Possible Role of Metformin as an Immune Modulator in the Tumor Microenvironment of Ovarian Cancer




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Potential for Immunoregulation by Metformin in the Ovarian Tumor Microenvironment
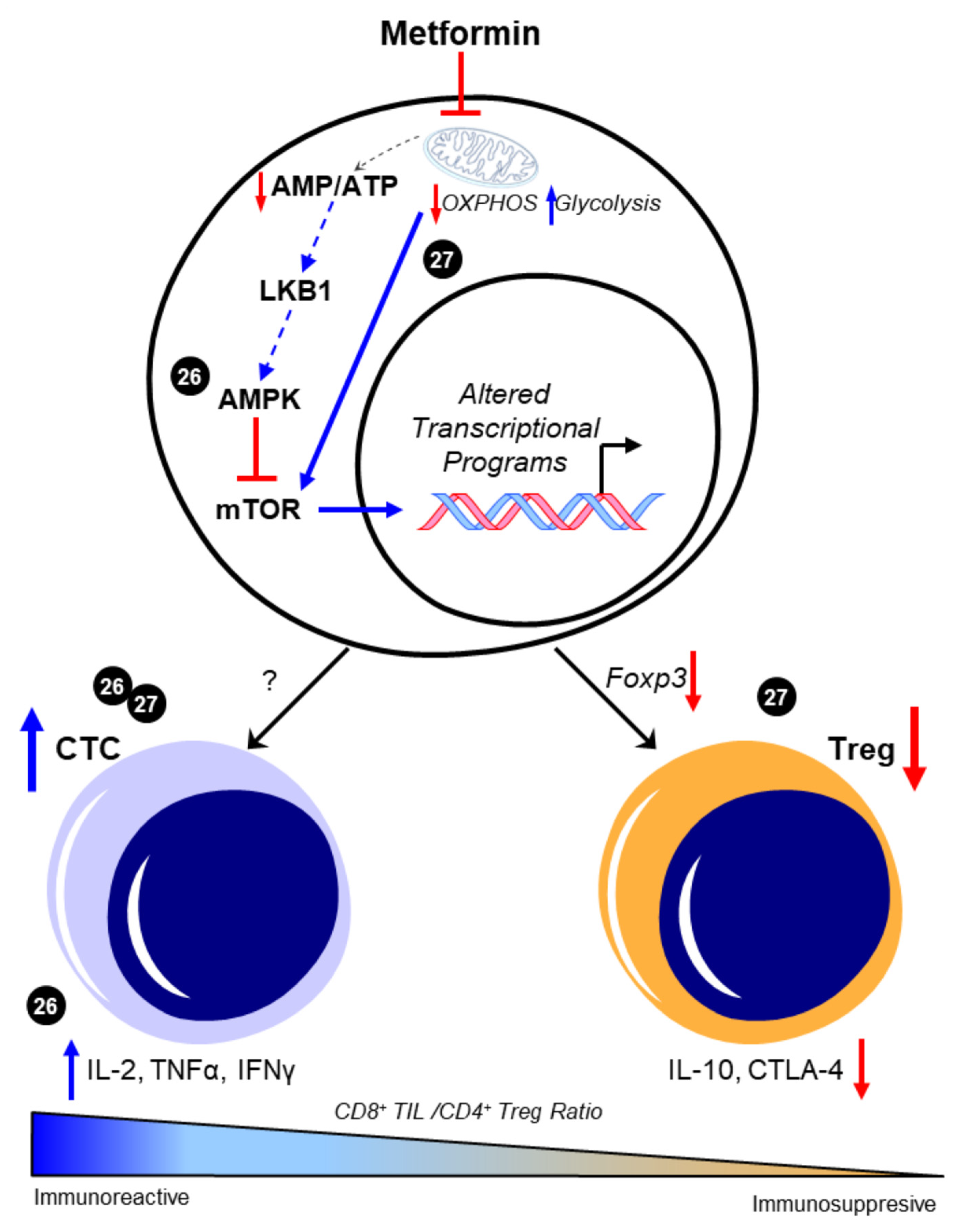
2.1. Metformin and T Cells—An Overview
2.2. Metformin and T Cells—Metabolic Targets and T Cell Phenotypes
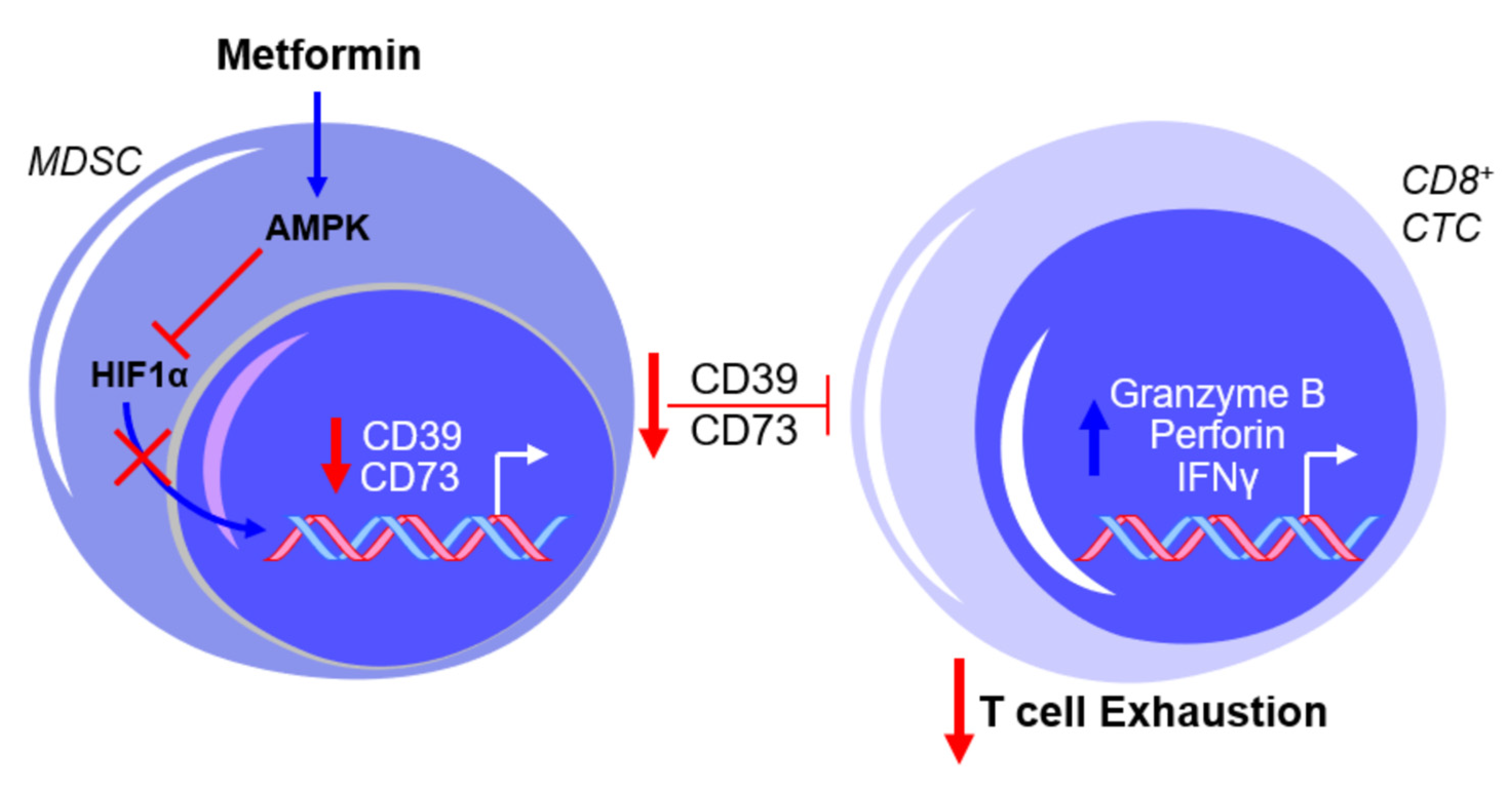
2.3. Metformin and Myeloid-Derived Suppressor Cells (MDSCs)—An Overview
2.4. Metformin and MDSCs—Clinical Implications and Limitations
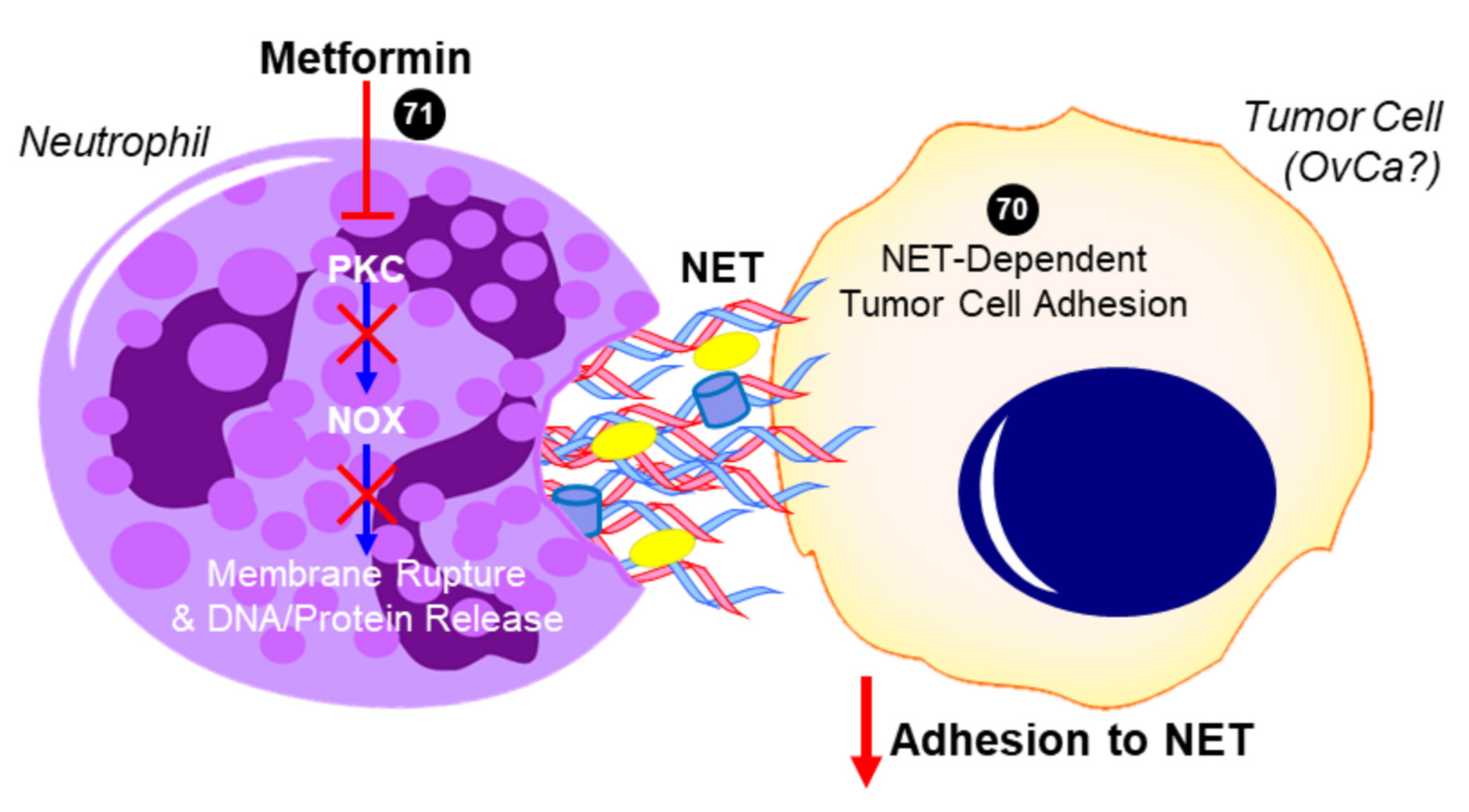
2.5. Metformin and Neutrophils—Neutrophil:Lymphocyte Ratio (NLR)
2.6. Metformin and Neutrophils—Neutrophil Extracellular Trap (NET)
2.7. Metformin and Macrophage Polarization
3. Considerations for Metformin on Immune Cell Metabolism
3.1. Impact of Metabolism on Immune Cell Differentiation and Function
3.2. Metformin, AMPK, Glycolysis, and Ovarian Cancer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | 5′ AMP-activated protein kinase |
| CTC | cytotoxic (CD8+) T lymphocytes |
| CD4 | cluster of differentiation 4 |
| CD8 | cluster of differentiation 8 |
| CD25 | cluster of differentiation 25 |
| CD39 | cluster of differentiation 39 |
| CD73 | cluster of differentiation 73 |
| CoCl2 | cobalt chloride |
| CTLA4 | cytotoxic T lymphocyte antigen-4 |
| Foxp3 | forkhead box P3 |
| GLUT1 | glucose transporter 1 |
| HGSOC | high-grade serous ovarian cancer |
| HIF1α | hypoxia-inducible factor 1-alpha |
| HMGB1 | high mobility group box 1 |
| IFNγ | interferon gamma |
| IL-2 | interleukin 2 |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IL-17 | interleukin 17 |
| LKB1 | liver kinase B1 |
| MDSC | myeloid-derived suppressor cell |
| mTOR | mammalian target of rapamycin |
| NET | neutrophil extracellular trap |
| NF-κB | nuclear factor kappa B |
| NOX | NADPH oxidase |
| OvCa | ovarian cancer |
| OXPHOS | oxidative phosphorylation |
| PAD4 | peptidylarginine deiminase 4 |
| PARP | poly(ADP-ribose) polymerase |
| PD1 | programmed death-1 |
| PFK-2 | phosphofructokinase 2 |
| PKCβII | protein kinase C beta-II |
| T2DM | type 2 diabetes mellitus |
| TAM | tumor-associated macrophage |
| TIL | tumor-infiltrating lymphocyte |
| TIM3 | T cell immunoglobulin and mucin-domain containing-3 |
| TGF-β | tumor growth factor beta |
| TME | tumor microenvironment |
| Tmem | memory T cell |
| TNFα | tumor necrosis factor alpha |
| Treg | regulatory T cell |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Doubeni, C.A.; Doubeni, A.R.; Myers, A.E. Diagnosis and management of ovarian cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar] [PubMed]
- Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Ovarian Cancer; SEER* State Database: Mortality—All COD, Aggregated with State, Total, U.S. (1969–2017); Underlying Mortality Data Provided by NCHS; Available online: www.seer.cancer.gov/statfacts/html/ovary.html; www.cdc.gov/nchs; (accessed on 10 November 2020).
- Winter, W.E.; Maxwell, G.L.; Tian, C.; Sundborg, M.J.; Rose, G.S.; Rose, P.G.; Rubin, S.C.; Muggia, F.M.; McGuire, W.P. Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage iv epithelial ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2008, 26, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Winter, W.E.; Maxwell, G.L.; Tian, C.; Carlson, J.W.; Ozols, R.F.; Rose, P.G.; Markman, M.; Armstrong, D.K.; Muggia, F.; McGuire, W.P. Prognostic factors for stage iii epithelial ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2007, 25, 3621–3627. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- van Baal, J.O.A.M.; van de Vijver, K.K.; Nieuwland, R.; van Noorden, C.J.F.; van Driel, W.J.; Sturk, A.; Kenter, G.G.; Rikkert, L.G.; Lok, C.A.R. The histophysiology and pathophysiology of the peritoneum. Tissue Cell 2017, 49, 95–105. [Google Scholar] [CrossRef]
- Taylor, K.N.; Eskander, R.N. PARP Inhibitors in epithelial ovarian cancer. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 145–158. [Google Scholar] [CrossRef]
- Banerjee, S.N.; Lord, C.J. First-line PARP inhibition in ovarian cancer—Standard of care for all? Nat. Rev. Clin. Oncol. 2020, 17, 136–137. [Google Scholar] [CrossRef]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Drew, Y. The development of PARP inhibitors in ovarian cancer: From bench to bedside. Br. J. Cancer 2015, 113, S3–S9. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.L.; McCormick, A.; McEwen, K.A.; Park, S.; Karrison, T.; Yamada, S.D.; Pannain, S.; Lengyel, E. Relationship of Type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol. 2012, 119, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Meuter, A.; Thapa, P.; Langstraat, C.; Giri, S.; Chien, J.; Rattan, R.; Cliby, W.A.; Shridhar, V. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer 2013, 119, 555–562. [Google Scholar]
- Shi, J.; Liu, B.; Wang, H.; Zhang, T.; Yang, L. Association of metformin use with ovarian cancer incidence and prognosis: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2019, 29, 140–146. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Higurashi, T.; Hosono, K.; Takahashi, H.; Komiya, Y.; Umezawa, S.; Sakai, E.; Uchiyama, T.; Taniguchi, L.; Hata, Y.; Uchiyama, S.; et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: A multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. 2016, 17, 475–483. [Google Scholar] [CrossRef]
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.M.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.J.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol. 2015, 212, 479.e1–479.e10. [Google Scholar] [CrossRef]
- Hart, P.C.; Kenny, H.A.; Grassl, N.; Watters, K.M.; Litchfield, L.M.; Coscia, F.; Blaženović, I.; Ploetzky, L.; Fiehn, O.; Mann, M.; et al. Mesothelial Cell HIF1α expression is metabolically downregulated by metformin to prevent oncogenic tumor-stromal crosstalk. Cell Rep. 2019, 29, 4086–4098. [Google Scholar] [CrossRef]
- Tebbe, C.; Chhina, J.; Dar, S.A.; Sarigiannis, K.; Giri, S.; Munkarah, A.R.; Rattan, R. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget 2014, 5, 4746–4764. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Z.-Y.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol. Cancer Ther. 2018, 17, 1291–1302. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, R.; Bai, Y.; Lu, X. The prognostic value of tumor-infiltrating T lymphocytes in ovarian cancer. Oncotarget 2017, 8, 15621–15631. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.-J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4 + CD25 + Regulatory T cells in the tumor microenvironment by metformin, a Type 2 diabetes drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.; Opipari, A.W., Jr.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef]
- Gerner, M.C.; Niederstaetter, L.; Ziegler, L.; Bileck, A.; Slany, A.; Janker, L.; Schmidt, R.L.J.; Gerner, C.; Del Favero, G.; Schmetterer, K.G. Proteome analysis reveals distinct mitochondrial functions linked to interferon response patterns in activated CD4+ and CD8+ T Cells. Front. Pharmacol. 2019, 10, 727. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2011, 122, 253–270. [Google Scholar] [CrossRef]
- Tamás, P.; MacIntyre, A.; Finlay, D.; Clarke, R.; Feijoo-Carnero, C.; Ashworth, A.; Cantrell, D. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. Eur. J. Immunol. 2009, 40, 242–253. [Google Scholar] [CrossRef]
- Maciver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The liver kinase B1 is a central regulator of t cell development, activation, and metabolism. J. Immunol. 2011, 187, 4187–4198. [Google Scholar] [CrossRef] [PubMed]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; van der Waart, A.B. Comprehensive Phenotyping of T cells using flow cytometry. Cytom. Part A 2019, 95, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Poffenberger, M.C.; Metcalfe-Roach, A.; Aguilar, E.; Chen, J.; Hsu, B.E.; Wong, A.H.; Johnson, R.M.; Flynn, B.; Samborska, B.; Ma, E.H.; et al. LKB1 deficiency in T cells promotes the development of gastrointestinal polyposis. Science 2018, 361, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Fan, J.; Zeng, Q.; Cai, J.; Chen, Y.; Chen, Z.; Wang, W.; Li, S.Y.; Cui, Q.; Yang, J.; et al. CD4 + T-cell endogenous cystathionine γ lyase–hydrogen sulfide attenuates hypertension by sulfhydrating liver kinase B1 to promote T regulatory cell differentiation and proliferation. Circulation 2020, 142, 1752–1769. [Google Scholar] [CrossRef]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.; Jones, R.G. The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; Depinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Horiuchi, T.; Sakata, N.; Narumi, Y.; Kimura, T.; Hayashi, T.; Nagano, K.; Liu, K.; Nishibori, M.; Tsukita, S.; Yamada, T.; et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J. Biol. Chem. 2017, 292, 8436–8446. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef]
- Li, G.; Liang, X.; Lotze, M.T. HMGB1: The central cytokine for all lymphoid cells. Front. Immunol. 2013, 4, 68. [Google Scholar] [CrossRef]
- Chandel, N.S.; Avizonis, D.; Reczek, C.R.; Weinberg, S.E.; Menz, S.; Neuhaus, R.; Christian, S.; Haegebarth, A.; Algire, C.; Pollak, M. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. 2016, 23, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 2018, 28, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; Van Rompuy, A.-S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-derived suppressor cells at diagnosis may discriminate between benign and malignant ovarian tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef]
- Baert, T.; Vankerckhoven, A.; Riva, M.; Van Hoylandt, A.; Thirion, G.; Holger, G.; Mathivet, T.; Vergote, I.; Coosemans, A. Myeloid derived suppressor cells: Key drivers of immunosuppression in ovarian cancer. Front. Immunol. 2019, 10, 1273. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on Tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2007, 4, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ke, Q.; Costa, M. Alterations of histone modifications by cobalt compounds. Carcinogenesis 2009, 30, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Trikha, P.; Plews, R.L.; Stiff, A.; Gautam, S.; Hsu, V.; Abood, D.; Wesolowski, R.; Landi, I.; Mo, X.; Phay, J.; et al. Targeting myeloid-derived suppressor cells using a novel adenosine monophosphate-activated protein kinase (AMPK) activator. OncoImmunology 2016, 5, e1214787. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, K.; Cai, Z.; Gupta, R.; Parajuli, N.; Fox-Talbot, K.; Darshan, M.S.; Gonzalez, F.J.; Semenza, G.L. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2012, 109, 10504–10509. [Google Scholar] [CrossRef]
- Kling, L.; Benck, U.; Breedijk, A.; Leikeim, L.; Heitzmann, M.; Porubsky, S.; Krämer, B.K.; Yard, B.A.; Kälsch, A.-I. Changes in CD73, CD39 and CD26 expression on T-lymphocytes of ANCA-associated vasculitis patients suggest impairment in adenosine generation and turn-over. Sci. Rep. 2017, 7, 11683. [Google Scholar] [CrossRef]
- Litchfield, L.M.; Mukherjee, A.; Eckert, M.A.; Johnson, A.; Mills, K.A.; Pan, S.; Shridhar, V.; Lengyel, E.; Romero, I.L. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 2015, 6, 23548–23560. [Google Scholar] [CrossRef]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Halle, S.; Halle, O.; Förster, R. Mechanisms and dynamics of T Cell-mediated cytotoxicity in vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- Yin, X.; Wu, L.; Yang, H.; Yang, H. Prognostic significance of neutrophil–lymphocyte ratio (NLR) in patients with ovarian cancer. Medicine 2019, 98, e17475. [Google Scholar] [CrossRef]
- Duman, T.T.; Aktas, G.; Atak, B.M.; Kocak, M.Z.; Erkus, E.; Savli, H. Neutrophil to lymphocyte ratio as an indicative of diabetic control level in type 2 diabetes mellitus. Afr. Health Sci. 2019, 19, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Schildkraut, J.M.; Schwingl, P.J.; Bastos, E.; Evanoff, A.; Hughes, C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet. Gynecol. 1996, 88, 554–559. [Google Scholar] [CrossRef]
- Kelly, C.C.J.; Lyall, H.; Petrie, J.R.; Gould, G.W.; Connell, J.M.; Sattar, N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 2453–2455. [Google Scholar] [CrossRef]
- Orio, F.; Palomba, S.; Cascella, T.; Di Biase, S.; Manguso, F.; Tauchmanovà, L.; Nardo, L.G.; Labella, D.; Savastano, S.; Russo, T.; et al. The increase of leukocytes as a new putative marker of low-grade chronic inflammation and early cardiovascular risk in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 2–5. [Google Scholar] [CrossRef]
- Ibáñez, L.; Jaramillo, A.M.; Ferrer, A.; de Zegher, F. High neutrophil count in girls and women with hyperinsulinaemic hyperandrogenism: Normalization with metformin and flutamide overcomes the aggravation by oral contraception. Hum. Reprod. 2005, 20, 2457–2462. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Lee, W.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.V.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Wang, X.; Deavers, M.; Patenia, R.; Bassett, R.L.; Mueller, P.; Ma, Q.; Wang, E.; Freedman, R.S. Monocyte/macrophage and T-cell infiltrates in peritoneum of patients with ovarian cancer or benign pelvic disease. J. Transl. Med. 2006, 4, 1–11. [Google Scholar] [CrossRef]
- Liu, J.; Geng, X.; Li, Y. Milky spots: Omental functional units and hotbeds for peritoneal cancer metastasis. Tumor Biol. 2016, 37, 5715–5726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; He, Y.-F.; Sun, X.J.; Li, Q.; Wang, W.; Zhao, A.M.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, S.; Sheng, L.; Zhu, J.; Gu, L.; Shen, H.; La, D.; Hambly, B.D.; Bao, S.; Di, W. Metformin inhibits the development and metastasis of ovarian cancer. Oncol. Rep. 2012, 28, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Wang, K.; Wu, L.; Duan, T. Interaction of monocytes/macrophages with ovarian cancer cells promotes angiogenesis in vitro. Cancer Sci. 2013, 104, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-F.; Chao, T.-T.; Su, Y.-F.; Hsu, C.-C.; Chien, C.-Y.; Chiu, K.-C.; Shiah, S.-G.; Lee, C.-H.; Liu, S.-Y.; Shieh, Y.-S. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget 2017, 8, 20706–20718. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. macrophage polarization in the development and progression of ovarian cancers: An overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef]
- Hu, Z.; Zou, Q.; Su, B. Regulation of T cell immunity by cellular metabolism. Front. Med. 2018, 12, 463–472. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, A.; Bi, Y.; Wang, Y.; Liu, G. Metabolic regulation of myeloid-derived suppressor cell function in cancer. Cells 2020, 9, 1011. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil metabolic shift during their lifecycle: Impact on their survival and activation. Int. J. Mol. Sci. 2019, 21, 287. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Stenmark, K.R. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin. Immunol. 2015, 27, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Hodeib, M.; Ogrodzinski, M.P.; Vergnes, L.; Reue, K.; Karlan, B.Y.; Lunt, S.Y.; Aspuria, P.P. Metformin induces distinct bioenergetic and metabolic profiles in sensitive versus resistant high grade serous ovarian cancer and normal fallopian tube secretory epithelial cells. Oncotarget 2018, 9, 4044–4060. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Chiyoda, T.; Liu, X.; Weigert, M.; Curtis, M.; Chiang, C.-Y.; Loth, R.; Lastra, R.; McGregor, S.M.; Locasale, J.W.; et al. SPHK1 is a novel target of metformin in ovarian cancer. Mol. Cancer Res. 2019, 17, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Marsin, A.-S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; MacIntyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-all cell stress and survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.-Y.; Temkin, S.M.; Fang, X. Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef]
- Laski, J.; Singha, B.; Wang, X.; Valdés, Y.R.; Collins, O.; Shepherd, T.G. Activated CAMKKβ-AMPK signaling promotes autophagy in a spheroid model of ovarian tumour metastasis. J. Ovarian Res. 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T Cell immunotherapy for cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Rajab, I.M.; Alebraheem, M.; Potempa, L.A. C-Reactive Protein and Cancer—Diagnostic and Therapeutic Insights. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, M.; Wohlmuth, C.; Waas, M.; Bernardini, M.Q.; Kislinger, T. High-throughput approaches for precision medicine in high-grade serous ovarian cancer. J. Hematol. Oncol. 2020, 13, 1–20. [Google Scholar] [CrossRef]
- Hijaz, M.; Chhina, J.; Mert, I.; Taylor, M.; Dar, S.; Al-Wahab, Z.; Ali-Fehmi, R.; Buekers, T.; Munkarah, A.R.; Rattan, R. Preclinical evaluation of olaparib and metformin combination in BRCA1 wildtype ovarian cancer. Gynecol. Oncol. 2016, 142, 323–331. [Google Scholar] [CrossRef]
- Han, Y.; Li, C.-W.; Hsu, J.-M.; Hsu, J.L.; Chan, L.-C.; Tan, X.; He, G.-J. Metformin reverses PARP inhibitors-induced epithelial-mesenchymal transition and PD-L1 upregulation in triple-negative breast cancer. Am. J. Cancer Res. 2019, 9, 800–815. [Google Scholar]
- Indraccolo, S.; Randon, G.; Zulato, E.; Nardin, M.; Aliberti, C.; Pomerri, F.; Casarin, A.; Nicoletto, M.O. Metformin: A modulator of bevacizumab activity in cancer? A case report. Cancer Biol. Ther. 2015, 16, 210–214. [Google Scholar] [CrossRef][Green Version]
- Marrone, K.A.; Zhou, X.; Forde, P.M.; Purtell, M.; Brahmer, J.R.; Hann, C.L.; Kelly, R.J.; Coleman, B.; Gabrielson, E.; Rosner, G.L.; et al. A randomized phase II Study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy-naïve advanced or metastatic nonsquamous non-small cell lung cancer. Oncologist 2018, 23, 859–865. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsogas, F.K.; Majerczyk, D.; Hart, P.C. Possible Role of Metformin as an Immune Modulator in the Tumor Microenvironment of Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 867. https://doi.org/10.3390/ijms22020867
Tsogas FK, Majerczyk D, Hart PC. Possible Role of Metformin as an Immune Modulator in the Tumor Microenvironment of Ovarian Cancer. International Journal of Molecular Sciences. 2021; 22(2):867. https://doi.org/10.3390/ijms22020867
Chicago/Turabian StyleTsogas, Faye K., Daniel Majerczyk, and Peter C. Hart. 2021. "Possible Role of Metformin as an Immune Modulator in the Tumor Microenvironment of Ovarian Cancer" International Journal of Molecular Sciences 22, no. 2: 867. https://doi.org/10.3390/ijms22020867
APA StyleTsogas, F. K., Majerczyk, D., & Hart, P. C. (2021). Possible Role of Metformin as an Immune Modulator in the Tumor Microenvironment of Ovarian Cancer. International Journal of Molecular Sciences, 22(2), 867. https://doi.org/10.3390/ijms22020867