; Tanaka, S.; Kawakami, A.; Iwasaki, M.; Katayama, K.; Rokuda, M.; Izutsu, H.; Ushijima, S.; Kaneko, Y.; et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann. Rheum. Dis. 2019, 78, 1305–1319. [Google Scholar] [CrossRef]
- Keystone, E.C.; Taylor, P.C.; Drescher, E.; Schlichting, D.E.; Beattie, S.D.; Berclaz, P.Y.; Lee, C.H.; Fidelus-Gort, R.K.; Luchi, M.E.; Rooney, T.P.; et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann. Rheum. Dis. 2015, 74, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 612–632. [Google Scholar] [CrossRef]
- Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome activation and IL-1v and IL-18 processing during infection. Trends Immunol 2011, 32, 110–116. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Wesche, H.; Korherr, C.; Kracht, M.; Falk, W.; Resch, K.; Martin, M.U. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP Kinases). J. Biol. Chem. 1997, 272, 7727–7731. [Google Scholar] [CrossRef]
- Chomarat, P.; Vannier, E.; Dechanet, J.; Rissoan, M.C.; Banchereau, J.; Dinarello, C.A.; Miossec, P. Balance of IL-1 receptor antagonist/IL-1 beta in rheumatoid synovium and its regulation by IL-4 and IL-10. J. Immunol. 1995, 154, 1432–1439. [Google Scholar]
- Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology 2004, 43 (Suppl. S3), iii2–iii9. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.L.; Imazio, M.; Cremer, P.; Brucato, A.; Abbate, A.; Fang, F.; Insalaco, A.; LeWinter, M.; Lewis, B.S.; Lin, D.; et al. A phase 3 trial of interleukin-1 trap rilonacept in recurrent pericarditis. N. Engl. J. Med. 2021, 384, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Ben-Cherit, E.; Foeldvari, I.; Amarilyo, G.; Ozdogan, H.; Vanderschueren, S.; Marzan, K.; Kahlenberg, J.M.; Dekker, E.; De Benedetti, F.; et al. Long-term efficacy and safety of canakinumab in patients with colchicine-resistant familial Mediterranean fever: Results from the randomised phase III CLUSTER trial. Ann. Rheum. Dis. 2020, 79, 1362–1369. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Schechtman, J.; Bennett, R.; Handel, M.L.; Burmester, G.R.; Tesser, J.; Modafferi, D.; Poulakos, J.; Sun, G. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multi-center, placebo-controlled trial. Arthritis Rheum. 2003, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Bingham, S.J.; Seto, Y.; McGonagle, D.; Bejarano, V.; White, J.; Emery, P. Lack of response to anakinra in rheumatoid ar-thritis following failure of tumor necrosis factor blockade. Arthritis Rheum. 2004, 50, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, K.; Ushio, S.; Okura, T.; Kobayasi, S.; Taniai, M.; Kunikata, T.; Murakami, T.; Sanou, O.; Kojima, H.; Fujii, M.; et al. Purification and characterization of the human in-terleukin-18 receptor. J. Biol. Chem. 1997, 72, 25737–25742. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Radstake, T.R.; Lubberts, E.; van den Bersselaar, L.A.; van Riel, P.L.; van Lent, P.L.E.M.; Barrera, P.; van den Berg, W.B. Association of interleukin-18 expression with enhanced levels of both interleukin-1beta and tumor necrosis factor alpha in knee synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2003, 48, 339–347. [Google Scholar] [CrossRef]
- Dai, S.-M.; Nishioka, K.; Yudoh, K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: Comparison with IL1β and tumour necrosis factor α. Ann. Rheum. Dis. 2004, 63, 1379–1386. [Google Scholar] [CrossRef]
- Krumm, B.; Meng, X.; Xiang, Y.; Deng, J. Identification of small molecule inhibitors of Interleukin-18. Sci. Rep. 2017, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Bacchi, M.; Bertolino, M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273. [Google Scholar] [CrossRef] [PubMed]
- Rex, D.A.B.; Agarwal, N.; Keshava Prasad, T.S.K.; Richard, K.; Kandasamy, R.K.; Subbannayya, Y.; Pinto, S.M. A comprehensive pathway map of IL-18-mediated signaling. J. Cell Commun. Signal. 2020, 14, 257–266. [Google Scholar] [CrossRef]
- Nozaki, Y.; Ri, J.; Sakai, K.; Niki, K.; Kinoshita, K. Inhibition of the IL-18 receptor signaling pathway ameliorates disease in a murine model of rheumatoid arthritis. Cells 2019, 9, 11. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Foxwell, B.; Brennan, F.; Feldmann, M. Defining therapeutic targets by using adenovirus: Blocking NF-kappaB inhibits both inflammatory and destructive mechanisms in rheumatoid synovium but spares anti-inflammatory mediators. Proc. Natl. Acad. Sci. USA 1999, 96, 5668–5873. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and in-duces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, P. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Front. Immunol. 2017, 24, 475. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Talabot-Ayer, D.; Lamacchia, C.; Toy, D.; Seemayer, C.A.; Sébastien Viatte, S.; Finckh, A.; Smith, D.E.; Gabay, C. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009, 60, 738–749. [Google Scholar]
- Talabot-Ayer, D.; McKee, T.; Gindre, P.; Bas, S.; Baeten, D.L.; Gabay, C.; Palmer, G. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine 2012, 79, 32–37. [Google Scholar] [CrossRef][Green Version]
- Hong, Y.S.; Moon, S.J.; Joo, Y.B.; Jeon, C.H.; Cho, M.L.; Ju, J.H.; Oh, H.J.; Heo, Y.J.; Park, S.H.; Kim, H.Y.; et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J. Korean Med. Sci. 2011, 26, 1132–1139. [Google Scholar] [CrossRef]
- Tang, S.; Huang, H.; Hu, F.; Zhou, W.; Guo, J.; HuJiang, H.; Mu, R.; Li, Z. Increased IL-33 in synovial fluid and paired serum is associated with disease activity and autoantibodies in rheumatoid arthritis. Clin. Dev. Immunol. 2013, 2013, 985301. [Google Scholar] [CrossRef]
- Xiangyang, Z.; Lutian, Y.; Lin, Z.; Liping, X.; Hui, S.; Jing, L. Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis. Cytokine 2012, 58, 6–9. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Y.; Chen, H.; Liu, X.; Li, M. Blocking interleukin-33 alleviates the joint inflammation and inhibits the development of collagen-induced arthritis in mice. J. Immunol. Res. 2020, 2020, 4297354. [Google Scholar] [CrossRef]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Prasad, T.S.K.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal. 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Endo, Y.; Hirahara, K.; Iinuma, T.; Shinoda, K.; Tumes, D.J.; Asou, H.K.; Matsugae, N.; Obata-Ninomiya, K.; Yamamoto, H.; Motohashi, S.; et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity 2015, 42, 294–308. [Google Scholar] [CrossRef]
- Norris, M.S.; McConnell, T.J.; Mannie, M.D. Interleukin-2 promotes antigenic reactivity of rested T cells but prolongs the postactivational refractory phase of activated T cells. Cell Immunol. 2001, 211, 51–60. [Google Scholar] [CrossRef]
- Tebib, J.G.; Boughaba, T.H.; Letroublon, M.C.; Bienvenu, J.; Noel, E.; Armanet, P.; Colson, F.; Roullet, A.; Bouvier, M. Serum IL-2 level in rheumatoid arthritis: Correlation with joint destruction and disease progression. Eur. Cytokine Netw. 1991, 2, 239–243. [Google Scholar] [PubMed]
- Li, B.; Guo, Q.; Wang, Y.; Su, R.; Gao, C.; Zhao, J.; Li, X.; Wang, C. Increased serum Interleukin-2 levels are associated with abnormal peripheral blood natural killer cell levels in patients with active rheumatoid arthritis. Mediat. Inflamm. 2020, 2020, 6108342. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.C.; Symons, J.A.; Duff, G.W. Serum interleukin-2-receptor in rheumatoid arthritis: A prognostic indicator of disease activity? J. Autoimmun. 1988, 1, 353–361. [Google Scholar] [CrossRef]
- Firestein, G.S.; Xu, W.D.; Townsend, K.; Broide, D.; Alvaro-Gracia, J.; Glasebrook, A.; Zvaifler, N.J. Cytokines in chronic inflammatory arthritis. J. Exp. Med. 1988, 168, 1573–1586. [Google Scholar] [CrossRef]
- Makuch, S.; Więcek, K.; Woźniak, M. The immunomodulatory and anti-inflammatory effect of curcumin on immune cell populations, cytokines, and in vivo models of rheumatoid arthritis. Pharmaceuticals 2021, 4, 309. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Lorenzon, R.; Cacoub, P.; Pham, H.P.; Pitoiset, F.; El Soufi, K.; RIbet, C.; Bernard, C.; Aractingi, S.; Banneville, B.; et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum. Dis. 2019, 78, 209–217. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cytokine-Receptor- Major Signaling Molecules | Proposed Roles in Rheumatoid Arthritis | Clinical Application |
|---|---|---|
| TNF-TNFR 1/2- NF-kB, MAPKs, PI3K | Osteoclastogenesis, TNFR1; Treg inhibition, Proinflammatory cytokine production TNFR2; Treg activation Epigenomic modification (acetylation/methylation) memTNF; protective against arthritis | Widely used |
| IL-6-IL-6R- gp130, JAK1/ 2, Tyk2, STAT1/3, PI3K, SHP-2, ERK | Osteoclastogenesis Proinflammatory cytokine production Autoantibody production Th17 differentiation, Treg inhibition | Widely used (anti-receptor antibody) |
| IL-33-ST2- IL-1-RacP, MyD88, IRAKs, TRAF6, NF-kB, MAPKs, AP1 | Proinflammatory cytokine production Activation of mast cells, Tregs, Th2, and ILC2 | Unreported |
| IL-1β- IL-1R, MyD88, IRAKs, TRAF6, NF-kB MAPKs, AP1 | Inflammation Th17 differentiation, Treg inhibition | Modest or negative |
| IL-18-IL-18Rα/18Rβ MyD88, IRAKs, NF-kB IL-18–IL-18BP | Inflammation Neutralization | Unreported |
| IL-23–IL-12Rβ1/IL-23R- Tyk2, JAK2, STAT3/4 | Activation of Th17, NKT, and ILC3 cells Cytokine production (IL-17, TNF-α, GM-CSF) | Did not meet primary endpoint |
| IL-17–IL-17R ACT1, TRAF6, NF-kB, MAPKs | Proinflammatory cytokine production Osteoclastogenesis Activation of synovial fibroblasts, macrophages | Did not meet primary endpoint |
| IL-7–IL-7R JAK1/3, STAT3/5, PI3K, AKT | Differentiation, expansion of Th17 cells Treg differentiation Osteoclastogenesis | Unreported |
| IL-21 JAK1/3, STAT1/3/5 | Autocrine amplification of Th17 cells Th17 differentiation, Treg inhibition | Phase I/IIa |
| GM-CSF-GM-CSFR- JAK2, STAT3/5 | Macrophage activation Proinflammatory cytokine production | Phase III |
| IL-2–IL-2R- JAK1/2/3, STAT3/5, SHC-1, ERK | Late phase: arthritogenic Activation of ILC2, NK cells, Th17 cells, IL-33 production Early phase: Anti-arthritogenic via IFN-γ Low dose IL-2 Treg activation | Phase I/IIa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10922. https://doi.org/10.3390/ijms222010922
Kondo N, Kuroda T, Kobayashi D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. International Journal of Molecular Sciences. 2021; 22(20):10922. https://doi.org/10.3390/ijms222010922
Chicago/Turabian StyleKondo, Naoki, Takeshi Kuroda, and Daisuke Kobayashi. 2021. "Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis" International Journal of Molecular Sciences 22, no. 20: 10922. https://doi.org/10.3390/ijms222010922
APA StyleKondo, N., Kuroda, T., & Kobayashi, D. (2021). Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. International Journal of Molecular Sciences, 22(20), 10922. https://doi.org/10.3390/ijms222010922