Comparative Analysis of Transcriptome and sRNAs Expression Patterns in the Brachypodium distachyon—Magnaporthe oryzae Pathosystems

 , , , and
, , , and
Abstract
1. Introduction
2. Results
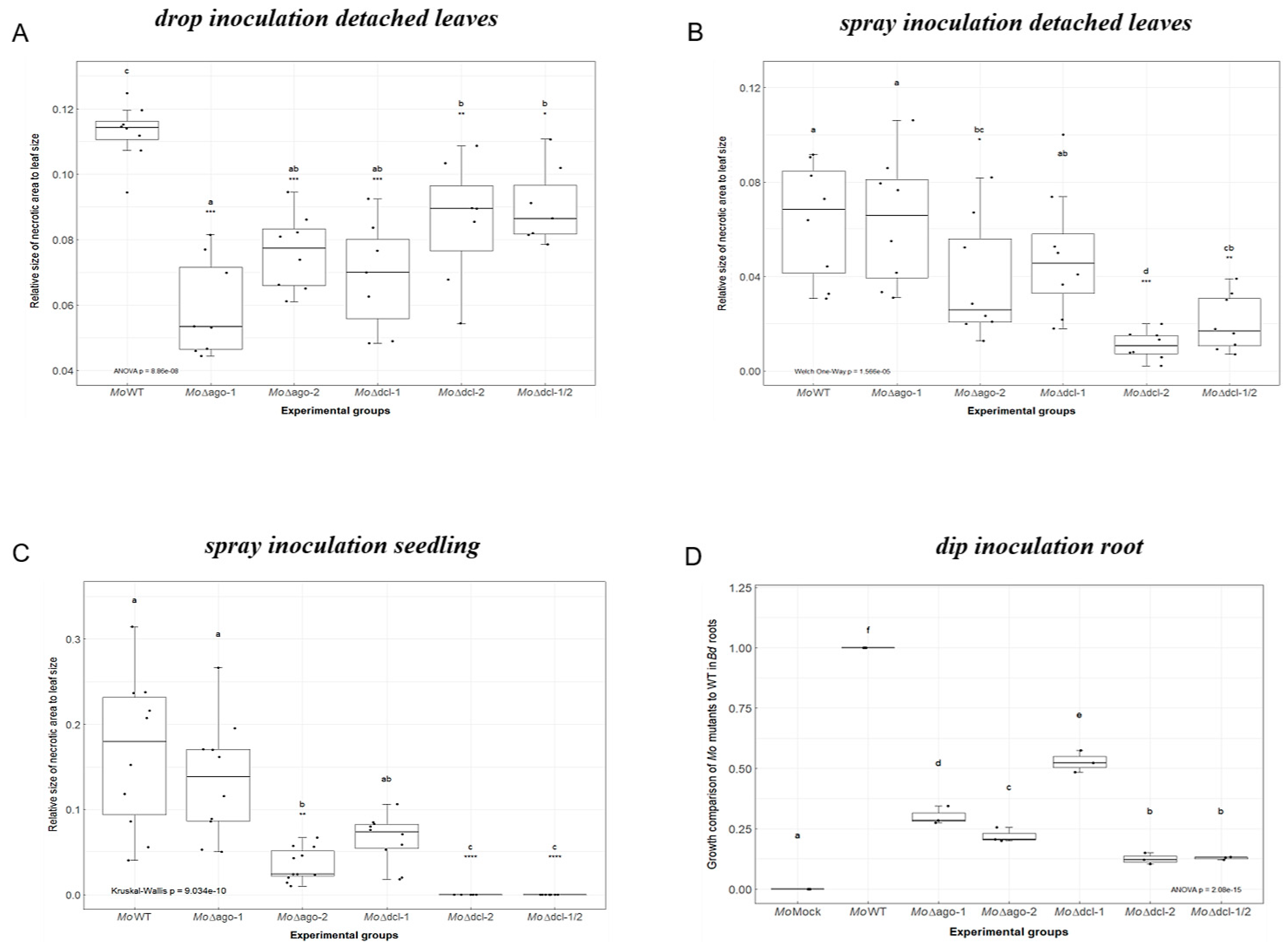
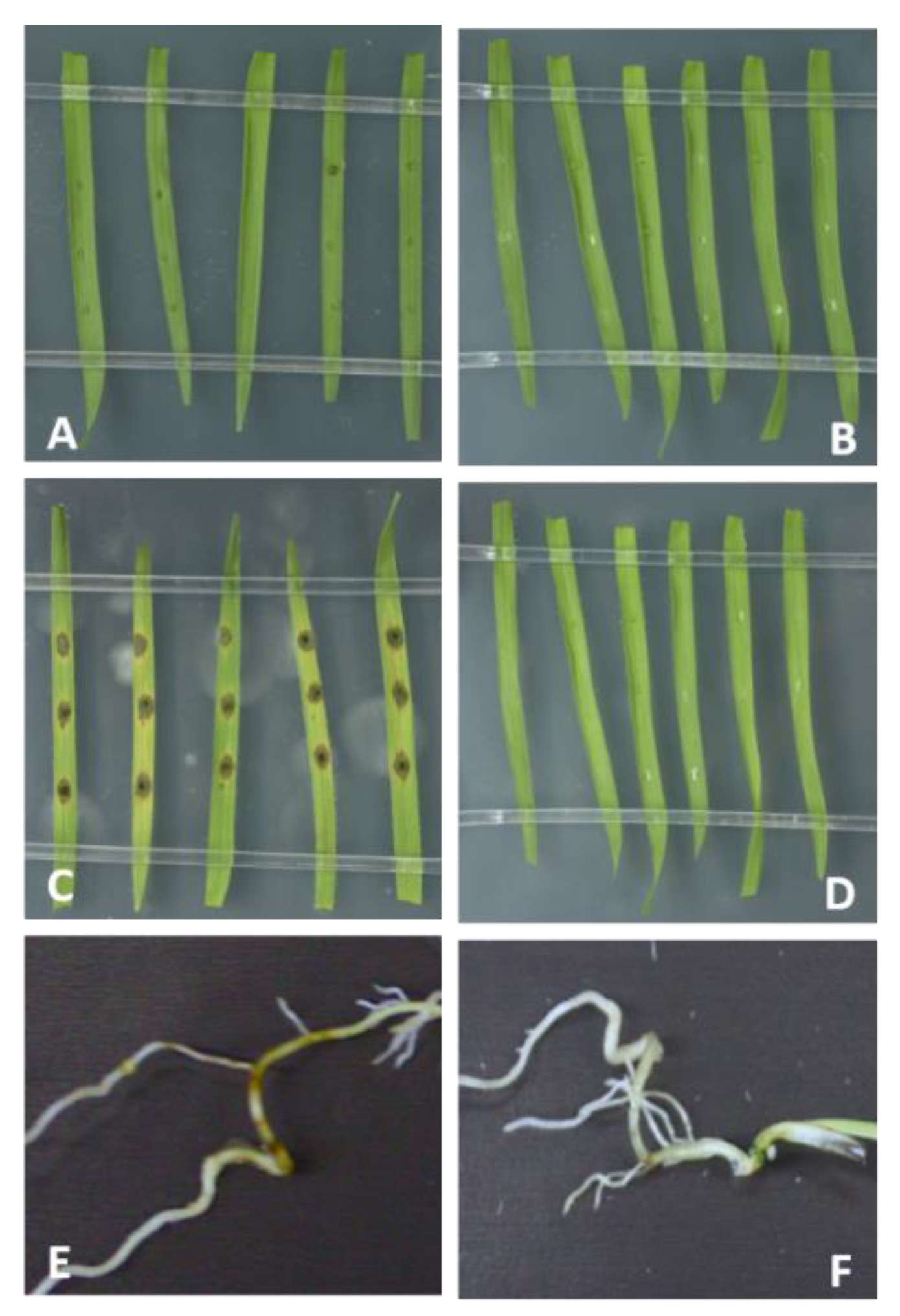
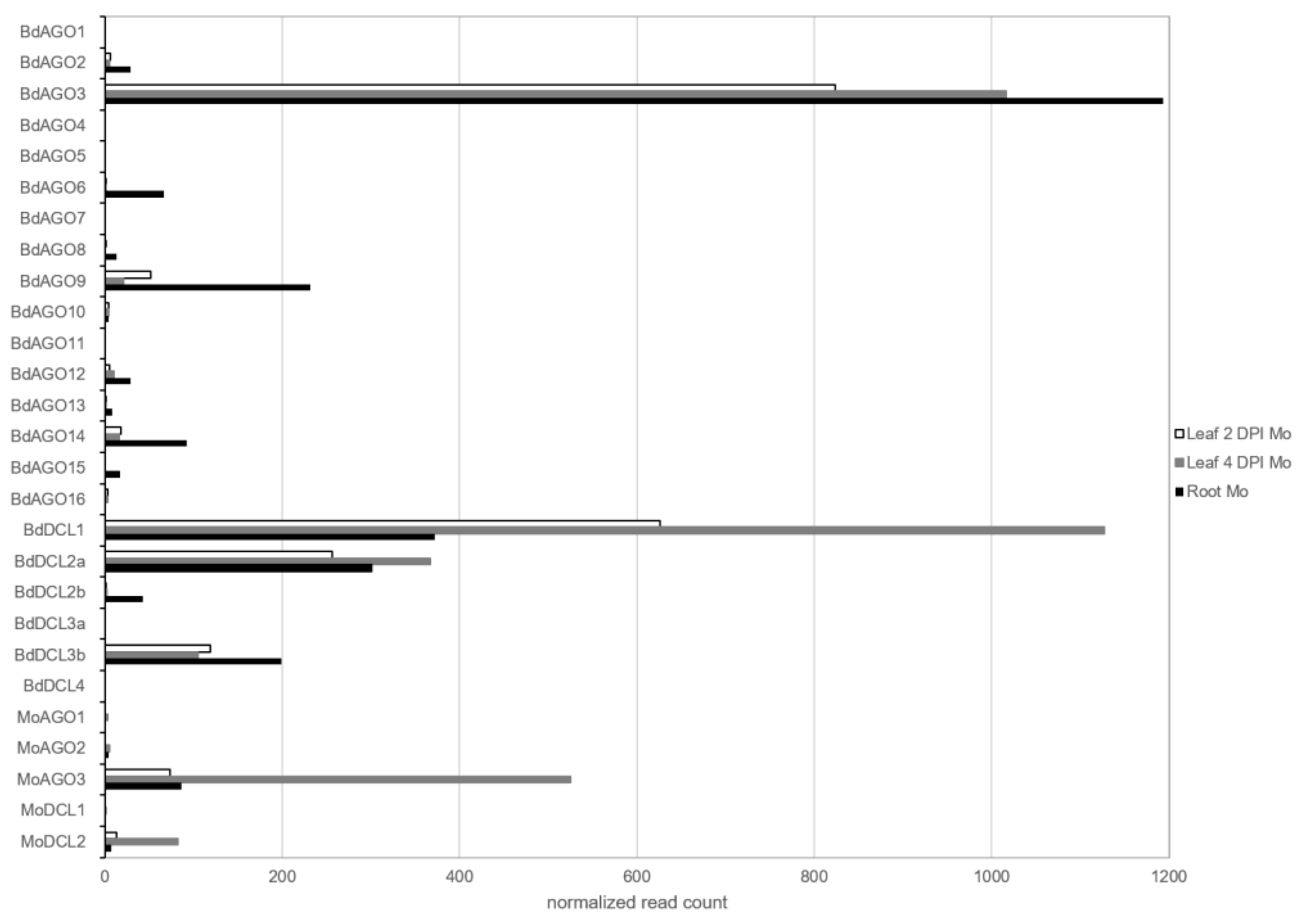
2.1. Requirement for Functional RNAi Genes in the Interaction of Brachypodium distachyon Bd21-3 and Magnaporthe oryzae 70-15
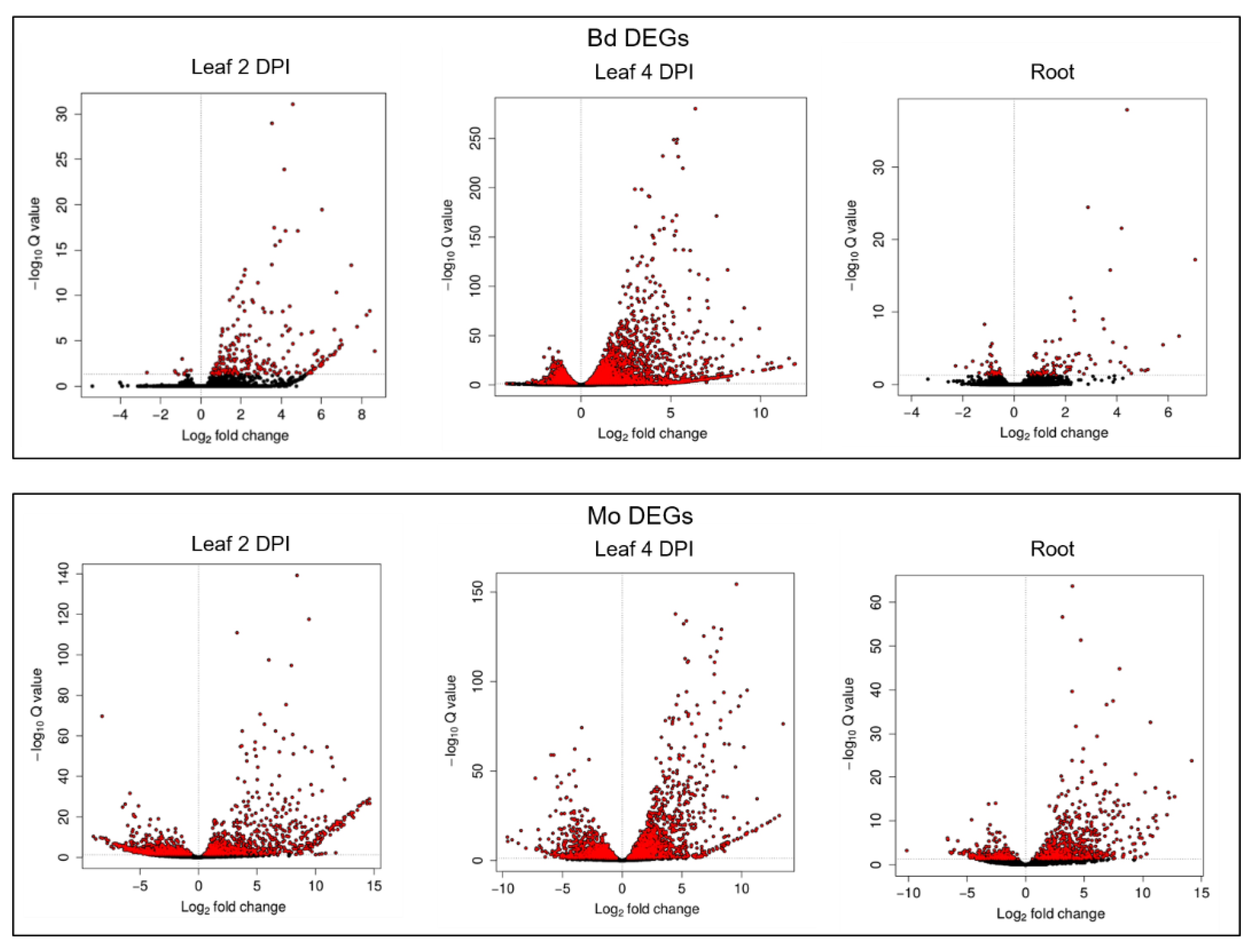
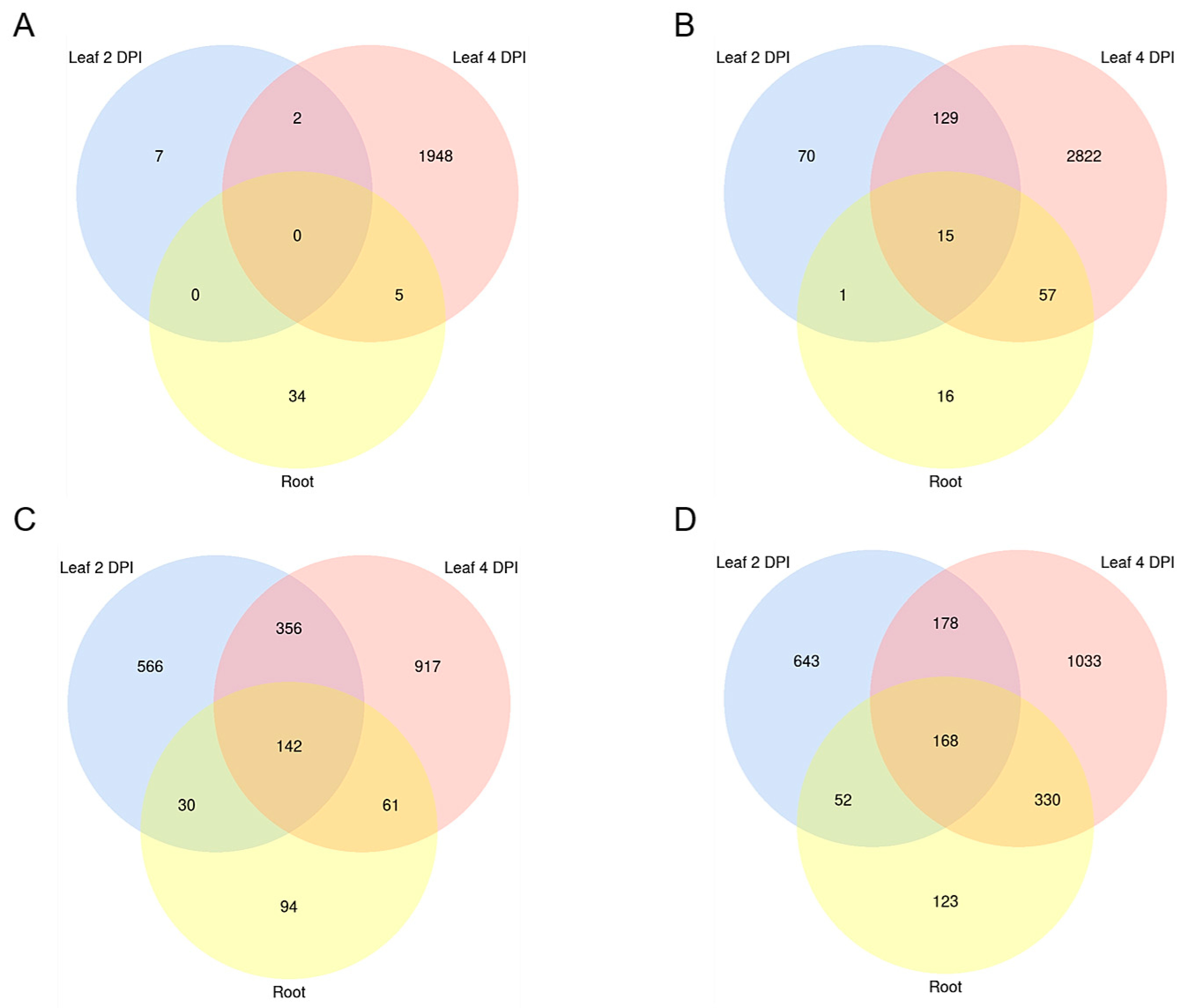
2.2. Differentially Expressed Genes (DEGs) in the Early Stages of Leaf and Root Infections
2.3. Gene Ontology Enrichment (GOE) and Defense-Related Gene Expression in Mo-Infected Bd
2.4. GOE and Gene Expression in Mo during Bd Infection
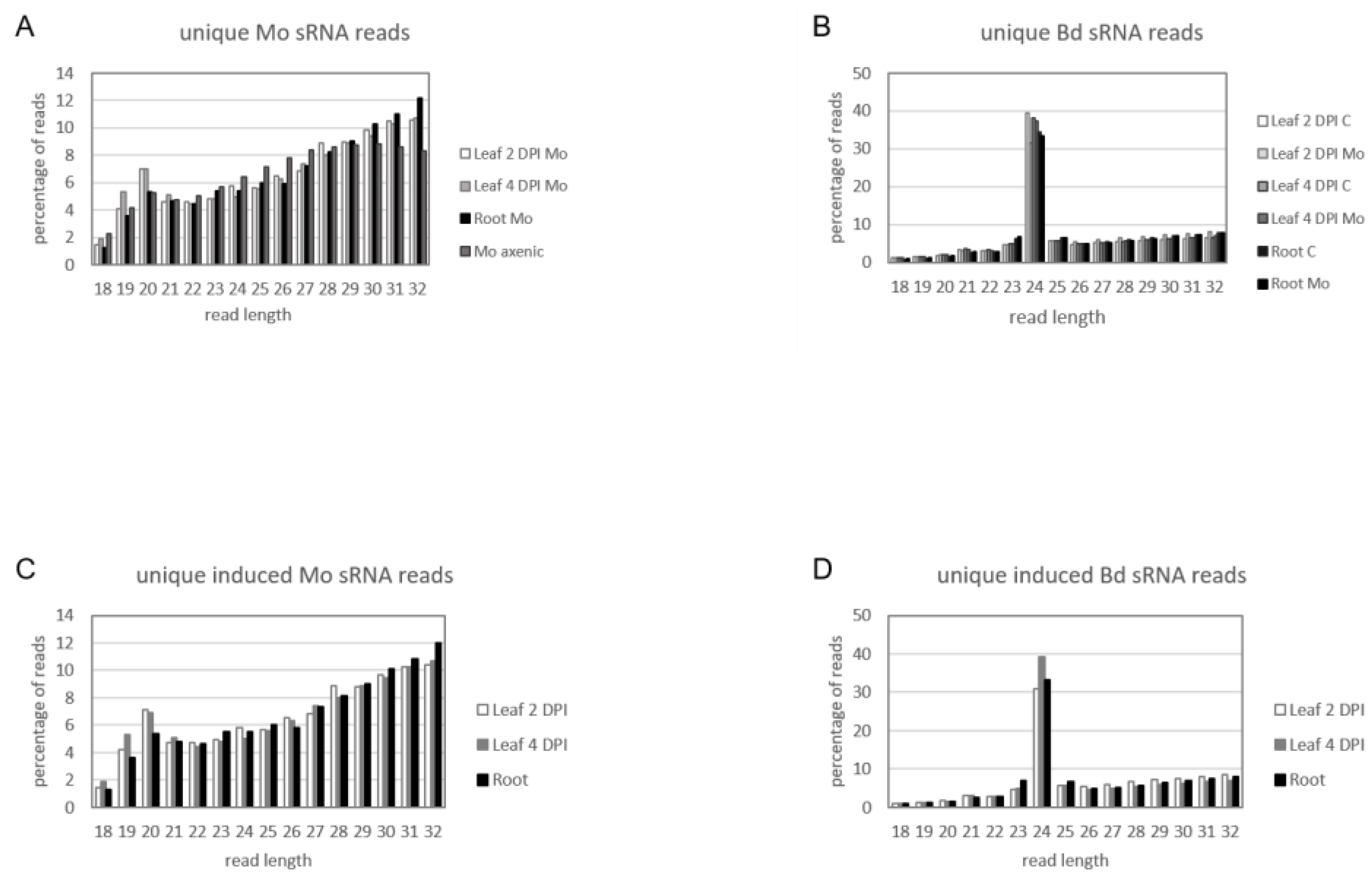
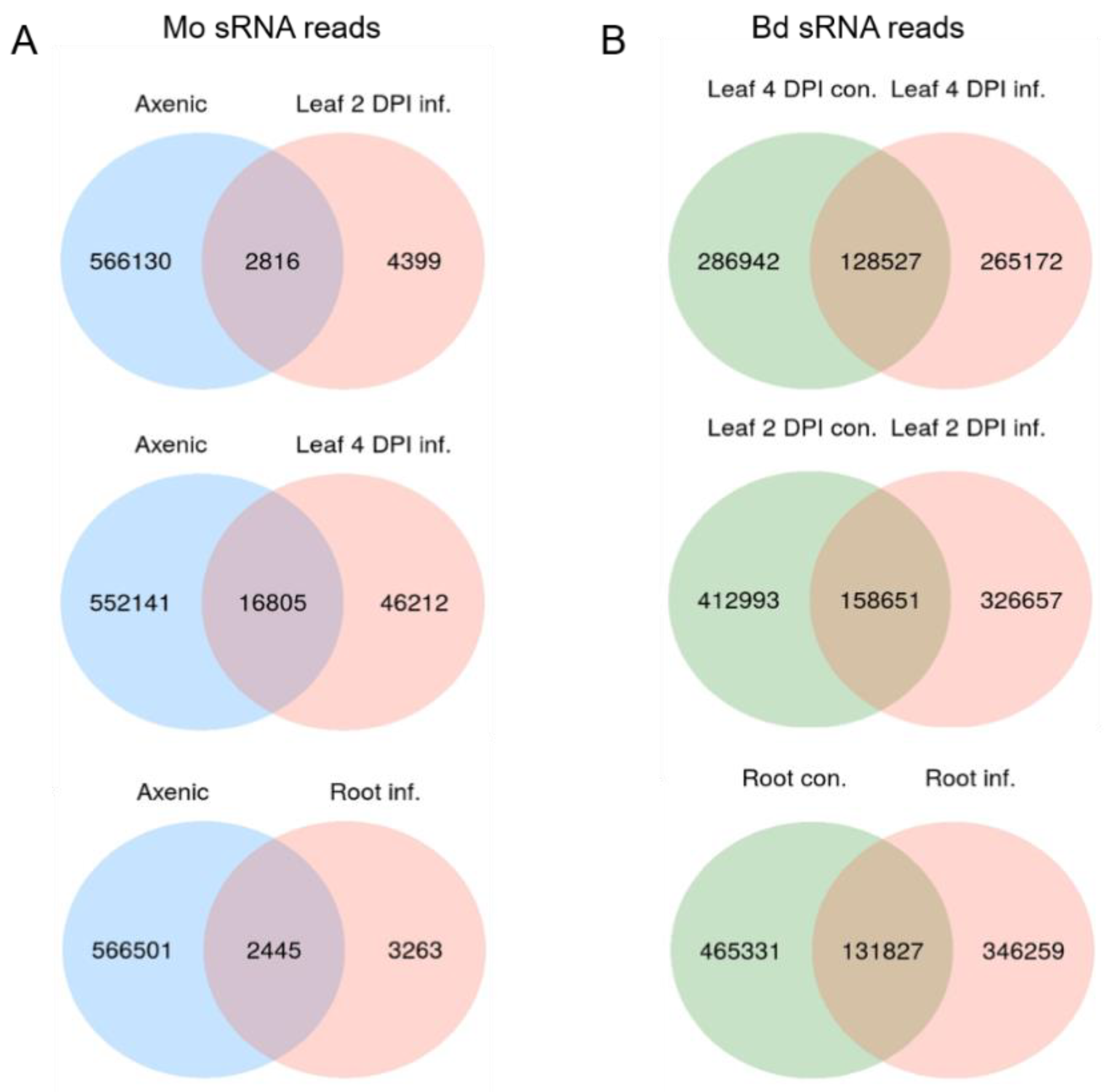
2.5. sRNA Reprogramming in Bd and Mo at Early Infection Stages
2.6. Infection-Related Bd miRNAs
2.7. In Silico Prediction of Mo-sRNA Targeting Bd Transcripts
2.8. In Silico Prediction of Bd-sRNAs Targeting Mo Transcripts
3. Discussion
4. Materials and Methods
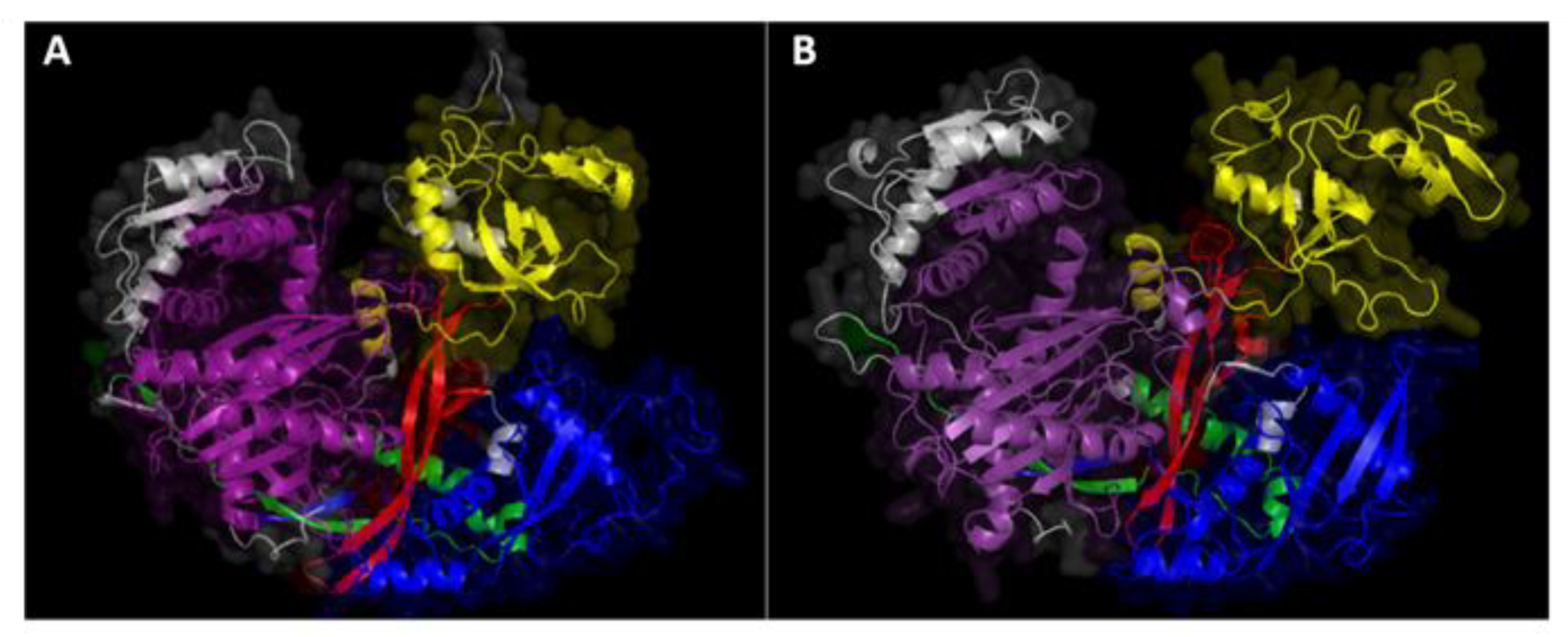
4.1. AGO and DCL Protein Analysis and 3D Structure Modeling
4.2. Mo Mutants Cultivation and Inoculation
4.3. Sample Preparation from Mo–Bd Interaction Sequencing
4.4. RNA Extraction, Library Preparation and Sequencing
4.5. Transcriptome Analysis
4.6. sRNA Analysis, Prediction of Endo- and Cross-Kingdom sRNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012. [Google Scholar] [CrossRef]
- Donofrio, N.M.; Hu, J.; Mitchell, T.K.; Wilson, R.A. Facilitating the fungus: Insights from the genome of the rice blast fungus, Magnaporthe Oryzae. Genom. Plant-Assoc. Fungi Monocot Pathog. 2014. [Google Scholar] [CrossRef]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012. [Google Scholar] [CrossRef] [PubMed]
- Sesma, A.; Osbourn, A.E. The rice leaf blast pathogen undergoes developmental processes typical of root-infecting fungi. Nature 2004. [Google Scholar] [CrossRef]
- Wilson, R.A.; Talbot, N.J. Under pressure: Investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol. 2009, 7, 185–195. [Google Scholar] [CrossRef]
- Nalley, L.; Tsiboe, F.; Durand-Morat, A.; Shew, A.; Thoma, G. Economic and environmental impact of rice blast pathogen (Magnaporthe oryzae) alleviation in the United States. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Asibi, A.E.; Chai, Q.; Coulter, J.A. Rice blast: A disease with implications for global food security. Agronomy 2019, 9, 451. [Google Scholar] [CrossRef]
- Osés-Ruiz, M.; Talbot, N.J. Cell cycle-dependent regulation of plant infection by the rice blast fungus Magnaporthe oryzae. Commun. Integr. Biol. 2017, 10, e1372067. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.J. Having a blast: Exploring the pathogenicity of Magnaporthe grisea. Trends Microbiol. 1995, 3, 9–16. [Google Scholar] [CrossRef]
- Marcel, S.; Paszkowski, U.; Sawers, R.; Oakeley, E.; Angliker, H. Tissue-adapted invasion strategies of the rice blast fungus Magnaporthe oryzae. Plant Cell 2010, 22, 3177–3187. [Google Scholar] [CrossRef]
- Routledge, A.P.M.; Shelley, G.; Smith, J.V.; Talbot, N.J.; Draper, J.; Mur, L.A.J. Magnaporthe grisea interactions with the model grass Brachypodium distachyon closely resemble those with rice (Oryza sativa). Mol. Plant Pathol. 2004. [Google Scholar] [CrossRef]
- Parker, D.; Beckmann, M.; Enot, D.P.; Overy, D.P.; Rios, Z.C.; Gilbert, M.; Talbot, N.; Draper, J. Rice blast infection of Brachypodium distachyon as a model system to study dynamic host/pathogen interactions. Nat. Protoc. 2008. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Powell, J.J.; Schneebeli, K.; Hsia, M.M.; Gardiner, D.M.; Bragg, J.N.; McIntyre, C.L.; Manners, J.M.; Ayliffe, M.; Watt, M.; et al. Brachypodium as an emerging model for cereal-pathogen interactions. Ann. Bot. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Garvin, D.F.; Leong, O.M.; Hayden, D.M. Agrobacterium-mediated transformation and inbred line development in the model grass Brachypodium distachyon. Plant Cell. Tissue Organ Cult. 2006. [Google Scholar] [CrossRef]
- Brutnell, T.P.; Bennetzen, J.L.; Vogel, J.P. Brachypodium distachyon and Setaria viridis: Model Genetic Systems for the Grasses. Annu. Rev. Plant Biol. 2015, 66, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Molnar, A.; Melnyk, C.W.; Bassett, A.; Hardcastle, T.J.; Dunn, R.; Baulcombe, D.C. Small silencing RNAs in plants are mobile and direct epigenetic modification in recipient cells. Science 2010, 328, 872–875. [Google Scholar] [CrossRef]
- Borges, F.; Martienssen, R.A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 2015. [Google Scholar] [CrossRef]
- Kehr, J.; Kragler, F. Long distance RNA movement. New Phytol. 2018. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998. [Google Scholar] [CrossRef]
- Vaucheret, H.; Fagard, M. Transcriptional gene silencing in plants: Targets, inducers and regulators. Trends Genet. 2001. [Google Scholar] [CrossRef]
- Castel, S.E.; Martienssen, R.A. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D. RNA silencing in plants. Nature 2004. [Google Scholar] [CrossRef] [PubMed]
- Vaucheret, H.; Vazquez, F.; Crété, P.; Bartel, D.P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004. [Google Scholar] [CrossRef] [PubMed]
- Kadotani, N.; Nakayashiki, H.; Tosa, Y.; Mayama, S. RNA silencing in the phytopathogenic fungus Magnaporthe oryzae. Mol. Plant-Microbe Interact. 2003. [Google Scholar] [CrossRef]
- Murphy, D.; Dancis, B.; Brown, J.R. The evolution of core proteins involved in microRNA biogenesis. BMC Evol. Biol. 2008. [Google Scholar] [CrossRef]
- Raman, V.; Simon, S.A.; Demirci, F.; Nakano, M.; Meyers, B.C.; Donofrio, N.M. Small RNA functions are required for growth and development of magnaporthe oryzae. Mol. Plant-Microbe Interact. 2017, 30, 517–530. [Google Scholar] [CrossRef]
- Nunes, C.C.; Gowda, M.; Sailsbery, J.; Xue, M.; Chen, F.; Brown, D.E.; Oh, Y.Y.; Mitchell, T.K.; Dean, R.A. Diverse and tissue-enriched small RNAs in the plant pathogenic fungus, Magnaporthe oryzae. BMC Genom. 2011, 12, 1–20. [Google Scholar] [CrossRef]
- Wei, B.; Cai, T.; Zhang, R.; Li, A.; Huo, N.; Li, S.; Gu, Y.Q.; Vogel, J.; Jia, J.; Qi, Y.; et al. Novel microRNAs uncovered by deep sequencing of small RNA transcriptomes in bread wheat (Triticum aestivum L.) and Brachypodium distachyon (L.) Beauv. Funct. Integr. Genom. 2009. [Google Scholar] [CrossRef]
- Wang, H.L.V.; Dinwiddie, B.L.; Lee, H.; Chekanova, J.A. Stress-induced endogenous siRNAs targeting regulatory intron sequences in Brachypodium. RNA 2015. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.; Huan, Q.; Chong, K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genom. 2009, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.R.; Schmidt, S.A.; Park, S.; Jeong, D.H.; Accerbi, M.; Green, P.J. Analysis of Brachypodium miRNA targets: Evidence for diverse control during stress and conservation in bioenergy crops. BMC Genom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, E.; Verelst, W.; Horner, D.S.; Gianfranceschi, L.; Piccolo, V.; Inzé, D.; Pè, M.E.; Mica, E. Addressing the role of micrornas in reprogramming leaf growth during drought stress in brachypodium distachyon. Mol. Plant 2013, 6, 423–443. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Schmidt, S.A.; Rymarquis, L.A.; Park, S.; Ganssmann, M.; German, M.A.; Accerbi, M.; Zhai, J.; Fahlgren, N.; Fox, S.E.; et al. Parallel analysis of rna ends enhances global investigation of micrornas and target rnas of brachypodium distachyon. Genome Biol. 2013, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Šečić, E.; Zanini, S.; Kogel, K.H. Further Elucidation of the Argonaute and Dicer Protein Families in the Model Grass Species Brachypodium distachyon. Front. Plant Sci. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Buck, A.H.; Coakley, G.; Simbari, F.; McSorley, H.J.; Quintana, J.F.; Le Bihan, T.; Kumar, S.; Abreu-Goodger, C.; Lear, M.; Harcus, Y.; et al. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Lamonte, G.; Philip, N.; Reardon, J.; Lacsina, J.R.; Majoros, W.; Chapman, L.; Thornburg, C.D.; Telen, M.J.; Ohler, U.; Nicchitta, C.V.; et al. Translocation of sickle cell erythrocyte MicroRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe 2012. [Google Scholar] [CrossRef]
- Garcia-Silva, M.R.; Cura Das Neves, R.F.; Cabrera-Cabrera, F.; Sanguinetti, J.; Medeiros, L.C.; Robello, C.; Naya, H.; Fernandez-Calero, T.; Souto-Padron, T.; De Souza, W.; et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol. Res. 2014. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016. [Google Scholar] [CrossRef]
- Cai, Q.; Qiao, L.; Wang, M.; He, B.; Lin, F.M.; Palmquist, J.; Huang, S.D.; Jin, H. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.; Zhang, Z.; Kaloshian, I.; Huang, H.D.; Jin, H. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiberg, A.; Dellota, E.; Yamane, D.; Jin, H. Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 2017. [Google Scholar] [CrossRef]
- Zanini, S.; Šečić, E.; Jelonek, L.; Kogel, K.H. A bioinformatics pipeline for the analysis and target prediction of rna effectors in bidirectional communication during plant–microbe interactions. Front. Plant Sci. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Kershaw, M.J.; Talbot, N.J. Genome-wide functional analysis reveals that infection-associated fungal autophagy is necessary for rice blast disease. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.C.; Dagdas, Y.F.; Gupta, Y.K.; Mentlak, T.A.; Yi, M.; Martinez-Rocha, A.L.; Saitoh, H.; Terauchi, R.; Talbot, N.J.; Valent, B. Two distinct secretion systems facilitate tissue invasion by the rice blast fungus Magnaporthe oryzae. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Cenik, E.S.; Zamore, P.D. Argonaute proteins. Curr. Biol. 2011, 21, R446–R449. [Google Scholar] [CrossRef]
- Song, J.J.; Smith, S.K.; Hannon, G.J.; Joshua-Tor, L. Crystal structure of argonaute and its implications for RISC slicer activity. Science 2004, 305, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niu, D.D.; Carbonell, A.; Wang, A.; Lee, A.; Tun, V.; Wang, Z.; Carrington, J.C.; Chang, C.E.A.; Jin, H. ARGONAUTE PIWI domain and microRNA duplex structure regulate small RNA sorting in Arabidopsis. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Raman, V.; Simon, S.A.; Romag, A.; Demirci, F.; Mathioni, S.M.; Zhai, J.; Meyers, B.C.; Donofrio, N.M. Physiological stressors and invasive plant infections alter the small RNA transcriptome of the rice blast fungus, Magnaporthe oryzae. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Kadotani, N.; Murata, T.; Nguyen, B.Q.; Adachi, Y.; Nakayashiki, H. Transcriptional control and protein specialization have roles in the functional diversification of two dicer-like proteins in Magnaporthe oryzae. Genetics 2008. [Google Scholar] [CrossRef] [PubMed]
- Chuang, R.Y.; Weaver, P.L.; Liu, Z.; Chang, T.H. Requirements of the DEAD-box protein Ded1p for messenger RNA translation. Science 1997, 275, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Spassov, D.S.; Jurecic, R. The PUF Family of RNA-binding Proteins: Does Evolutionarily Conserved Structure Equal Conserved Function? IUBMB Life 2003, 55, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Sabnam, N.; Roy Barman, S. WISH, a novel CFEM GPCR is indispensable for surface sensing, asexual and pathogenic differentiation in rice blast fungus. Fungal Genet. Biol. 2017, 105, 37–51. [Google Scholar] [CrossRef]
- Egan, M.J.; Wang, Z.Y.; Jones, M.A.; Smirnoff, N.; Talbot, N.J. Generation of reactive oxygen species by fungal NADPH oxidases is required for rice blast disease. Proc. Natl. Acad. Sci. USA 2007, 104, 11772–11777. [Google Scholar] [CrossRef]
- Galhano, R.; Illana, A.; Ryder, L.S.; Rodríguez-Romero, J.; Demuez, M.; Badaruddin, M.; Martinez-Rocha, A.L.; Soanes, D.M.; Studholme, D.J.; Talbot, N.J.; et al. Tpc1 is an important Zn(II)2Cys6transcriptional regulator required for polarized growth and virulence in the rice blast fungus. PLoS Pathog. 2017, 13, e1006516. [Google Scholar] [CrossRef]
- Choi, J.; Kim, K.S.; Rho, H.S.; Lee, Y.H. Differential roles of the phospholipase C genes in fungal development and pathogenicity of Magnaporthe oryzae. Fungal Genet. Biol. 2011, 48, 445–455. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chai, R.; Qiu, H.; Jiang, H.; Mao, X.; Wang, Y.; Liu, F.; Sun, G. An S-(hydroxymethyl)glutathione dehydrogenase is involved in conidiation and full virulence in the rice blast fungus Magnaporthe oryzae. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Tucker, S.L.; Besi, M.I.; Galhano, R.; Franceschetti, M.; Goetz, S.; Lenhert, S.; Osbourn, A.; Sesma, A. Common genetic pathways regulate organ-specific infection-related development in the rice blast fungus. Plant Cell 2010, 22, 953–972. [Google Scholar] [CrossRef]
- Kawano, T. Roles of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell Rep. 2003. [Google Scholar] [CrossRef]
- Powell, J.J.; Carere, J.; Sablok, G.; Fitzgerald, T.L.; Stiller, J.; Colgrave, M.L.; Gardiner, D.M.; Manners, J.M.; Vogel, J.P.; Henry, R.J.; et al. Transcriptome analysis of Brachypodium during fungal pathogen infection reveals both shared and distinct defense responses with wheat. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-Y.; Wang, G.-L.; Chen, L.L.; Kim, H.-S.; Pi, L.-Y.; Holsten, T.; Gardner, J.; Wang, B.; Zhai, W.-X.; Zhu, L.-H.; et al. A receptor Kinase-Like Protein Encoded by the Rice Disease Ressitance Gene, Xa21. Science 1995, 270, 1804–1806. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmüller, R. Wrky transcription factors jack of many trades in plants. Plant Signal. Behav. 2014. [Google Scholar] [CrossRef] [PubMed]
- Murozuka, E.; Massange-Sánchez, J.A.; Nielsen, K.; Gregersen, P.L.; Braumann, I. Genome wide characterization of barley NAC transcription factors enables the identification of grain-specific transcription factors exclusive for the Poaceae family of monocotyledonous plants. PLoS ONE 2018. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.F.A.; Ali, N.M.; Ismail, I.; Murad, A.M.A. Analysis of miRNAs targeting transcription factors in Persicaria minor induced by Fusarium oxysporum. AIP Conf. Proc. 2016. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Wang, Z.; Guo, X.; Wang, F.; Xia, X.J.; Zhou, J.; Shi, K.; Yu, J.Q.; Zhou, Y.H. Microarray and genetic analysis reveals that csa-miR159b plays a critical role in abscisic acid-mediated heat tolerance in grafted cucumber plants. Plant Cell Environ. 2016, 39, 1790–1804. [Google Scholar] [CrossRef]
- Liu, X.H.; Lu, J.P.; Zhang, L.; Dong, B.; Min, H.; Lin, F.C. Involvement of a Magnaporthe grisea serine/threonine kinase gene, MgATG1, in Appressorium turgor and pathogenesis. Eukaryot. Cell 2007, 6, 997–1005. [Google Scholar] [CrossRef]
- Park, C.H.; Chen, S.; Shirsekar, G.; Zhou, B.; Khang, C.H.; Songkumarn, P.; Afzal, A.J.; Ning, Y.; Wang, R.; Bellizzi, M.; et al. The magnaporthe oryzae effector avrpiz-t targets the RING E3 ubiquitin ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in ricesW OA. Plant Cell 2012. [Google Scholar] [CrossRef]
- Bossche, H.V.; Lauwers, W.; Willemsens, G.; Marichal, P.; Cornelissen, F.; Cools, W. Molecular basis for the antimycotic and antibacterial activity of N-substituted imidazoles and triazoles: The inhibition of isoprenoid biosynthesis. Pestic. Sci. 1984. [Google Scholar] [CrossRef]
- Koch, A.; Kumar, N.; Weber, L.; Keller, H.; Imani, J.; Kogel, K.H. Host-induced gene silencing of cytochrome P450 lanosterol C14α-demethylase-encoding genes confers strong resistance to Fusarium species. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Biedenkopf, D.; Furch, A.; Weber, L.; Rossbach, O.; Abdellatef, E.; Linicus, L.; Johannsmeier, J.; Jelonek, L.; Goesmann, A.; et al. An RNAi-Based Control of Fusarium graminearum Infections Through Spraying of Long dsRNAs Involves a Plant Passage and Is Controlled by the Fungal Silencing Machinery. PLoS Pathog. 2016. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Höfle, L.; Werner, B.T.; Imani, J.; Schmidt, A.; Jelonek, L.; Kogel, K.H. SIGS vs. HIGS: A study on the efficacy of two dsRNA delivery strategies to silence Fusarium FgCYP51 genes in infected host and non-host plants. Mol. Plant Pathol. 2019. [Google Scholar] [CrossRef]
- Margis, R.; Fusaro, A.F.; Smith, N.A.; Curtin, S.J.; Watson, J.M.; Finnegan, E.J.; Waterhouse, P.M. The evolution and diversification of Dicers in plants. FEBS Lett. 2006. [Google Scholar] [CrossRef]
- Feller, A.; MacHemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011. [Google Scholar] [CrossRef]
- Kang, J.; Park, J.; Choi, H.; Burla, B.; Kretzschmar, T.; Lee, Y.; Martinoia, E. Plant ABC Transporters. Arab. Book/Am. Soc. Plant Biol. 2011, 9, e0153. [Google Scholar] [CrossRef]
- Afzal, Z.; Howton, T.C.; Sun, Y.; Mukhtar, M.S. The roles of aquaporins in plant stress responses. J. Dev. Biol. 2016, 9. [Google Scholar] [CrossRef]
- Riley, K.J.; Yario, T.A.; Steitz, J.A. Association of argonaute proteins and microRNAs can occur after cell lysis. RNA 2012. [Google Scholar] [CrossRef]
- Carbonell, A. Plant ARGONAUTES: Features, functions, and unknowns. Methods Mol. Biol. 2017. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.P.; Leroy, C.; Sander, C. MView: A web-compatible database search or multiple alignment viewer. Bioinformatics 1998. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Mei, Q.; Chen, M. PSI: A Comprehensive and Integrative Approach for Accurate Plant Subcellular Localization Prediction. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Bioinforma 1992. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System, version 1.8; Schrödinger LLC: New York, NY, USA, 2002. [Google Scholar]
- Vogel, J.; Hill, T. High-efficiency Agrobacterium-mediated transformation of Brachypodium distachyon inbred line Bd21-3. Plant Cell Rep. 2008. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol. Plant. 1962. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. [Google Scholar] [CrossRef]
- Kolde, R. Package ‘pheatmap’. Bioconductor 2015, 7, 790. [Google Scholar]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005. [Google Scholar] [CrossRef]
- Andrews, S. FastQC:—A quality control tool for high throughput sequence data. Babraham Bioinforma 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 11 January 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011. [Google Scholar] [CrossRef]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinforma 2010. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011. [Google Scholar] [CrossRef]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved placement of multi-mapping small RNAs. G3 Genes Genomes Genet. 2016. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.K.; Winnenburg, R.; Urban, M.; Rawlings, C.; Koehler, J.; Hammond-Kosack, K.E. The Pathogen-Host Interactions Database (PHI-base) provides insights into generic and novel themes of pathogenicity. Mol. Plant-Microbe Interact. 2006. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Setup | Total Mo Genes (Up) | Total Mo Genes (Down) | Total Bd Genes (Up) | Total Bd Genes (Down) |
|---|---|---|---|---|
| Leaf 2 DPI | 1041 | 1094 | 224 | 9 |
| Leaf 4 DPI | 1710 | 1476 | 3023 | 1955 |
| Root | 673 | 327 | 89 | 39 |
| Gene | Description | Log2FC- Leaf 2 DPI | Log2FC- Leaf 4 DPI | Log2FC- Root |
|---|---|---|---|---|
| BdiBd21-3.3G0465100 | ABC transporter a family member 2-like | 1.15 | 3.38 | 0.79 |
| BdiBd21-3.3G0464900 | ABC transporter a family member 7-like | 2.64 | 0.71 | |
| BdiBd21-3.2G0605400 | ABC transporter b family member 4-like | 2.15 | 5.08 | |
| BdiBd21-3.2G0550500 | Pleiotropic drug resistance protein 3-like | 1.78 | 6.82 | 1.51 |
| BdiBd21-3.5G0159100 | Anthranilate synthase component ii | 0.76 | 2.75 | 0.88 |
| BdiBd21-3.3G0344500 | Chitinase 1 | 2.70 | ||
| BdiBd21-3.2G0371800 | Cytochrome p450 71c4 | 1.32 | 2.12 | |
| BdiBd21-3.1G0952300 | Disease resistance response | 2.07 | ||
| BdiBd21-3.3G0144800 | Protein kinase xa21 | 1.77 | 2.41 | 1.24 |
| BdiBd21-3.2G0545400 | LRR receptor-like serine threonine-protein kinase gso1 | 1.59 | 1.79 | |
| BdiBd21-3.2G0632400 | Receptor-like protein kinase hsl1-like | 3.15 | ||
| BdiBd21-3.1G0713100 | Proton-coupled amino acid transporter 3-like | 0.85 | 1.97 | 0.97 |
| BdiBd21-3.2G0114800 | Pathogenesis related protein | 1.34 | 1.25 | |
| BdiBd21-3.1G0165000 | Pathogenesis-related protein 1 | 6.06 | 8.19 | |
| BdiBd21-3.4G0068000 | Pathogenesis-related protein 10 | 3.47 | 3.75 | |
| BdiBd21-3.4G0043000 | Pathogenesis-related protein 5 | 2.91 | ||
| BdiBd21-3.1G0772600 | Pathogenesis-related protein class i | 5.29 | 4.47 | |
| BdiBd21-3.1G0772700 | Pathogenesis-related protein prb1-2-like | 3.41 | 6.42 | |
| BdiBd21-3.3G0422200 | PR3-like 1 | 2.63 | ||
| BdiBd21-3.3G0639500 | PR3-like 2 | 3.96 | ||
| BdiBd21-3.4G0025400 | PR5-like | 1.37 | ||
| BdiBd21-3.2G0600500 | PR5-like | 2.28 | ||
| BdiBd21-3.1G0875700 | Pathogenesis-related protein Bet v I family | 2.14 | ||
| BdiBd21-3.1G0054700 | NAC1 transcription factor | 2.49 | ||
| BdiBd21-3.3G0652700 | MYB-related protein myb4-like | 3.33 | ||
| BdiBd21-3.2G0688100 | Probable WRKY transcription factor 33-like | 2.69 | ||
| BdiBd21-3.2G0615100 | Probable WRKY transcription factor 51-like | 1.45 | 3.44 | |
| BdiBd21-3.3G0669400 | Ethylene-responsive transcription factor 1a | 2.16 | ||
| BdiBd21-3.4G0171000 | Multicopper oxidase family expressed | 4.35 | 9.93 | 2.35 |
| BdiBd21-3.4G0387000 | Cationic peroxidase spc4-like | 3.17 | 3.72 | |
| BdiBd21-3.1G0233900 | Peroxidase | 2.11 | ||
| BdiBd21-3.2G0563800 | Secologanin synthase-like | 0.57 | 0.54 | 1.08 |
| Gene Stable ID | Description | Log2FC- Leaf 2 DPI | Log2FC- Leaf 4 DPI | Log2FC- Root |
|---|---|---|---|---|
| MGG_00750 | Cytochrome b-245 heavychain subunit beta | 2.23 | 2.12 | 2.80 |
| MGG_01081 | Peroxin 14/17 | 1.05 | ||
| MGG_01092 | Homocitrate synthase | 1.35 | ||
| MGG_01748 | Putative protease | 1.28 | ||
| MGG_02074 | Potassium/sodium efflux P-type ATPase | 1.28 | 2.55 | |
| MGG_03356 | Ricin B lectin:Parallel beta-helix | 7.08 | 5.06 | |
| MGG_04202 | MAS3 protein | 2.27 | 2.75 | |
| MGG_04212 | L-ornithine 5-monooxygenase | 2.81 | 3.46 | 3.17 |
| MGG_04301 | Pwl2 protein (PWL2) gene | 8.53 | ||
| MGG_04545 | Cytochrome c peroxidase, mitochondrial | 3.26 | 0.95 | |
| MGG_06011 | S-(Hydroxymethyl)glutathione dehydrogenase | 2.53 | ||
| MGG_06648 | Hsp70 (LHS1) gene | 1.25 | ||
| MGG_07514 | 3-oxoacyl-[acyl-carrier-protein] reductase | 1.49 | ||
| MGG_07971 | Calcium-transporting ATPase 1 | 1.80 | ||
| MGG_08315 | 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 | 8.83 | 7.94 | |
| MGG_08409 | Cellulose-growth-specific protein | 3.78 | ||
| MGG_09022 | Transmembrane CFEM domain-containing protein | 5.41 | 7.42 | 7.88 |
| MGG_09559 | Autophagy-related protein 9 | 1.07 | ||
| MGG_09956 | PRO41 protein | 1.93 | 1.91 | 2.62 |
| MGG_10097 | Intracellular hyphae protein 1 | 5.42 | ||
| MGG_10510 | Ribonuclease T2 | 3.90 | ||
| MGG_10730 | Potassium/sodium efflux P-type ATPase | 4.75 | ||
| MGG_11882 | Sensor protein zRas | 1.60 | 3.15 | |
| MGG_11899 | SH3 domain-containing protein | 1.93 | 1.52 | |
| MGG_15370 | Metalloproteinase | 11.86 | ||
| MGG_15972 | AVR-pik/pikm/pikp | 14.63 | 6.33 |
| Setup | Cluster Number | miRNA | Cluster RPM | Mature miRNA Name |
|---|---|---|---|---|
| Leaf 2 DPI | 3086 | Bdi-miR159b | 118.27 | miR159b-3p.2 |
| miR159b-3p.1 | ||||
| 3421 | Bdi-MIR531 | 15.03 | MIR531 | |
| 6495 | Bdi-MIR156b | 2.61 | miR156b-3p | |
| 7687 | Bdi-miR9481b | 19.12 | miR9481b -5p | |
| miR9481b-3p | ||||
| Leaf 4 DPI | 7744 | Bdi-MIR156h | 13.79 | MIR156h-5p |
| 3312 | Bdi-MIR159b | 184.31 | miR159b-3p.1 | |
| miR159b-3p.2 | ||||
| miR159b-5p.1 | ||||
| miR159b-5p.2 | ||||
| 3162 | Bdi-MIR171d | 0.58 | MIR171d-3p | |
| 7470 | Bdi-MIR529 | 30.80 | MIR529-5p | |
| 2384 | Bdi-miR7723a | 13.02 | miR7723a-3p | |
| 10592 | Bdi-MIR156d | 8.31 | MIR156d-5p | |
| Root | 8229 | Bdi-MIR156i | 2.87 | MIR156i-5p |
| MIR156i -3p | ||||
| 5121 | Bdi-MIR168 | 310.27 | MIR168-5p | |
| 9081 | Bdi-MIR156d | 1.15 | miR156d-5p | |
| 4330 | bdi-MIR9484 | 3.90 | MIR9484 |
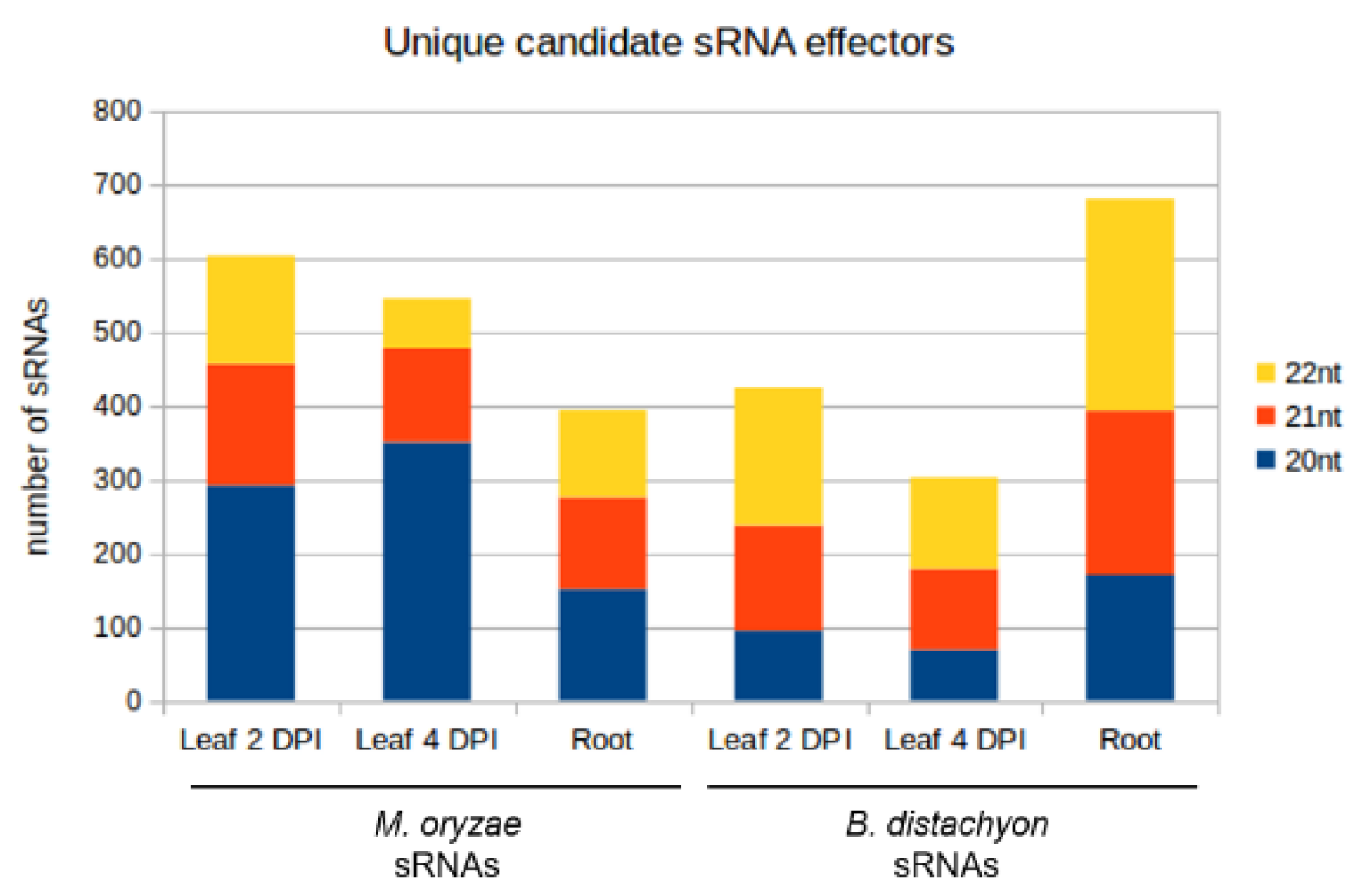
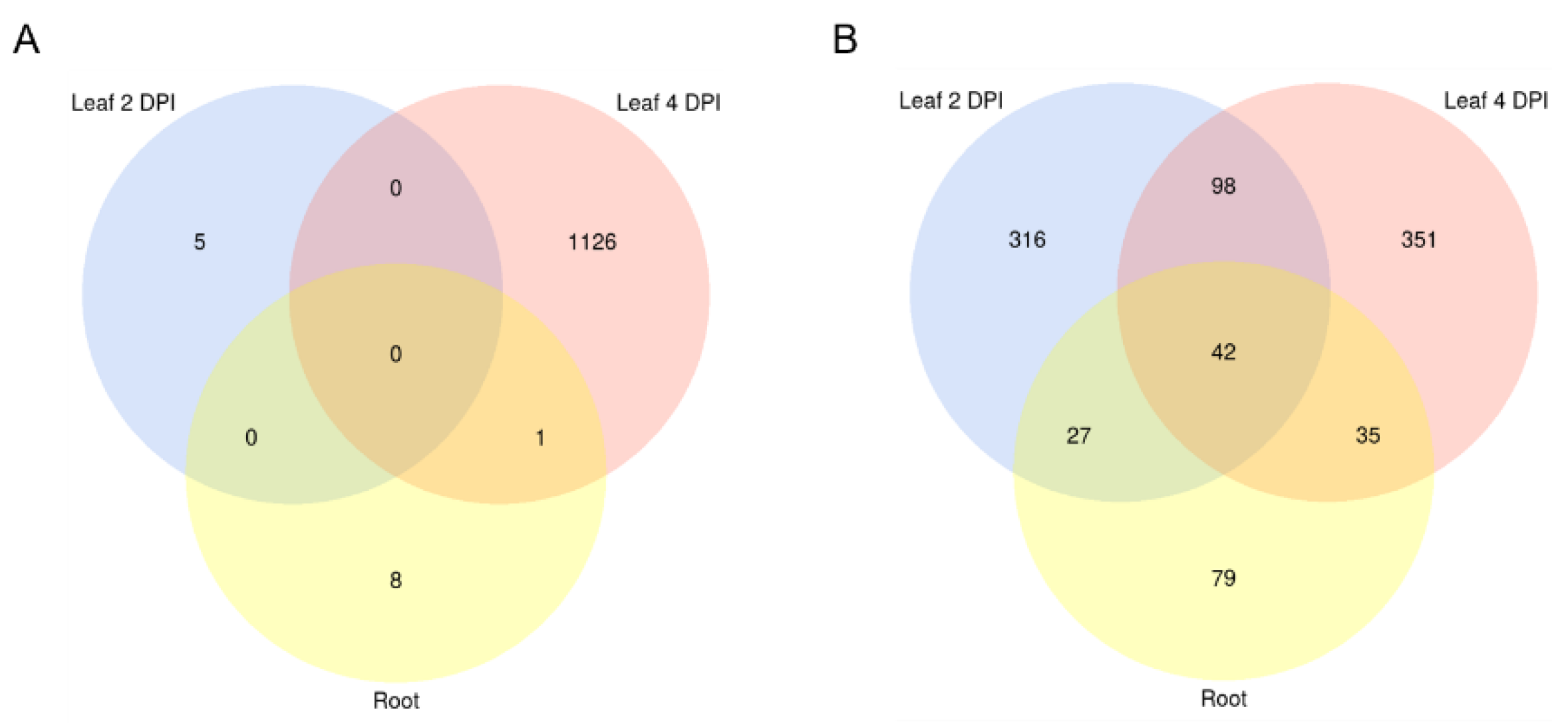
| Setup | Number of sRNA Candidates | Number of sRNA Candidates with Downregulated Targets | Number of Targets Predicted | Number of Targets Downregulated | ||
|---|---|---|---|---|---|---|
| Mo sRNAs | Leaf 2 DPI | 604 | 7 | 25106 | 5 | Bd mRNAs |
| Leaf 4 DPI | 546 | 490 | 25415 | 1128 | ||
| Root | 394 | 14 | 17335 | 10 | ||
| Bd sRNAs | Leaf 2 DPI | 424 | 314 | 4621 | 484 | Mo mRNAs |
| Leaf 4 DPI | 302 | 236 | 4431 | 527 | ||
| Root | 681 | 263 | 6730 | 183 |
| Setup | sRNA | RPM I | Target | Log2FC | Description |
|---|---|---|---|---|---|
| Leaf 2 DPI | TTTCGACGCTGCCCTGACTT | 31.9 | BdiBd21-3.1G0045900.1 | −0.49 | Bowman–Birk type trypsin inhibitor |
| TTTCGACGCTGCCCTGACTT | 31.9 | BdiBd21-3.4G0610700.1 | −1.25 | probable apyrase 3 | |
| GGTTATCATCGTCCCAGCCC | 15.9 | BdiBd21-3.4G0347500.1 | −0.70 | abscisic stress-ripening protein 3 | |
| Leaf 4 DPI | TGGCAGCGGCGCAGGATCTCG | 8.5 | BdiBd21-3.5G0309700.1 | −1.39 | ABC transporter B family member 19 |
| CAATCGTTGTCTGGCATTGA | 83.4 | BdiBd21-3.4G0207200.1 | −0.72 | ABC transporter F family member 5 | |
| TTGTGTCCAAGCGTTCTGAAA | 13.3 | BdiBd21-3.2G0019400.1 | −0.88 | ABC transporter G family member 7 | |
| TGATTAAGGAGAAGCGGGGG | 6.0 | BdiBd21-3.5G0286800.1 | −0.95 | auxin-responsive protein SAUR71 | |
| TCGCTTTGGCGGCGCGCCGGC | 27.8 | BdiBd21-3.1G0937800.1 | −1.15 | cytochrome P450 94B3 | |
| TCGCACTTCGCGGCGTTGGCG | 16.9 | BdiBd21-3.3G0497100.1 | −1.11 | cytokinin dehydrogenase 11 | |
| AGGGGCTACGATCTTTGAGAA | 18.1 | BdiBd21-3.3G0787100.1 | −1.13 | DexH-box ATP-dependent RNA helicase DExH15 | |
| TTTCGAGATTGGAAACGGCT | 12.1 | BdiBd21-3.2G0305700.1 | −0.69 | dicer homolog 3b | |
| TGACGGGATAGGTAAAGAACTA | 8.5 | BdiBd21-3.2G0095300.1 | −0.72 | G-type lectin S-receptor-like serine/threonine-protein kinase | |
| TAGATCGGTTGGTGTCGGGC | 109.9 | BdiBd21-3.1G0507400.1 | −0.66 | GATA transcription factor 21 | |
| TGGGCGGCGGTCATTTCGGC | 6.0 | BdiBd21-3.3G0739100.1 | −1.86 | peroxidase 1 | |
| AGAAGAATTTCATGCCGGCCAG | 8.5 | BdiBd21-3.1G0796400.1 | −0.71 | peroxidase 50 | |
| CCGGCATGAAATTCTTCTCGAA | 7.2 | BdiBd21-3.1G0093800.1 | −0.99 | photosystem I reaction center subunit III, chloroplastic | |
| TAGTAGGGCTGCAAGATCTA | 10.9 | BdiBd21-3.5G0116500.1 | −0.91 | photosystem I subunit PsaO | |
| TAGTTGAGTTCCGCCTGCTG | 58.0 | BdiBd21-3.1G0118800.1 | −0.60 | photosystem II D1 precursor processing protein PSB27-H2, chloroplastic | |
| TGGCTGTGAATTCGGCGAGGG | 59.2 | BdiBd21-3.5G0237900.1 | −0.62 | probable aquaporin PIP1-2 | |
| CCAATCGTTGTCTGGCATTGA | 105.1 | BdiBd21-3.2G0771500.1 | −1.33 | putative disease resistance protein RGA3 | |
| TTTCGGATAGAGGCACCCAA | 10.9 | BdiBd21-3.3G0396200.1 | −0.54 | putative disease resistance protein RGA4 | |
| TATCGTCGCGCAGTTGGTCG | 7.2 | BdiBd21-3.3G0461000.1 | −0.56 | RNA exonuclease 4 | |
| TGAGCCGGGGGTATAATCGG | 6.0 | BdiBd21-3.4G0029600.1 | −0.65 | stress-induced-phosphoprotein 1 | |
| TGATTCGGCGGCAGGTCTGGC | 14.5 | BdiBd21-3.1G0170300.1 | −1.17 | transcription factor bHLH25 | |
| TCGGTTTCGGCTTCTGGGGT | 10.9 | BdiBd21-3.2G0063400.1 | −1.19 | transcription factor NIGTH1 | |
| TAAATACCGTCCCGGCAAGG | 9.7 | BdiBd21-3.4G0018600.1 | −0.66 | wall-associated receptor kinase 5 | |
| TGACGGAGCTCGGCCTGGAA | 45.9 | BdiBd21-3.5G0304000.1 | −0.90 | wound-induced protein | |
| Root | TAGGGTGGCCTGAATTATAGT | 10.4 | BdiBd21-3.2G0378400.1 | −0.74 | glycine-rich cell wall structural protein 1 |
| AGTATTCCGTCGTCGCCGTA | 20.7 | BdiBd21-3.3G0257600.1 | −0.78 | xyloglucan endotransglycosylase/hydrolase protein 8 | |
| TGAACCAGCCGTTGAGTAAG | 10.4 | BdiBd21-3.3G0280200.1 | −0.55 | fatty acyl-CoA reductase 1 |
| Predicted Target | Target ID | Target Description | Log2FC | ||||
|---|---|---|---|---|---|---|---|
| Leaf 2 DPI | Leaf 4 DPI | Root | Leaf 2 DPI | Leaf 4 DPI | Root | ||
| X | X | X | MGG_02127 | alcohol oxidase | −5.46 | −2.53 | −3.11 |
| X | X | X | MGG_02695 | cysteine proteinase 1 | −4.72 | −1.35 | −2.46 |
| X | X | X | MGG_06494 | D-arabinitol 2-dehydrogenase | −5.52 | −1.16 | −3.21 |
| X | X | X | MGG_01386 | FAD dependent oxidoreductase superfamily protein | −5.19 | −2.16 | −4.51 |
| X | X | X | MGG_05981 | glutamine amidotransferase subunit pdxT | −4.79 | −2.34 | −4.25 |
| X | X | X | MGG_10400 | GPI-anchored cell wall beta−1,3-endoglucanase EglC | −0.99 | −2.05 | −2.11 |
| X | X | X | MGG_01361 | PHO85 cyclin-1 | −5.81 | −1.27 | −2.85 |
| X | MGG_15576 | DNA repair protein rhp51 | n.s. | −1.05 | n.s. | ||
| X | MGG_03587 | essential for mitotic growth 1 | n.s. | −0.60 | n.s. | ||
| X | X | MGG_04345 | cytochrome P450 17A1 | −4.40 | −1.06 | n.s. | |
| X | X | MGG_03201 | acetyl-coenzyme A synthetase | −1.39 | 0.86 | −1.84 | |
| X | MGG_09950 | C2H2 type zinc finger domain-containing protein | n.s. | −1.49 | n.s. | ||
| X | X | MGG_16901 | ATP-dependent RNA helicase DBP2 | n.s. | −1.04 | −1.18 | |
| X | X | X | MGG_07667 | autophagy-related protein 17 | −2.45 | −1.33 | n.s. |
| X | X | X | MGG_01391 | ent-kaurene oxidase | −3.90 | −2.44 | n.s. |
| X | X | X | MGG_11962 | G-protein coupled receptor | −5.75 | −3.42 | n.s. |
| X | X | X | MGG_04378 | integral membrane protein | −3.99 | −1.04 | n.s. |
| X | X | X | MGG_04935 | integral membrane protein | −3.58 | −1.40 | n.s. |
| X | X | X | MGG_03123 | MATE efflux family protein subfamily | −4.68 | −1.11 | n.s. |
| X | X | X | MGG_14872 | calpain-9 | −4.19 | −1.17 | n.s. |
| X | X | X | MGG_09460 | cell wall protein | −4.72 | −3.92 | n.s. |
| X | X | X | MGG_03186 | 1,4-alpha-glucan-branching enzyme | −1.52 | n.s. | −1.36 |
| X | X | X | MGG_14154 | RETRO5, retrotransposons MoTeR1s and MoTeR2 | −6.04 | −1.15 | n.s. |
| X | X | MGG_06393 | serine/threonine-protein kinase ATG1 | −3.24 | n.s. | n.s. | |
| X | X | MGG_04938 | C-3 sterol dehydrogenase/C-4 decarboxylase | n.s. | −1.85 | n.s. | |
| X | X | X | MGG_10568 | sterol 24-C-methyltransferase | n.s. | n.s. | −1.24 |
| X | X | MGG_06371 | pyruvate dehydrogenase E1 component subunit alpha | n.s. | n.s. | −1.75 | |
| Transcript ID | Gene ID | Description | Phenotype | Host Species | Reference Phi-Base |
|---|---|---|---|---|---|
| MGG_00063T0 | AGL1 | glycogen debranching enzyme | reduced_virulence | Os_Hv | PHI:3814 |
| MGG_00365T0 | MAGB | G alpha protein subunit | reduced_virulence | Os | PHI:83 |
| MGG_00620T0 | MoDac | GlcNAc-6-phosphate deacetylase | reduced_virulence | Os | PHI:5471 |
| MGG_01096T0 | SGA1 | vacuolar glucoamylase | loss_of_pathogenicity | Os_Hv | PHI:2138 |
| MGG_01180T0 | MoSCAD3 | short-chain specific acyl-CoA dehydrogenase | reduced_virulence | Os_Hv | PHI:8929 |
| MGG_01285T0 | Tpc1 | sranscription factor for Polarity Control 1 | reduced_virulence | Os_Hv | PHI:7317 |
| MGG_01819T0 | Gph1 | phosphorylase | loss_of_pathogenicity/reduced_virulence | Os_Hv | PHI:2062/3815 |
| MGG_02444T0 | MoPLC1 | modulator of calcium flux | loss_of_pathogenicity | Os | PHI:2057 |
| MGG_02457T0 | RHO2 | Rho GTPase | reduced_virulence | Os | PHI:8752 |
| MGG_02884T0 | MoFLP1 | fasciclin-like protein | reduced_virulence | Os_Hv | PHI:4231 |
| MGG_03148T0 | TDG4 | trigalactosyldiacylglycerol-4 | reduced_virulence | Os_Hv | PHI:3811 |
| MGG_03198T0 | TIG1 | histone deacetylation | loss_of_pathogenicity | Os_Hv | PHI:2002 |
| MGG_03670T0 | SPM1 | subtilisin-like proteinase Spm1 | reduced_virulence | Os_Hv | PHI:2117 |
| MGG_03860T0 | TPS1 | trehalose-6-phosphate synthase | loss_of_pathogenicity/reduced_virulence | Os_Hv | PHI:322/1064 |
| MGG_04895T0 | ICL1 | isocitrate lyase | reduced_virulence | Os | PHI:305 |
| MGG_05344T0 | MgSM1 | effector | increased_virulence | At | PHI:2118/5540 |
| MGG_06393T0 | ATG1 | autophagy-related protein 1 | loss_of_pathogenicity | Os_Hv | PHI:2035/2069/8612 |
| MGG_07667T0 | Moatg17 | autophagy-related protein 17 | loss_of_pathogenicity | Os_Hv | PHI:2083 |
| MGG_08054T0 | MoChi1 | chitinase 1 | reduced_virulence | Os_Hv | PHI:8753/8806 |
| MGG_08370T0 | gel3 | 1,3-beta-glucanosyltransferase | loss_of_pathogenicity | Os | PHI:6713 |
| MGG_09471T0 | NTH1 | neutral trehalase | reduced_virulence | Os_Hv | PHI:123/775/794 |
| MGG_10859T0 | MoLDS1 | heme peroxidase | reduced_virulence | Os | PHI:5189 |
| MGG_11862T0 | ABC4 | ABC transporter | reduced_virulence/loss_of_pathogenicity | Hv | PHI:1017/2067 |
| MGG_12122T0 | MoGSK1 | glycogen synthase kinase 1 | loss_of_pathogenicity | Os_Hv | PHI:7117 |
| MGG_12814T0 | MoAP1 | BZIP domain-containing protein | loss_of_pathogenicity | Os_Hv | PHI:2142 |
| MGG_17909T0 | ATG3 | autophagy-related protein 3 | loss_of_pathogenicity | Os | PHI:2071 |
| MGG_05287T0 | CON7 | transcription factor CON7 | loss_of_pathogenicity | Os_Hv | PHI:2039 |
| MGG_09055T0 | AvrPiz-t | avrpiz-tgene, effector protein | increased_virulence | Os | PHI:7896 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanini, S.; Šečić, E.; Busche, T.; Galli, M.; Zheng, Y.; Kalinowski, J.; Kogel, K.-H. Comparative Analysis of Transcriptome and sRNAs Expression Patterns in the Brachypodium distachyon—Magnaporthe oryzae Pathosystems. Int. J. Mol. Sci. 2021, 22, 650. https://doi.org/10.3390/ijms22020650
Zanini S, Šečić E, Busche T, Galli M, Zheng Y, Kalinowski J, Kogel K-H. Comparative Analysis of Transcriptome and sRNAs Expression Patterns in the Brachypodium distachyon—Magnaporthe oryzae Pathosystems. International Journal of Molecular Sciences. 2021; 22(2):650. https://doi.org/10.3390/ijms22020650
Chicago/Turabian StyleZanini, Silvia, Ena Šečić, Tobias Busche, Matteo Galli, Ying Zheng, Jörn Kalinowski, and Karl-Heinz Kogel. 2021. "Comparative Analysis of Transcriptome and sRNAs Expression Patterns in the Brachypodium distachyon—Magnaporthe oryzae Pathosystems" International Journal of Molecular Sciences 22, no. 2: 650. https://doi.org/10.3390/ijms22020650
APA StyleZanini, S., Šečić, E., Busche, T., Galli, M., Zheng, Y., Kalinowski, J., & Kogel, K.-H. (2021). Comparative Analysis of Transcriptome and sRNAs Expression Patterns in the Brachypodium distachyon—Magnaporthe oryzae Pathosystems. International Journal of Molecular Sciences, 22(2), 650. https://doi.org/10.3390/ijms22020650