Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis

 ,
, 

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
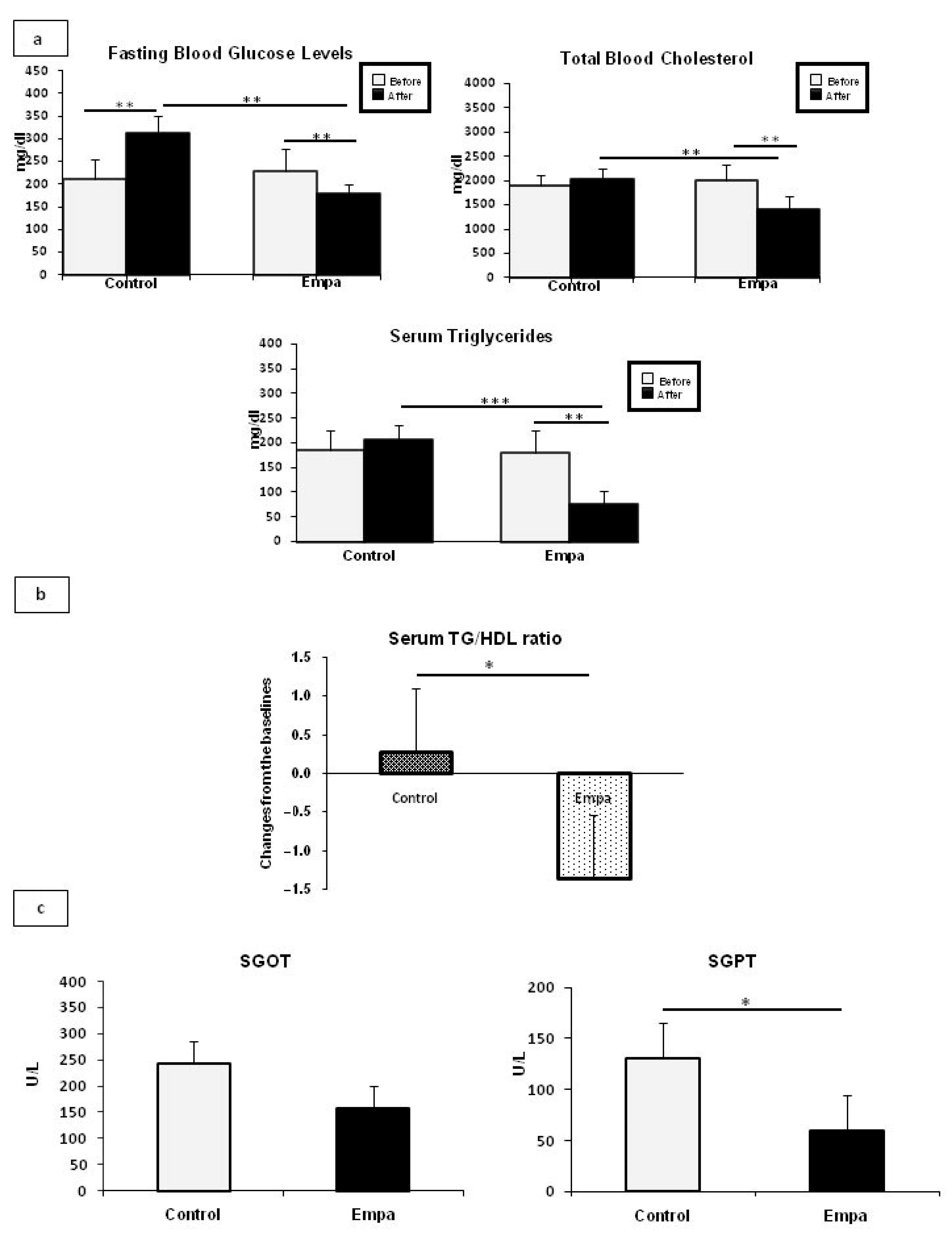
2.1. Empagliflozin Administration for Five Weeks Improves Fasting Blood Glucose and Lipid Profiles
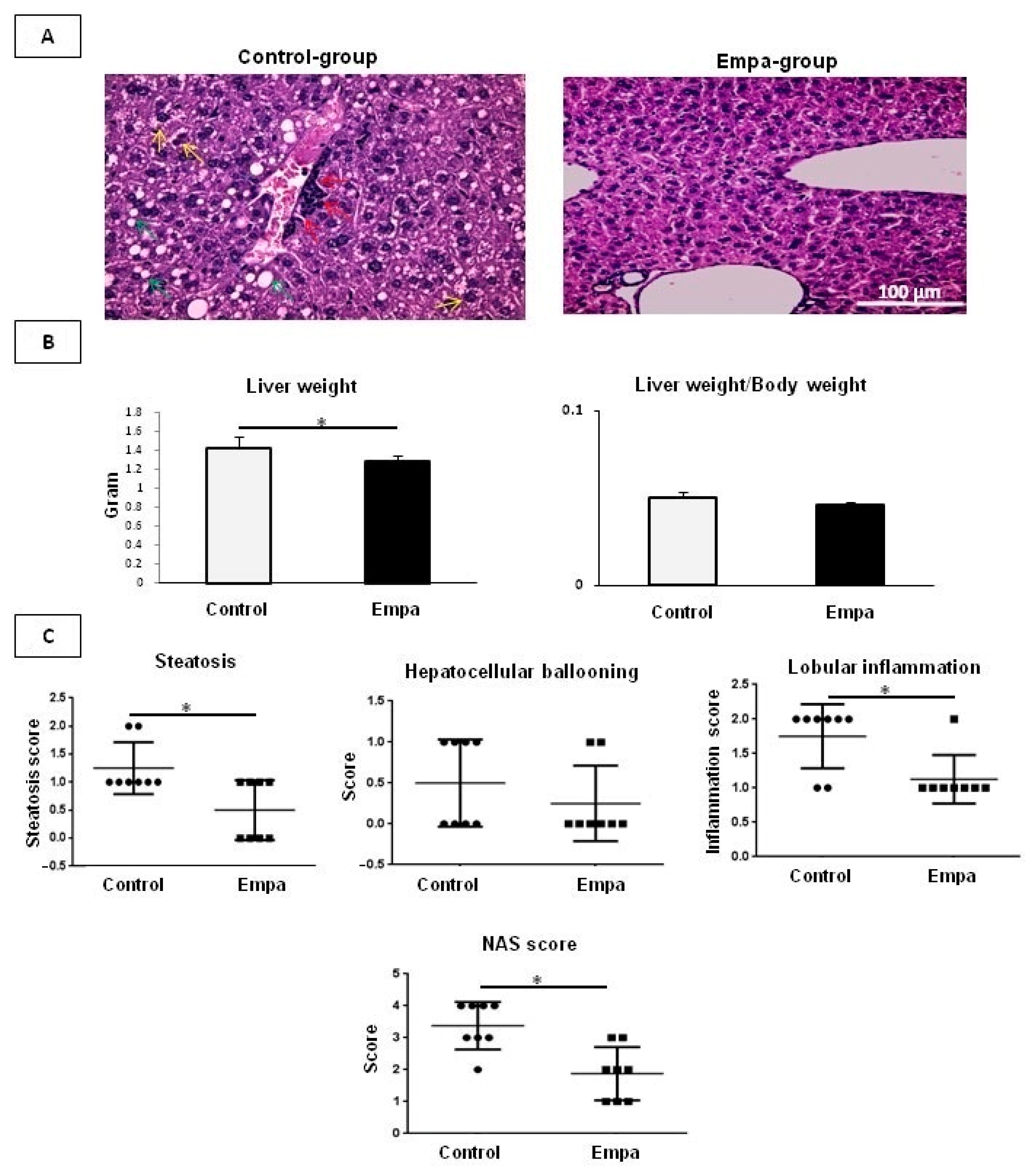
2.2. Empagliflozin Administration for Five Weeks Improves Hepatic Lipid Accumulation
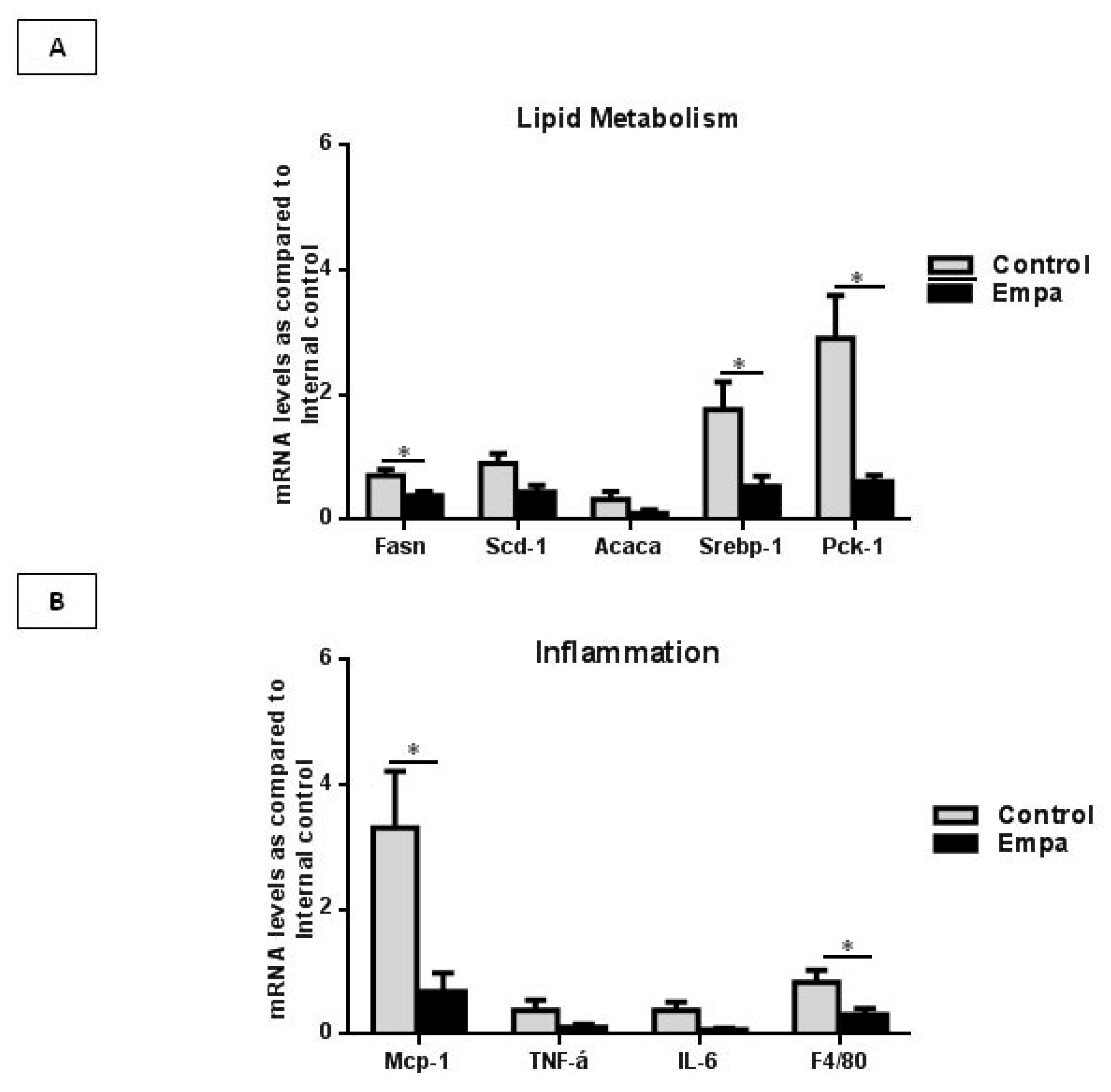
2.3. Empagliflozin Administration for Five Weeks Reduces the Expression of Lipogenic Enzymes and Inflammatory Markers
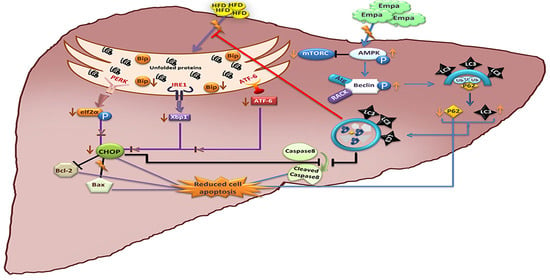
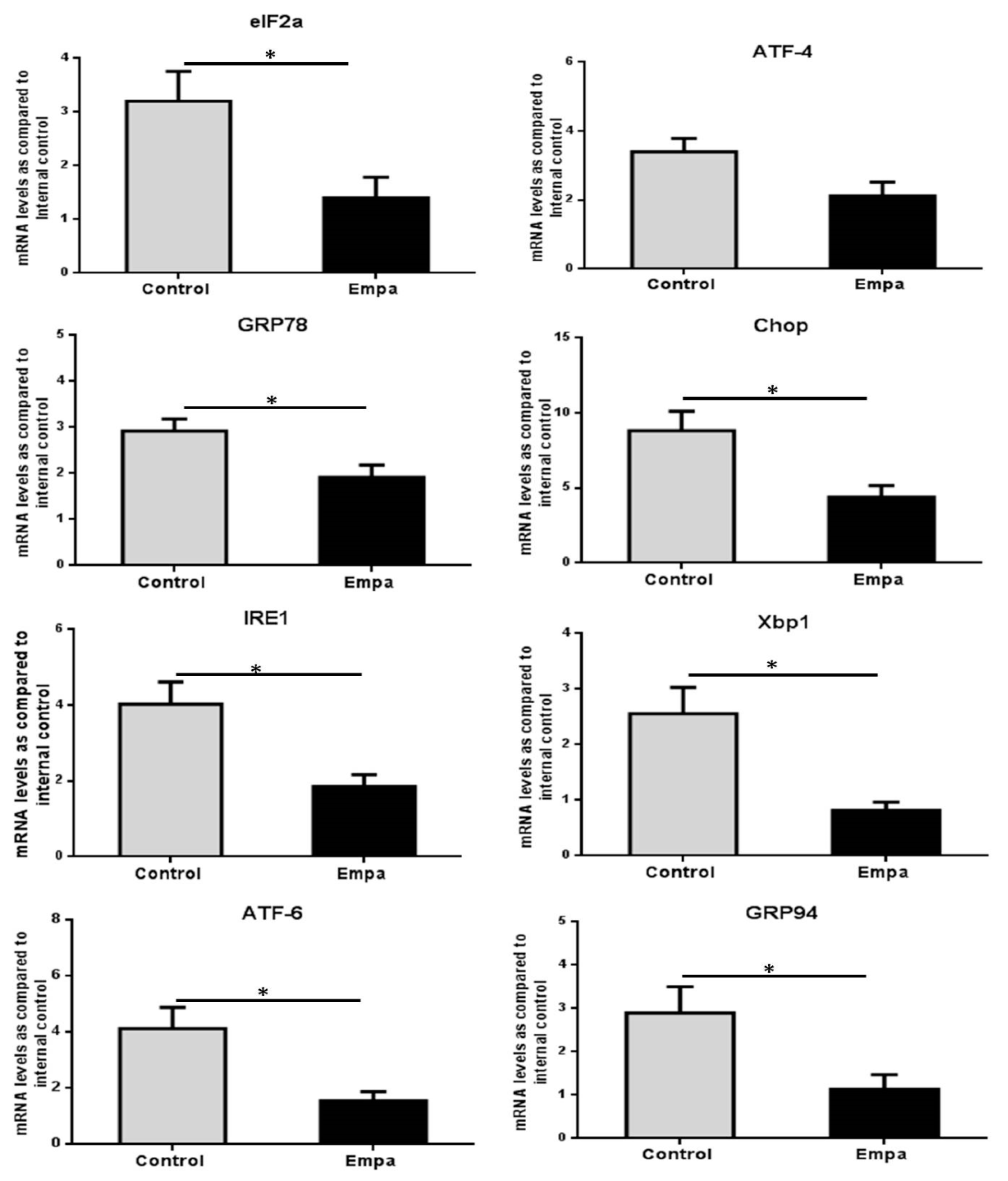
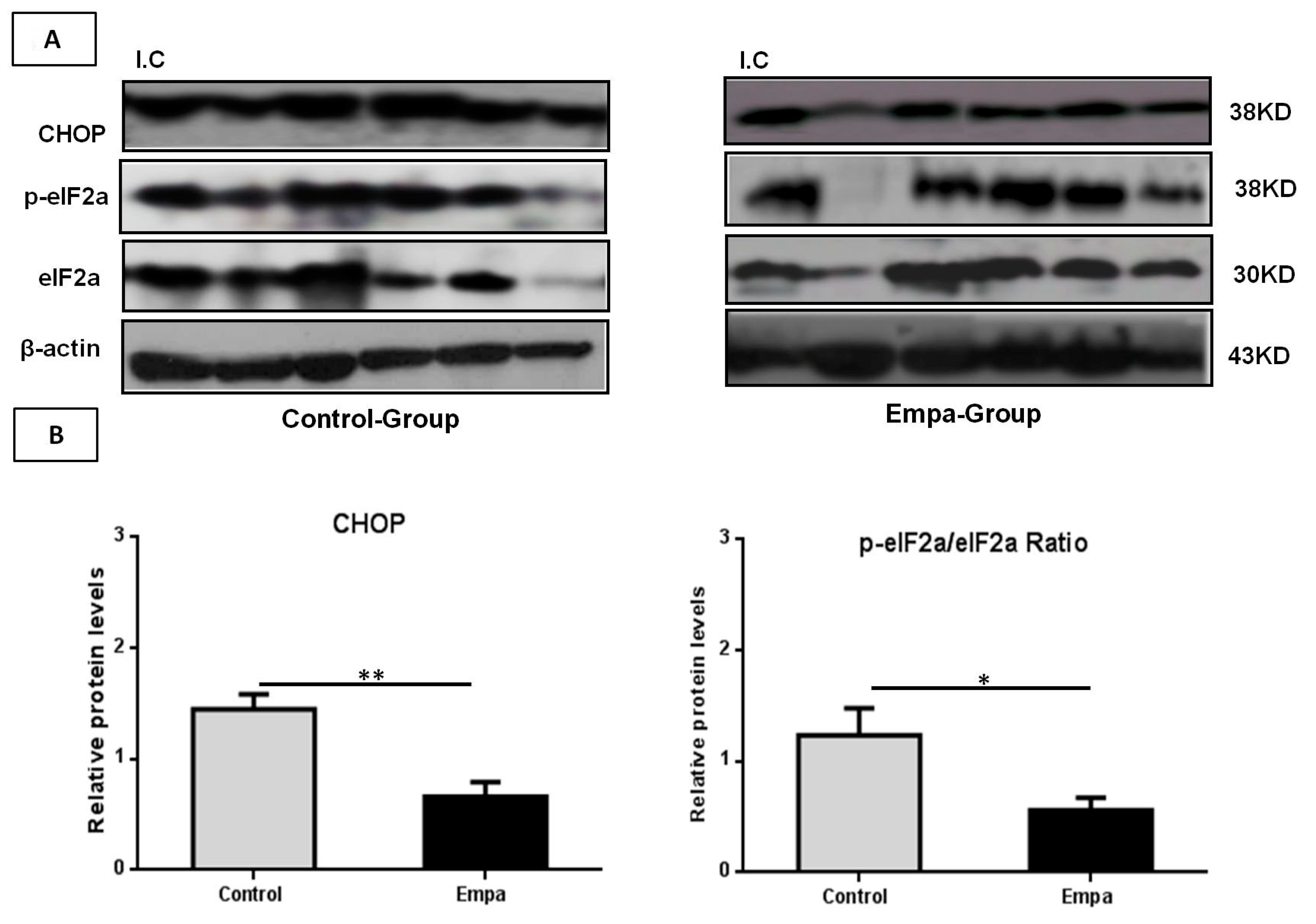
2.4. Empagliflozin Administration for Five Weeks Reduces the Expression of ER Stress Markers
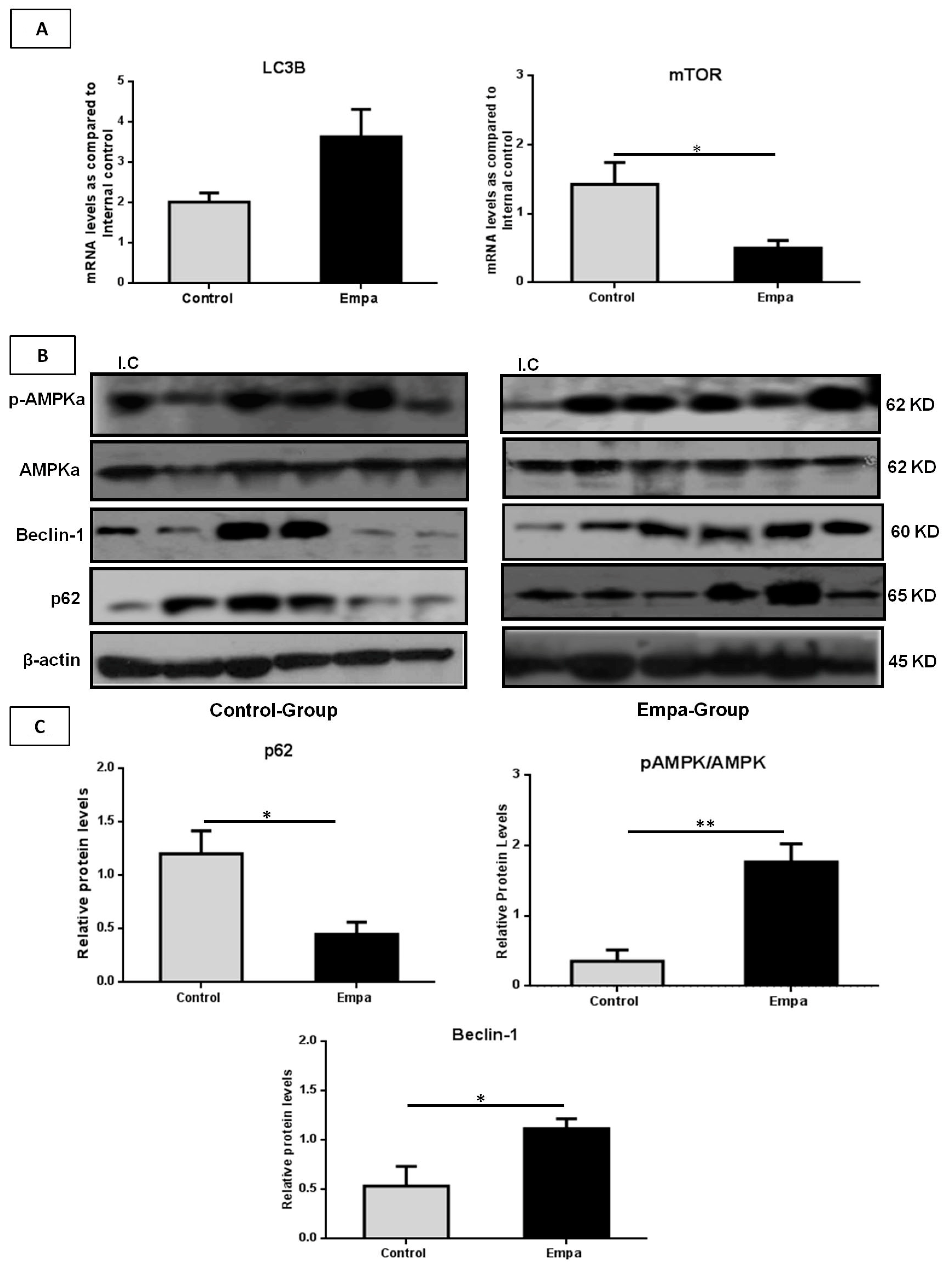
2.5. Empagliflozin Administration for Five Weeks Activates the Hepatic Autophagic Flux
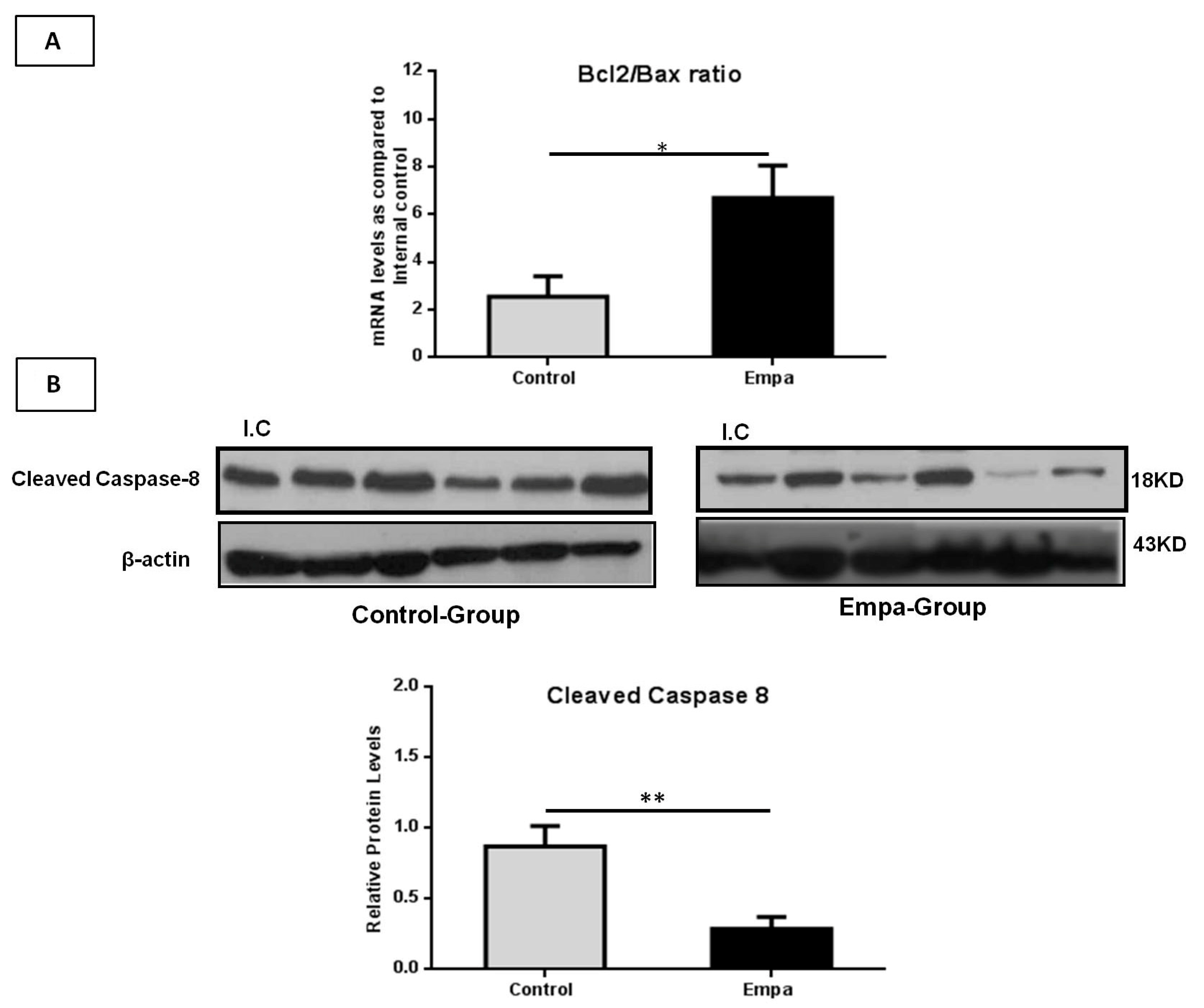
2.6. Empagliflozin Administration for Five Weeks Attenuates Hepatic Apoptosis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Protocol
4.3. Serum Analysis of Biochemical Parameters
4.4. RNA Extraction and Quantitative Real-Time PCR
4.5. Liver Histological Analysis
4.6. SDS-PAGE and Western-Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ApoE | Apolipoprotein E |
| ATF4 | Activating Transcription Factor 4 |
| AΤF6 | Activating Transcription Factor 6 |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| ACLY | ATP citrate lyase |
| Acc2 | Acetyl coA carboxylase 2 |
| AMPKa1 | AMP-activated protein kinase alpha 1 |
| AMPKa2 | AMP-activated protein kinase alpha 2 |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | BCL2-associated X protein |
| CHOP | C/EBP homologous protein |
| CVD | Cardiovascular disease |
| Dgat2 | Diacyl glycerol acetyltransferase 2 |
| DMT2 | Diabetes mellitus type 2 |
| Empa | Empagliflozin |
| ER | Endoplasmic reticulum |
| elF2α | Eukaryotic Initiation Factor 2 alpha |
| Fasn | Fatty acid synthase |
| F4/80 | EGF-like module-containing mucin-like hormone receptor-like 1 |
| FFA | Free fatty acid |
| GRP78 | Binding immunoglobulin protein |
| GRP94 | Glucose-regulated protein 94 |
| GPAT | Glycerol-3-phosphate acyltransferase |
| HFD | High-fat diet |
| HbA1c | Hemoglobin A1c |
| ICAM-1 | Intercellular adhesion molecule-1 |
| Il-6 | Interleukin 6 |
| Insig | Insulin-induced gene |
| IRE1α | Inositol-requiring enzyme 1 α |
| LC3B | Microtubule-Associated Protein 1 Light Chain 3B |
| mTOR | Mechanistic Target Of Rapamycin Kinase |
| Mcp-1 | Monocyte chemoattractant protein-1 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NAS | NAFLD Activity Score |
| Ppar-γ | Peroxisome proliferator-activated receptor gamma |
| Pck-1 | Phosphoenolpyruvate carboxykinase 1 |
| p62(Sqstm1) | Sequestosome 1 |
| qPCR | Quantitative polymerase chain reaction |
| ROS | Reactive oxygen species |
| SGLT-2i | Sodium-glucose cotransporter-2 inhibitors |
| Screbp-1 | Sterol regulatory element-binding protein 1 |
| Scd-1 | Stearoyl-CoA desaturase 1 |
| SGOT | Glutamic oxaloacetic transaminase |
| SGPT | Glutamic-pyruvic transaminase |
| TNF-α | Tumor necrosis factor alpha |
| UPR | Unfolded protein response |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| ΧΒΡ1 | X-box binding protein 1 |
| ZDF | Zucker diabetic fatty |
| γ-GT | Gamma-glutamyl transferase |
References
- Mantovani, A.; Scorletti, E.; Mosca, A.; Alisi, A.; Byrne, C.D.; Targher, G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism 2020, 11, 154170. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Tomlinson, J.W. Non-alcoholic fatty liver disease in common endocrine disorders. Eur. J. Endocrinol. 2013, 169, R27–R37. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Katsiki, N.; Perakakis, N.; Mantzoros, C. Effects of sodium-glucose co-transporter-2 (SGLT2) inhibitors on non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Ex quo et quo vadimus? Metabolism 2019, 98, iii–ix. [Google Scholar] [CrossRef]
- Ranjbar, G.; Mikhailidis, D.P.; Sahebkar, A. Effects of newer antidiabetic drugs on nonalcoholic fatty liver and steatohepatitis: Think out of the box! Metabolism 2019, 101, 154001. [Google Scholar] [CrossRef]
- Upadhyay, J.; Polyzos, S.A.; Perakakis, N.; Thakkar, B.; Paschou, S.A.; Katsiki, N.; Underwood, P.; Park, K.H.; Seufert, J.; Kang, E.S.; et al. Pharmacotherapy of type 2 diabetes: An update. Metabolism 2018, 78, 13–42. [Google Scholar] [CrossRef]
- Anastasilakis, A.D.; Sternthal, E.; Mantzoros, C.S. Beyond glycemic control: New guidance on cardio-renal protection. Metabolism 2019, 99, 113–115. [Google Scholar] [CrossRef]
- Dimitriadis, G.K.; Nasiri-Ansari, N.; Agrogiannis, G.; Kostakis, I.D.; Randeva, M.S.; Nikiteas, N.; Patel, V.H.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; et al. Empagliflozin improves primary haemodynamic parameters and attenuates the development of atherosclerosis in high fat diet fed APOE knockout mice. Mol. Cell. Endocrinol. 2019, 494, 110487. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef]
- Kahl, S.; Gancheva, S.; Strassburger, K.; Herder, C.; Machann, J.; Katsuyama, H.; Kabisch, S.; Henkel, E.; Kopf, S.; Lagerpusch, M.; et al. Empagliflozin Effectively Lowers Liver Fat Content in Well-Controlled Type 2 Diabetes: A Randomized, Double-Blind, Phase 4, Placebo-Controlled Trial. Diabetes Care 2020, 43, 298–305. [Google Scholar] [CrossRef]
- Sattar, N.; Fitchett, D.; Hantel, S.; George, J.T.; Zinman, B. Empagliflozin is associated with improvements in liver enzymes potentially consistent with reductions in liver fat: Results from randomised trials including the EMPA-REG OUTCOME(R) trial. Diabetologia 2018, 61, 2155–2163. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, B.W. Beneficial effect of anti-diabetic drugs for nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2020, 26, 430–443. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Guo, B.; Li, Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front. Genet. 2014, 5, 242. [Google Scholar] [CrossRef]
- Daems, C.; Welsch, S.; Boughaleb, H.; Vanderroost, J.; Robert, A.; Sokol, E.; Lysy, P.A. Early Treatment with Empagliflozin and GABA Improves beta-Cell Mass and Glucose Tolerance in Streptozotocin-Treated Mice. J. Diabetes Res. 2019, 2019, 2813489. [Google Scholar] [CrossRef]
- Hosokawa, K.; Takata, T.; Sugihara, T.; Matono, T.; Koda, M.; Kanda, T.; Taniguchi, S.; Ida, A.; Mae, Y.; Yamamoto, M.; et al. Ipragliflozin Ameliorates Endoplasmic Reticulum Stress and Apoptosis through Preventing Ectopic Lipid Deposition in Renal Tubules. Int. J. Mol. Sci. 2020, 21, 190. [Google Scholar] [CrossRef]
- Shibusawa, R.; Yamada, E.; Okada, S.; Nakajima, Y.; Bastie, C.C.; Maeshima, A.; Kaira, K.; Yamada, M. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci. Rep. 2019, 9, 9887. [Google Scholar] [CrossRef]
- Petito-da-Silva, T.I.; Souza-Mello, V.; Barbosa-da-Silva, S. Empaglifozin mitigates NAFLD in high-fat-fed mice by alleviating insulin resistance, lipogenesis and ER stress. Mol. Cell. Endocrinol. 2019, 498, 110539. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.R.; Carbajal, H.A.; Espeche, W.G.; Leiva Sisnieguez, C.E.; Balbin, E.; Dulbecco, C.A.; Aizpurua, M.; Marillet, A.G.; Reaven, G.M. Relation among the plasma triglyceride/high-density lipoprotein cholesterol concentration ratio, insulin resistance, and associated cardio-metabolic risk factors in men and women. Am. J. Cardiol. 2012, 109, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Katsuno, K.; Isaji, M.; Nagasawa, T.; Buehrer, B.; Walker, S.; Wilkison, W.O.; Cheatham, B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 2015, 5, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Qiang, S.; Nakatsu, Y.; Seno, Y.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Kamata, H.; et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Imajo, K.; Kato, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. The Selective SGLT2 Inhibitor Ipragliflozin Has a Therapeutic Effect on Nonalcoholic Steatohepatitis in Mice. PLoS ONE 2016, 11, e0146337. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef]
- Chiang, H.; Lee, J.C.; Huang, H.C.; Huang, H.; Liu, H.K.; Huang, C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and non-alcoholic fatty liver disease in mice. Br. J. Pharmacol. 2020, 177, 239–253. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Guirguis, E.; Thornby, K.A. A Systematic Review of Newer Antidiabetic Agents in the Treatment of Nonalcoholic Fatty Liver Disease. Ann. Pharmacother. 2020, 55, 65–79. [Google Scholar] [CrossRef]
- Shibata, M.-A.; Shibata, E.; Fujioka, S.; Harada-Shiba, M. Apolipoprotein E-knockout Mice as a Lifestyle-related Disease Model of Atherosclerosis and Non-alcoholic Fatty Liver Disease. Int. J. Lab. Med. Res. 2015, 1, 107. [Google Scholar] [CrossRef][Green Version]
- Schierwagen, R.; Maybuchen, L.; Zimmer, S.; Hittatiya, K.; Back, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef]
- Lee, P.C.; Ganguly, S.; Goh, S.Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 2018, 19, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE(-/-) mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Adingupu, D.D.; Gopel, S.O.; Gronros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.M.; Jonsson-Rylander, A.C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob(-/-) mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.; Hwang, S.; Cherrington, N.J.; Ryu, D.Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 2017, 8, 63370–63381. [Google Scholar] [CrossRef]
- Wang, L.; Chen, J.; Ning, C.; Lei, D.; Ren, J. Endoplasmic Reticulum Stress Related Molecular Mechanisms in Nonalcoholic Fatty Liver Disease (NAFLD). Curr. Drug Targets 2018, 19, 1087–1094. [Google Scholar] [CrossRef]
- Habeos, I.G.; Ziros, P.G.; Chartoumpekis, D.; Psyrogiannis, A.; Kyriazopoulou, V.; Papavassiliou, A.G. Simvastatin activates Keap1/Nrf2 signaling in rat liver. J. Mol. Med. (Berlin) 2008, 86, 1279–1285. [Google Scholar] [CrossRef]
- Rodrigues, G.; Moreira, A.J.; Bona, S.; Schemitt, E.; Marroni, C.A.; Di Naso, F.C.; Dias, A.S.; Pires, T.R.; Picada, J.N.; Marroni, N.P. Simvastatin Reduces Hepatic Oxidative Stress and Endoplasmic Reticulum Stress in Nonalcoholic Steatohepatitis Experimental Model. Oxid. Med. Cell. Longev. 2019, 2019, 3201873. [Google Scholar] [CrossRef]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111, 154299. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Liu, Y.L.; Yang, L.; Liu, P.; Zhang, Y.; Wang, X.Y. MiR-149 attenuates endoplasmic reticulum stress-induced inflammation and apoptosis in nonalcoholic fatty liver disease by negatively targeting ATF6 pathway. Immunol. Lett. 2020, 222, 40–48. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, W. The Sodium-Glucose Co-Transporter 2 Inhibitor, Empagliflozin, Protects against Diabetic Cardiomyopathy by Inhibition of the Endoplasmic Reticulum Stress Pathway. Cell. Physiol. Biochem. 2017, 41, 2503–2512. [Google Scholar] [CrossRef]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Holczer, M.; Hajdu, B.; Lorincz, T.; Szarka, A.; Banhegyi, G.; Kapuy, O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. Int. J. Mol. Sci. 2019, 20, 5543. [Google Scholar] [CrossRef]
- Kliosnky, D. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy 2016, 12, 443. [Google Scholar]
- Kassi, E.; Papavassiliou, A.G. Could glucose be a proaging factor ? J. Cell. Mol. Med. 2008, 12, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar] [CrossRef]
- Aragon-Herrera, A.; Feijoo-Bandin, S.; Santiago, M.O.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Pereira, T.M.C.; Garcia-Caballero, T.; Rodriguez-Segade, S.; Rodriguez, J.; et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem. Pharmacol. 2019, 170, 113677. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur. J. Heart Fail. 2020, 22, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Li, C.P.; Li, J.H.; He, S.Y.; Li, P.; Zhong, X.L. Roles of Fas/Fasl, Bcl-2/Bax, and Caspase-8 in rat nonalcoholic fatty liver disease pathogenesis. Genet. Mol. Res. 2014, 13, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakao, K. To die or not to die: Death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 893–906. [Google Scholar] [CrossRef]
- Hatting, M.; Zhao, G.; Schumacher, F.; Sellge, G.; Al Masaoudi, M.; Gassler, N.; Boekschoten, M.; Muller, M.; Liedtke, C.; Cubero, F.J.; et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013, 57, 2189–2201. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.H.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, L.; Yu, S.; Shen, W.; Cai, C.; Yu, H. Inhibition of stearoyl-coenzyme A desaturase 1 ameliorates hepatic steatosis by inducing AMPK-mediated lipophagy. Aging 2020, 12, 7350–7362. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Girelli, S.; Scopa, C.; Buonocore, G.; Longini, M.; Balduini, W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010, 6, 366–377. [Google Scholar] [CrossRef]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Shirakami, Y.; Maruta, A.; Ideta, T.; Miyazaki, T.; Kochi, T.; Sakai, H.; Tanaka, T.; Seishima, M.; Shimizu, M. Preventive effects of the sodium glucose cotransporter 2 inhibitor tofogliflozin on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic mice. Oncotarget 2017, 8, 58353–58363. [Google Scholar] [CrossRef]
- David-Silva, A.; Esteves, J.V.; Morais, M.; Freitas, H.S.; Zorn, T.M.; Correa-Giannella, M.L.; Machado, U.F. Dual SGLT1/SGLT2 Inhibitor Phlorizin Ameliorates Non-Alcoholic Fatty Liver Disease and Hepatic Glucose Production in Type 2 Diabetic Mice. Diabetes Metab. Syndr. Obes. 2020, 13, 739–751. [Google Scholar] [CrossRef]
- Kim, J.H.; Moon, J.S.; Byun, S.J.; Lee, J.H.; Kang, D.R.; Sung, K.C.; Kim, J.Y.; Huh, J.H. Fatty liver index and development of cardiovascular disease in Koreans without pre-existing myocardial infarction and ischemic stroke: A large population-based study. Cardiovasc. Diabetol. 2020, 19, 51. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. https://doi.org/10.3390/ijms22020818
Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, Chatzigeorgiou A, Kalotychou V, Randeva MS, Chatha K, et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. International Journal of Molecular Sciences. 2021; 22(2):818. https://doi.org/10.3390/ijms22020818
Chicago/Turabian StyleNasiri-Ansari, Narjes, Chrysa Nikolopoulou, Katerina Papoutsi, Ioannis Kyrou, Christos S. Mantzoros, Georgios Kyriakopoulos, Antonios Chatzigeorgiou, Vassiliki Kalotychou, Manpal S. Randeva, Kamaljit Chatha, and et al. 2021. "Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis" International Journal of Molecular Sciences 22, no. 2: 818. https://doi.org/10.3390/ijms22020818
APA StyleNasiri-Ansari, N., Nikolopoulou, C., Papoutsi, K., Kyrou, I., Mantzoros, C. S., Kyriakopoulos, G., Chatzigeorgiou, A., Kalotychou, V., Randeva, M. S., Chatha, K., Kontzoglou, K., Kaltsas, G., Papavassiliou, A. G., Randeva, H. S., & Kassi, E. (2021). Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. International Journal of Molecular Sciences, 22(2), 818. https://doi.org/10.3390/ijms22020818