Spectral Probe for Electron Transfer and Addition Reactions of Azide Radicals with Substituted Quinoxalin-2-Ones in Aqueous Solutions



Abstract
1. Introduction
4TU+(S●) + 4TU(=S) ⇆ (4TUS∴S4TU)+
2TU+(S●) → 2TUS● + H+
2TU+(S●) + 2TU(=S) ⇆ (2TUS∴S2TU)


2. Results
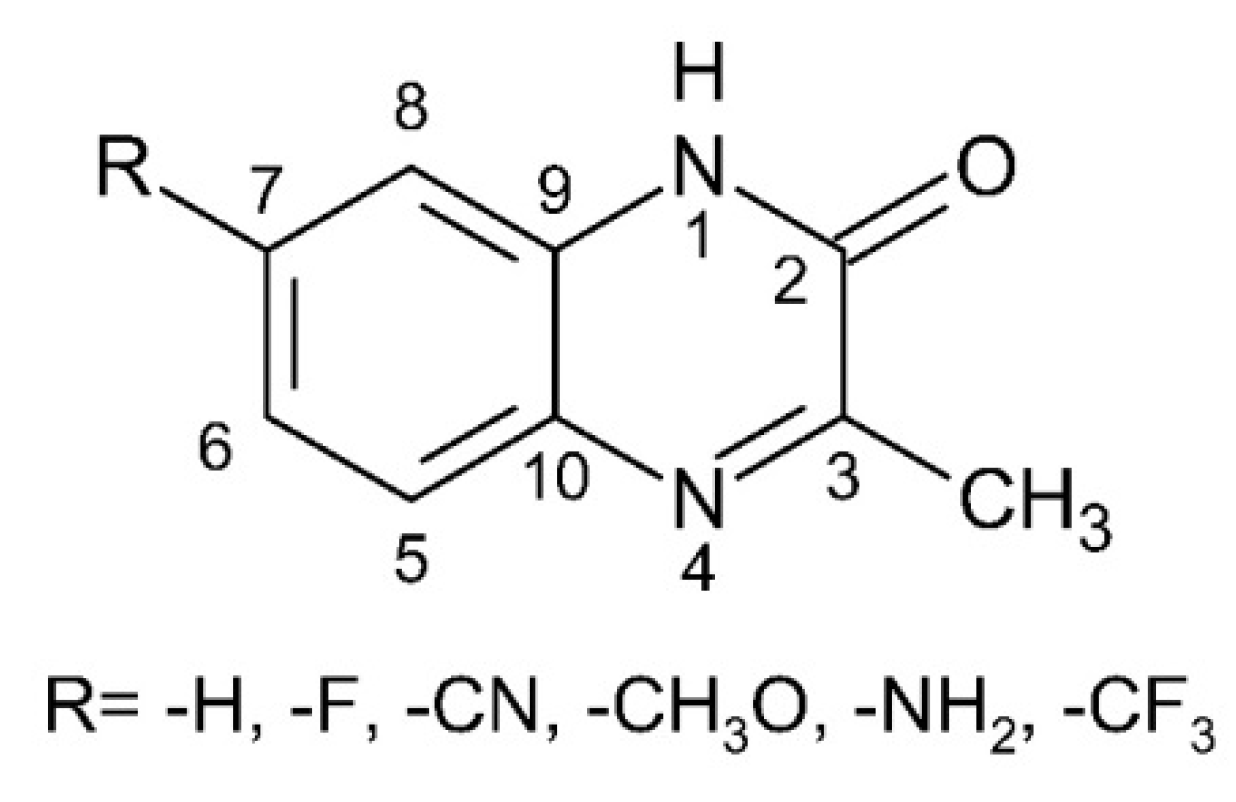
2.1. Reactions of SO4●− and N3● with 3-Methyl-1H-Quinoxalin-2-One (3-MeQ) and Its 7-substituted Derivatives (7-R-3MeQ)–Absorption Spectra
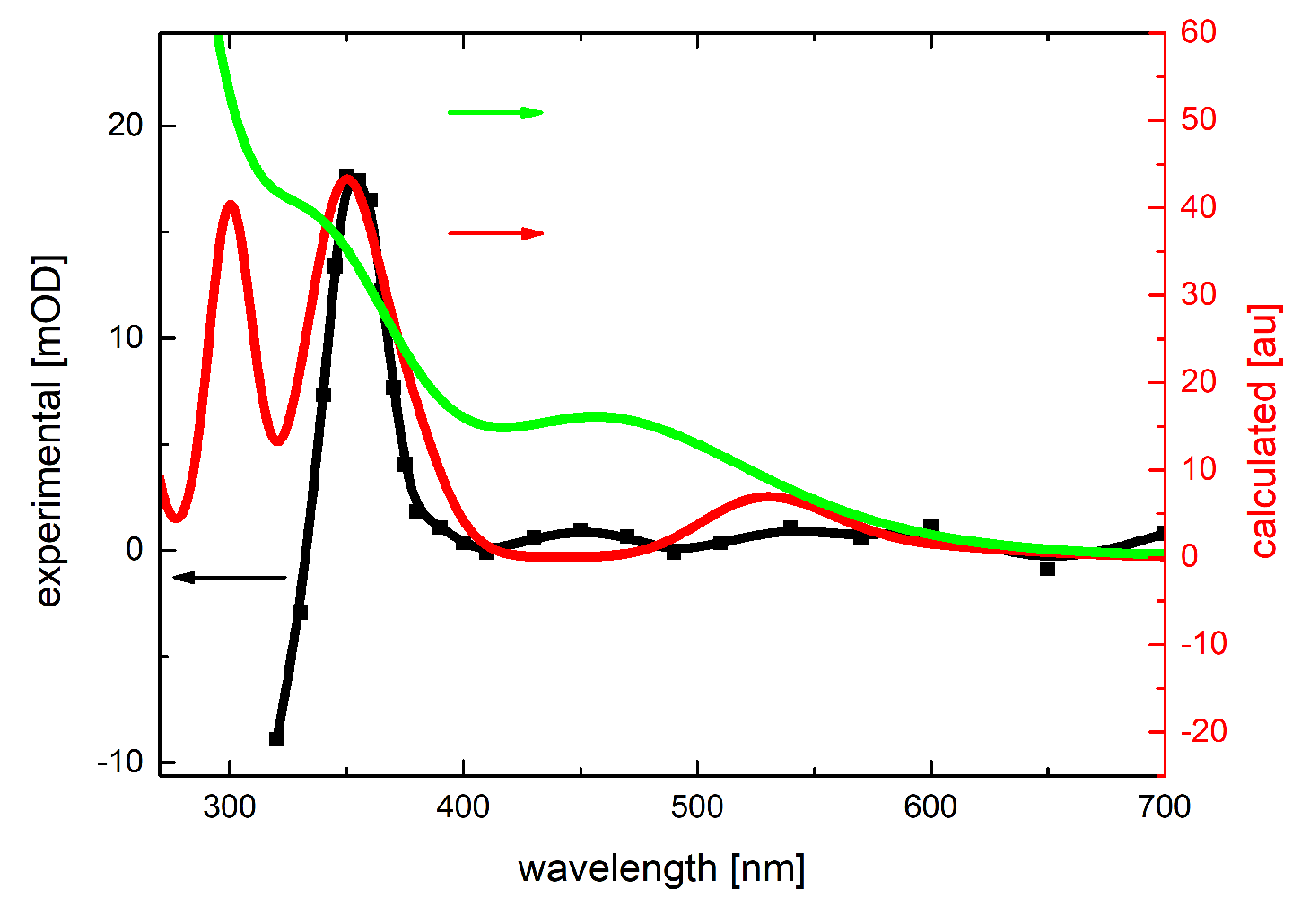
2.1.1. Absorption Spectra Recorded after Reaction of N3● and SO4●− with 3-MeQ
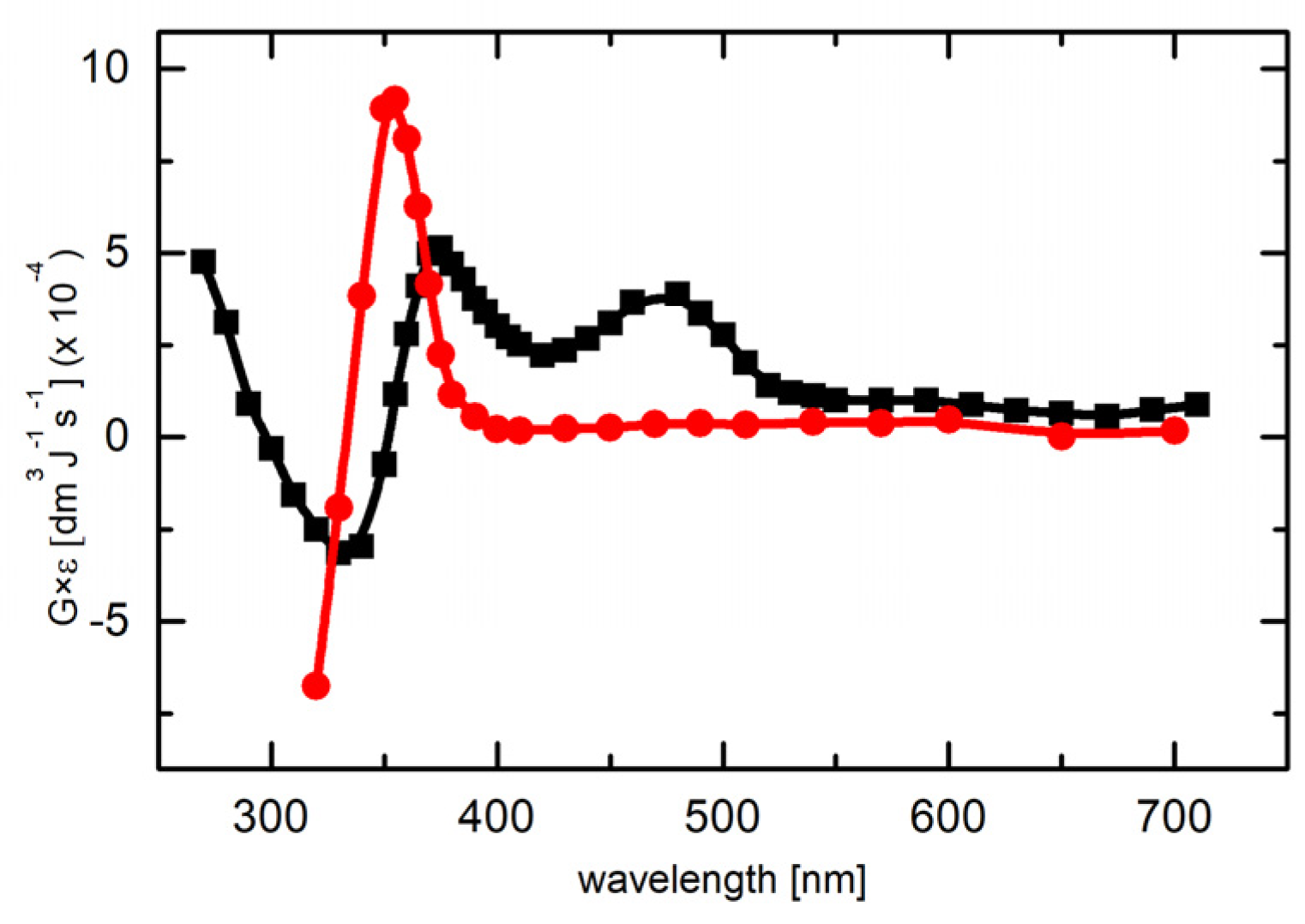
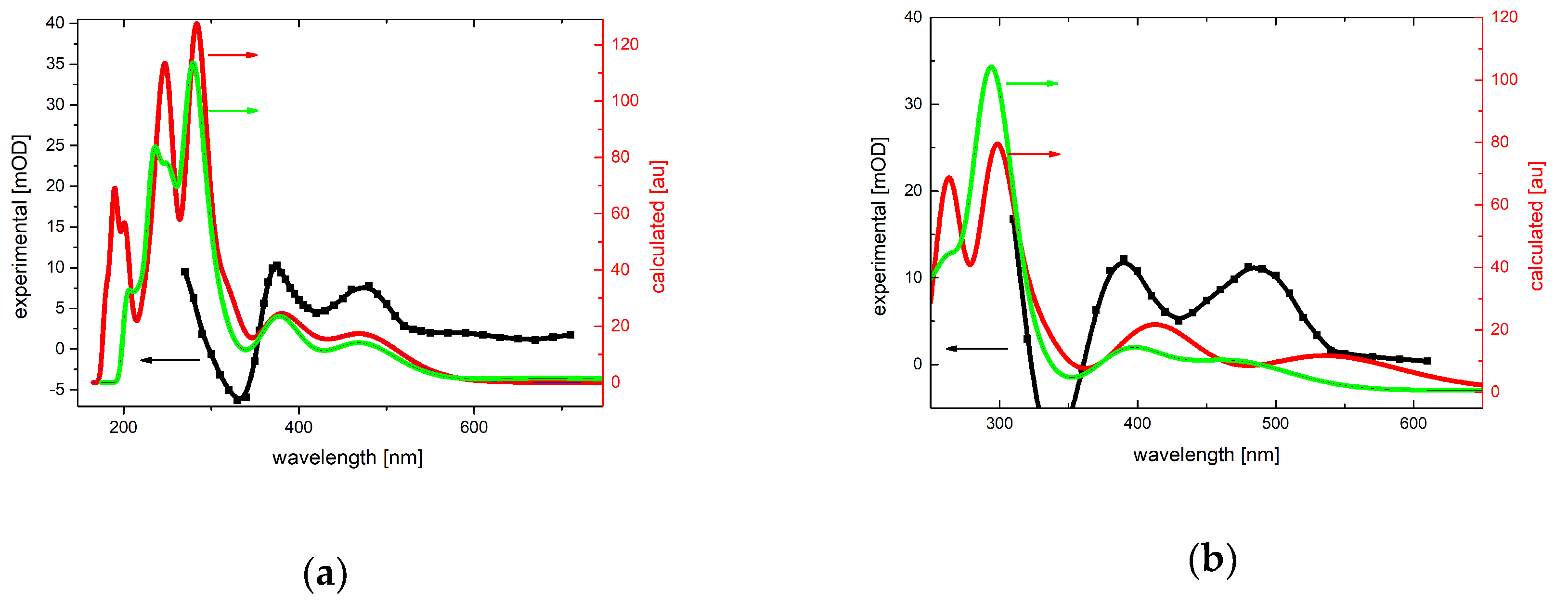
2.1.2. Absorption Spectra Recorded after Reaction of N3● and SO4●− with 7-R-3-MeQ Derivatives
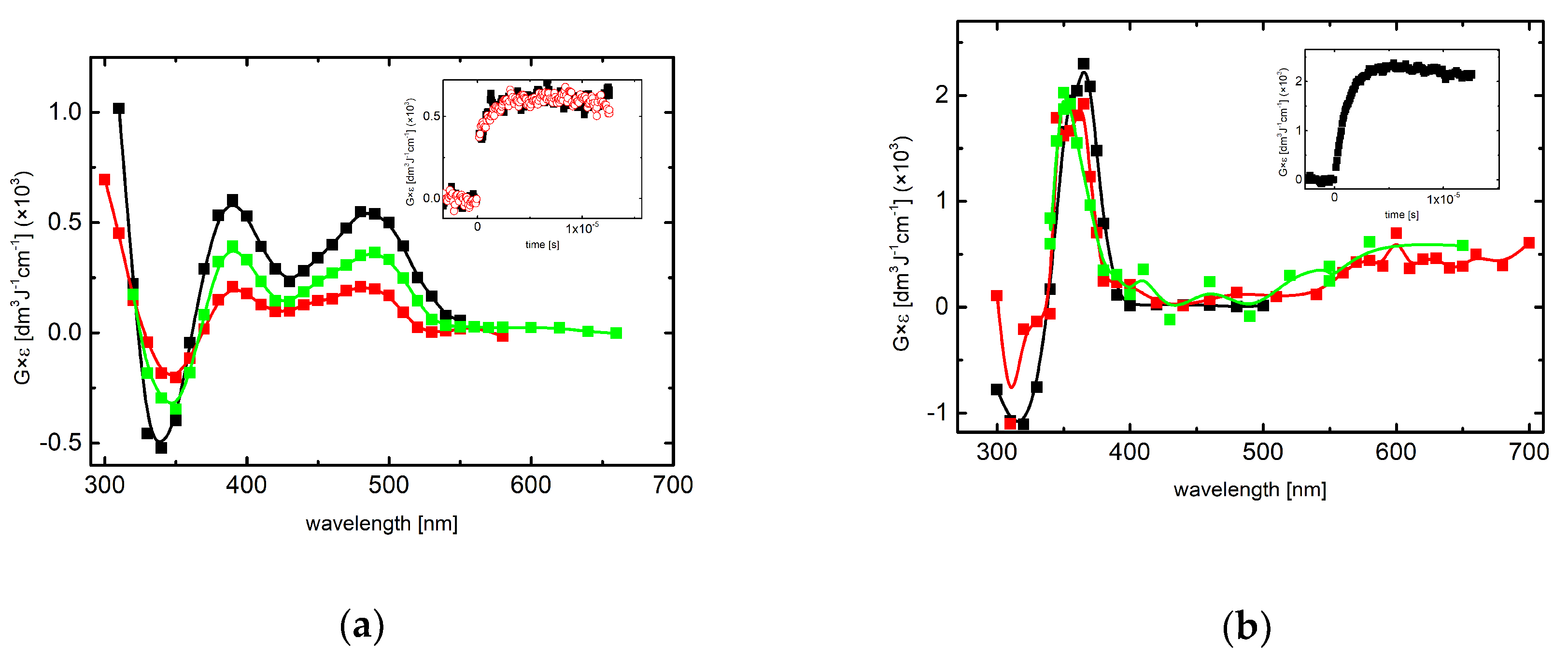
2.2. Reactions of Tl2+ and CO3●− with 7-OCH3-3MeQ–Absorption Spectra
2.2.1. Absorption Spectra Recorded in Slightly Acidic and Neutral Solutions
2.2.2. Absorption Spectra Recorded in Alkaline Solutions
2.3. Reactions of N3● with 3-Methyl-1H-Quinoxalin-2-one (3-MeQ) and Its 7-Substituted Derivatives (7-R-3MeQ–Kinetics
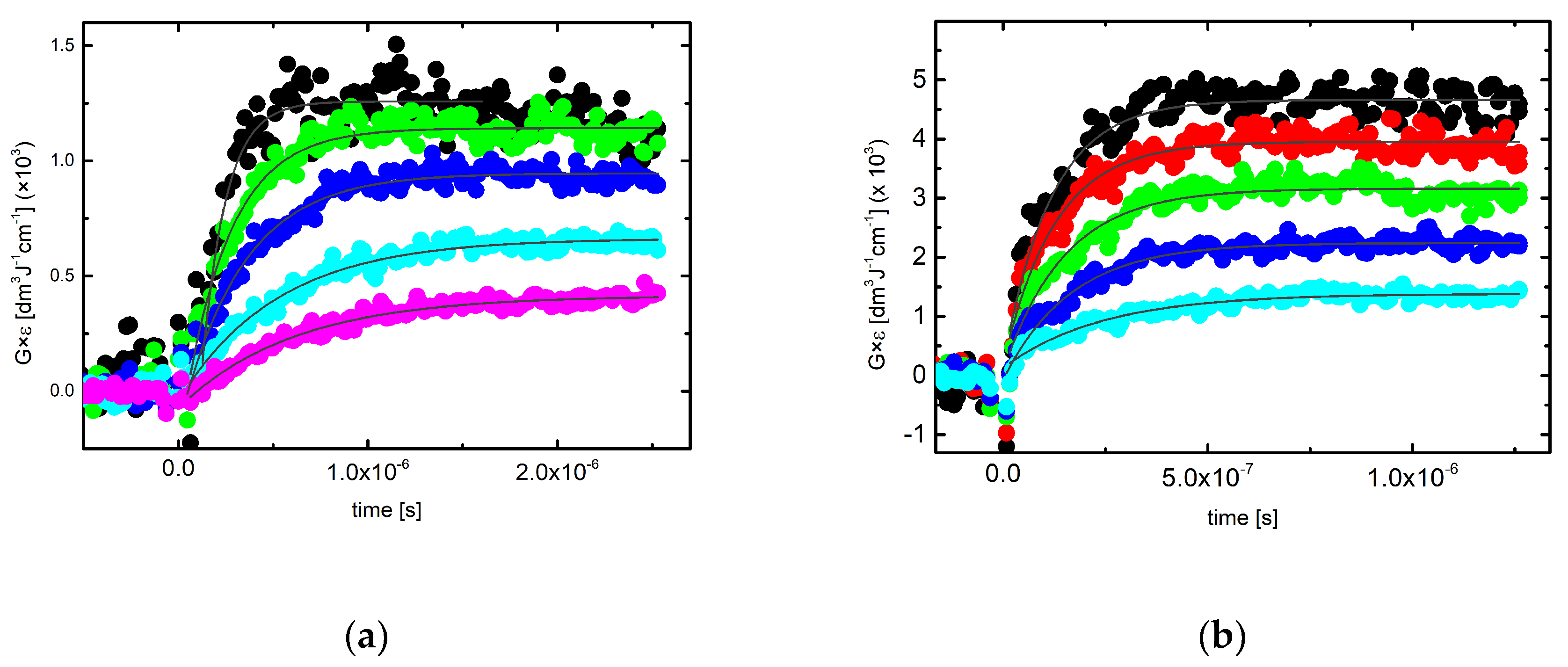
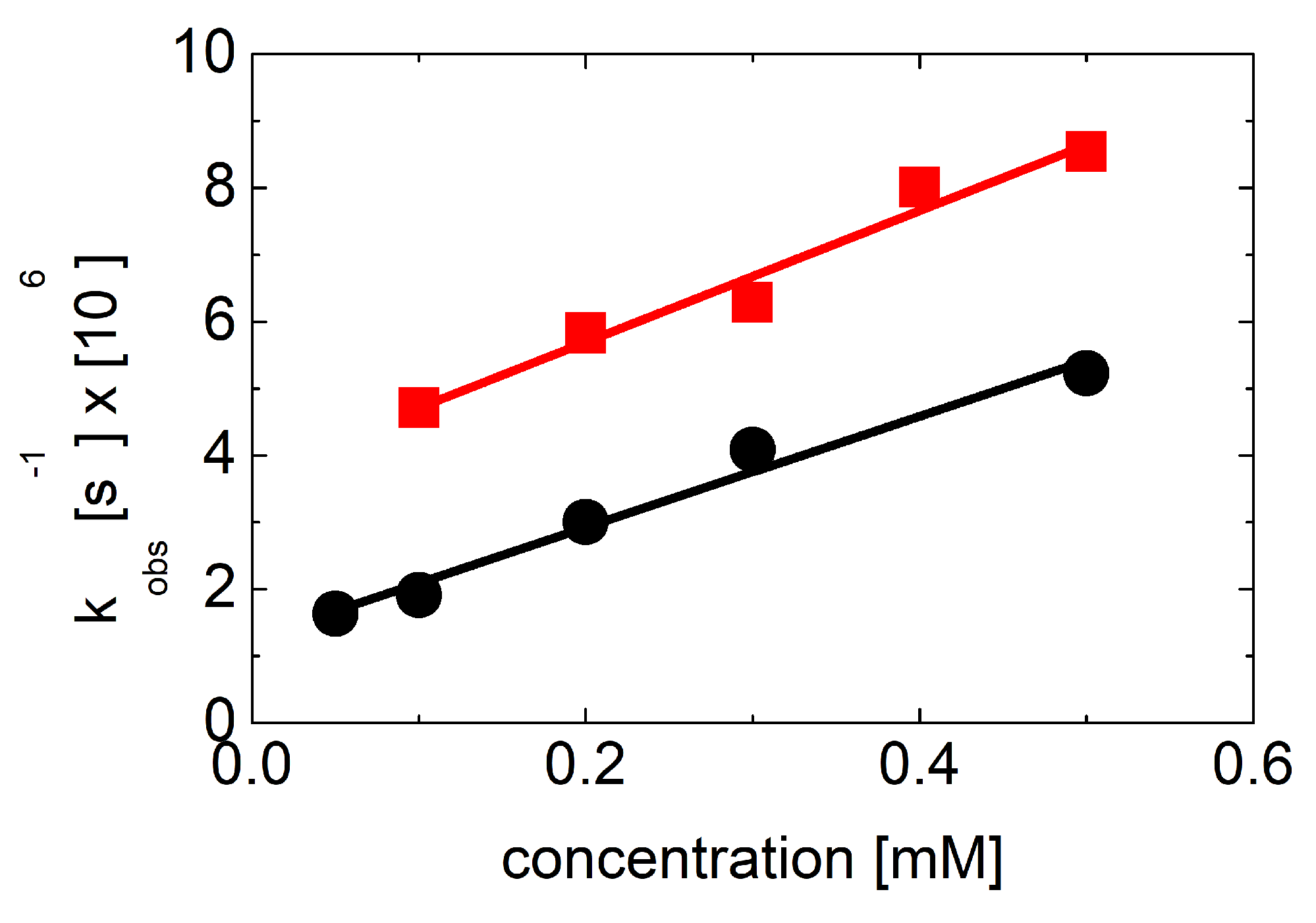
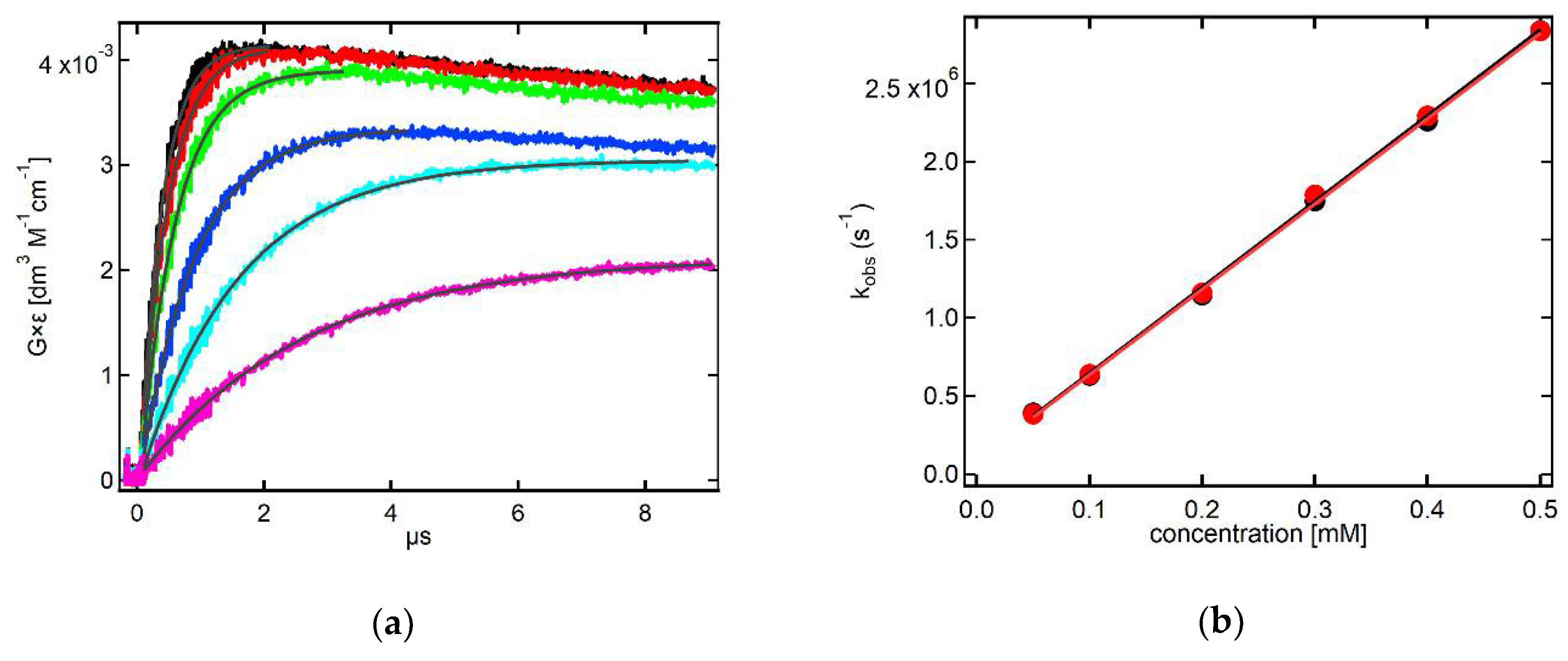
2.3.1. Kinetics Recorded in Solutions Containing 3-MeQ and 7-R-3-MeQ Derivatives with Electron-Withdrawing Substituents
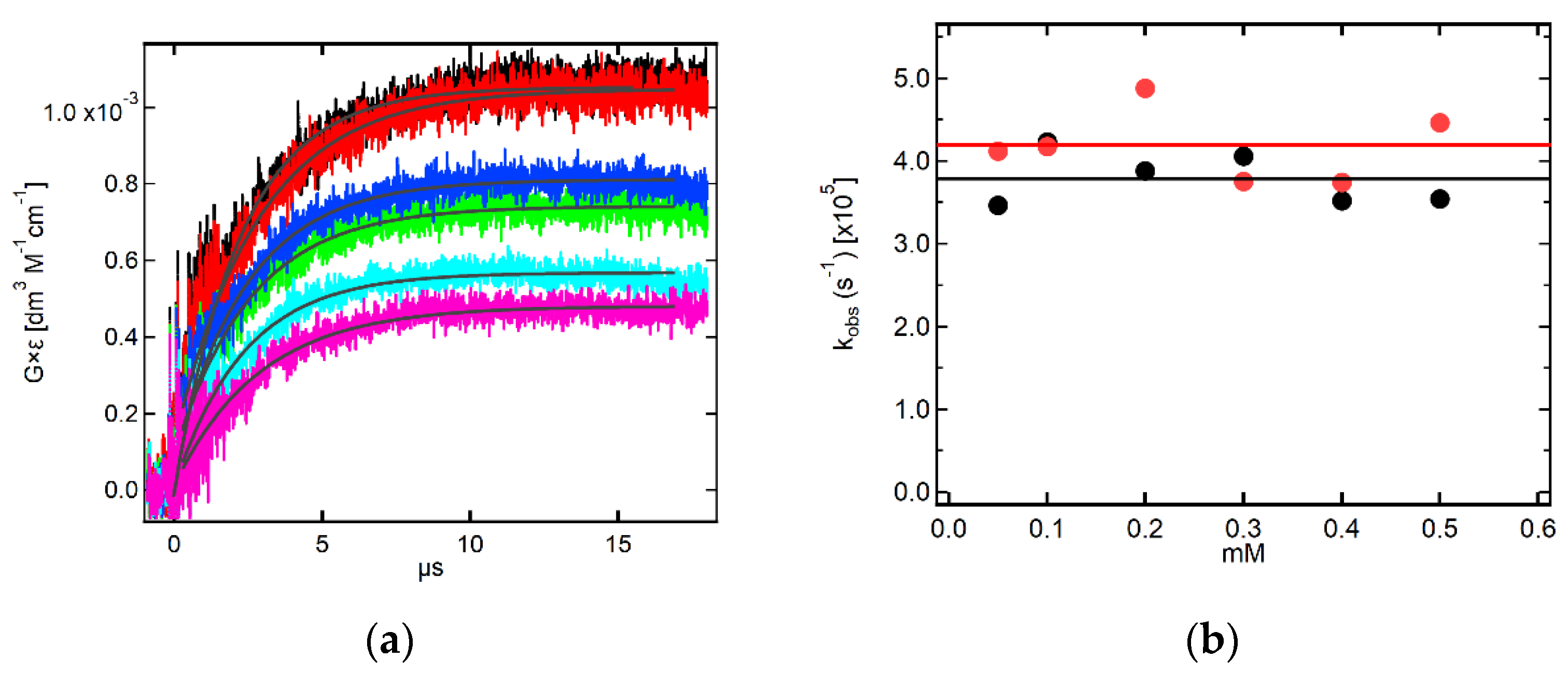
2.3.2. Kinetics Recorded in Neutral Solutions Containing 7-OCH3-3-MeQ
2.3.3. Kinetics Recorded in Alkaline Solutions Containing 7-OCH3-3-MeQ
2.4. Theoretical Calculations of Radical Cations and Their Deprotonated Forms Derived from 3-MeQ and 7-R-3-MeQ Derivatives
2.5. Theoretical Calculations of N3● Adducts to 3-MeQ and 7-R-3-MeQ Derivatives
3. Discussion
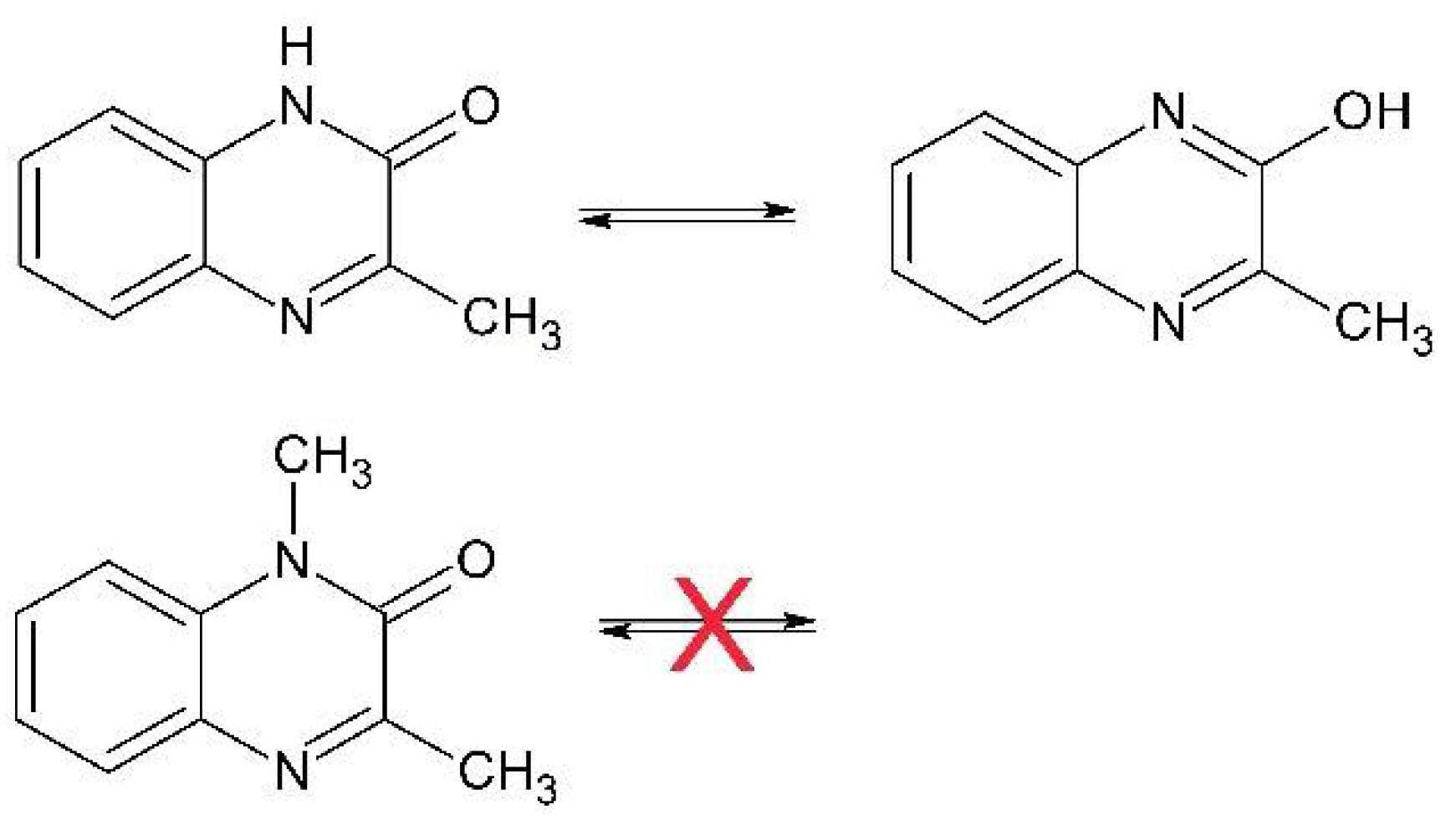
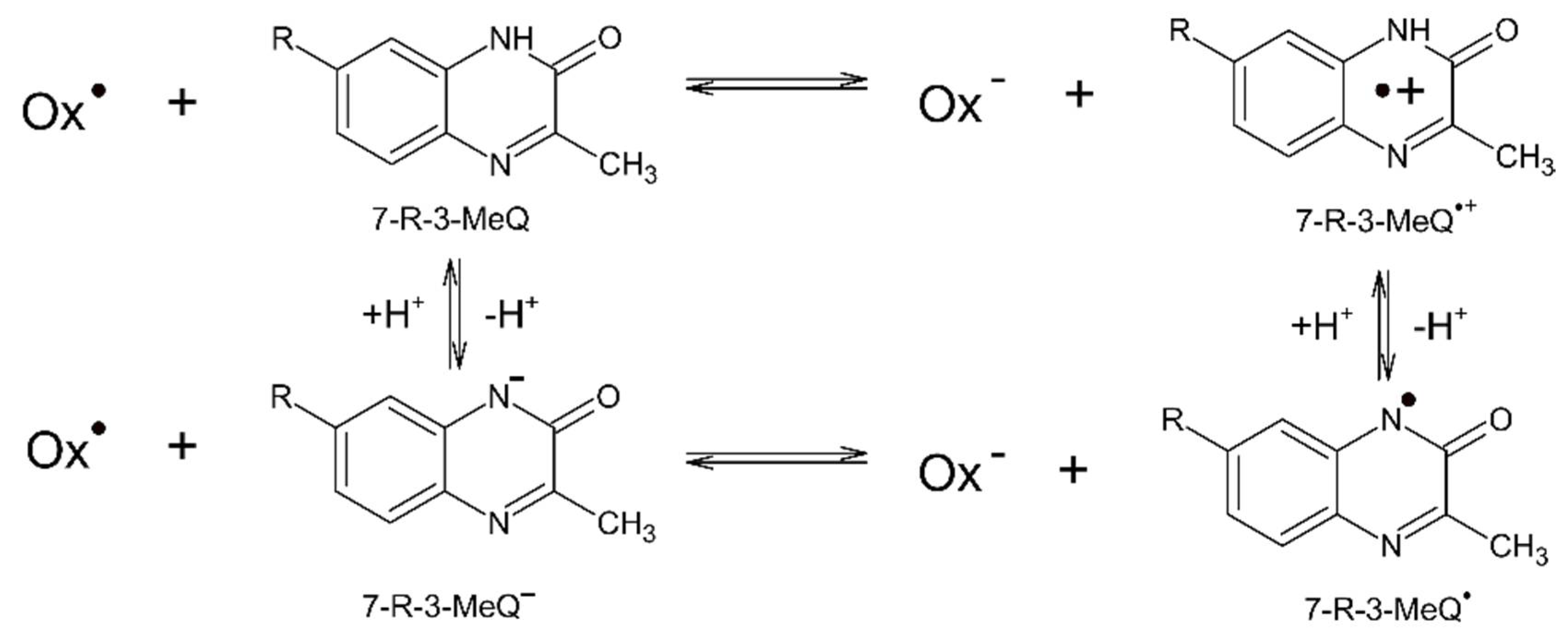
3.1. Reaction Pathways Involving One-Electron Oxidants and 3-MeQ/7-R-3-MeQ Derivatives
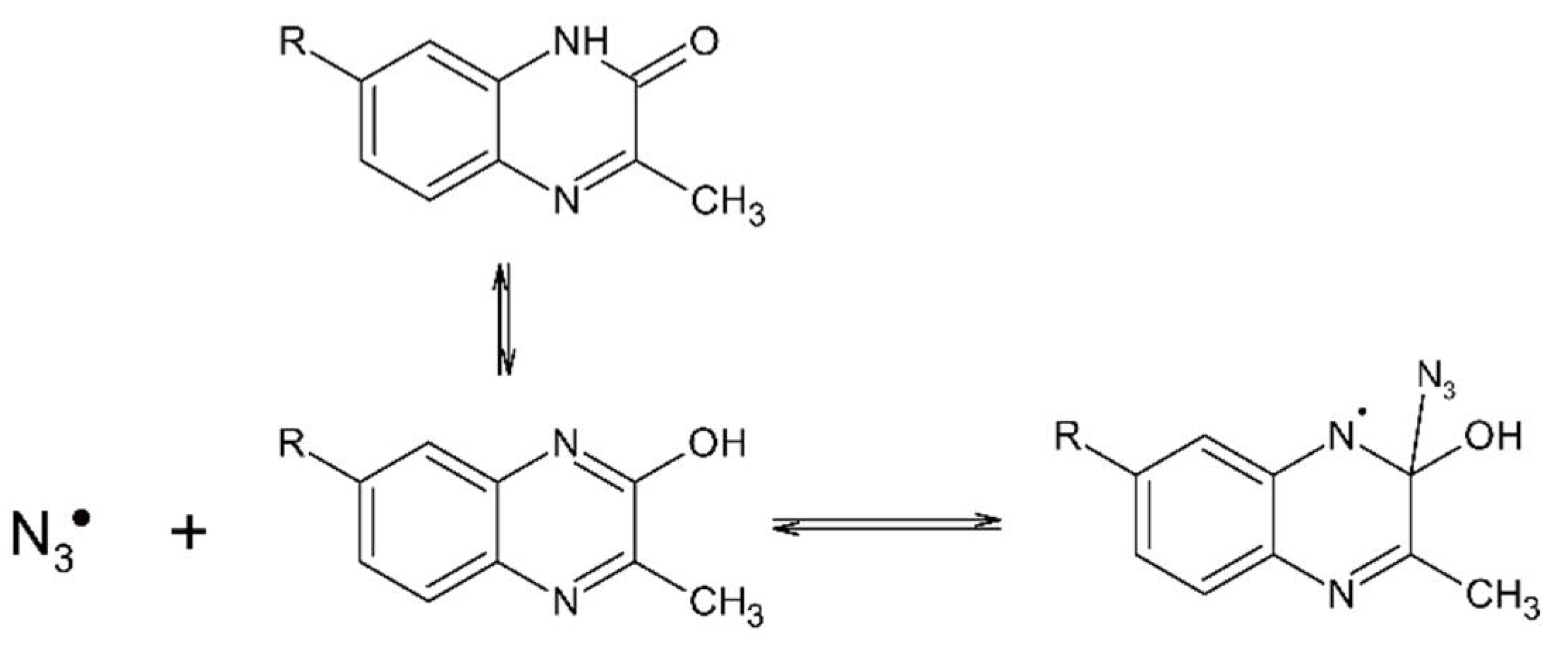
3.2. Reaction Pathway Involving N3● and 3-MeQ/7-R-3-MeQ Derivatives with Electron Withdrawing Substituents (R)
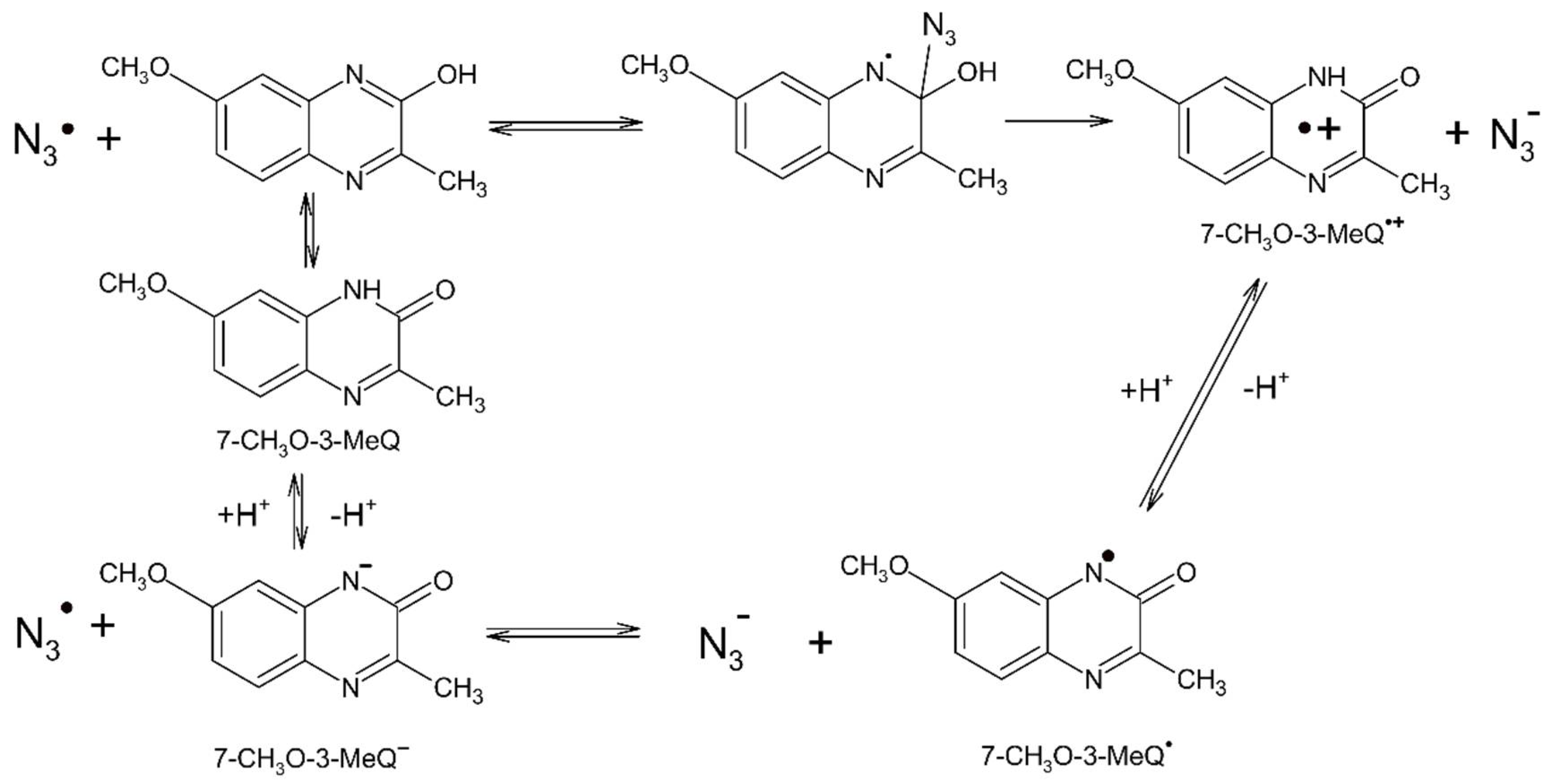
3.3. Reaction Pathways Involving N3● and 7-OCH3-3-MeQ
4. Materials and Methods
4.1. Chemicals
4.2. Synthesis of 7-Substituted 3-Methyl-2(1H)-Quinoxalin-2-Ones
4.3. Preparation of Solutions
4.4. Pulse Radiolysis Instrumentation
4.5. Pulse Radiolysis Experiments
4.5.1. Dosimetry
4.5.2. Selective Generation of the Primary Reactive Species
4.5.3. Generation of the Selective Oxidizing Radicals and Metal Ions
4.6. Theoretical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NHE | Normal Hydrogen Electrode |
| ESR | Electron Spin Resonance |
| PMT | Photomultiplier |
| PC | Personal Computer |
| DFT | Density Functional Theory |
| TD-DFT | Time Dependent Density Functional Theory |
| pVTZ | Polarized-Valence-Triple-Zeta |
| NBO | Natural Bond Orbital |
References
- Buxton, G.V.; Janovsky, I. Mechanism of the Oxidation of Iron(II) by the Azide Radical. J. Chem. Soc. Faraday Trans. I 1976, 72, 1884–1886. [Google Scholar] [CrossRef]
- Eriksen, T.E.; Lind, J.; Merenyi, G. The reactivity of the azide radical (N3) towards dioxygen species. Radiochem. Radioanal. Lett. 1981, 48, 405–410. [Google Scholar]
- Alfassi, Z.B.; Harriman, A.; Huie, R.E.; Mosseri, S.; Neta, P. The Redox Potential of the Azide/Azidyl Couple. J. Phys Chem. 1987, 91, 2120–2122. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Schuler, R.H. Reaction of Azide Radicals with Aromatic Compounds. Azide as a Selective Oxidant. J. Phys. Chem. 1985, 89, 3359–3363. [Google Scholar] [CrossRef]
- Qin, L.; Tripathi, G.N.R.; Schuler, R.H. Radiation Chemical Studies of the Oxidation of Aniline in Aqueous Solution. Z. Naturforsch. 1985, 40a, 1026–1039. [Google Scholar] [CrossRef]
- Brede, O.; Schwinn, J.; Leistner, S.; Naumov, S.; Sprinz, H. Reactivity and Selectivity of Reactions of Small Radicals with a Multifunctional Heterocyclic Molecule: 3-(Mercaptoethyl)chinazoline-2,4-(1H,3H)dione. J. Phys. Chem. A 2001, 105, 119–127. [Google Scholar] [CrossRef]
- Hermann, R.; Dey, G.R.; Naumov, S.; Brede, O. Thiol radical cations and thiyl radicals as direct products of the free electron transfer from aromatic thiols to n-butyl chloride radical cations. Phys. Chem. Chem. Phys. 2000, 2, 1213–1220. [Google Scholar] [CrossRef]
- Kishore, K.; Dey, G.R.; Naik, D.B. Nature of transient species formed during pulse radiolysis of 4-mercaptopyridine in aqueous solutions: Formation of a dimer radical species by one-electron reduction reaction. Res. Chem. Intermed. 2002, 28, 29–39. [Google Scholar] [CrossRef]
- Naik, D.B.; Kishore, K. Nature of sulfur centered radicals formed during pulse radiolysis of 2-mercaptopyridine in aqueous solutions. Res. Chem. Intermed. 2002, 28, 603–617. [Google Scholar] [CrossRef]
- Shen, X.; Lind, J.; Merenyi, G. One-electron Oxidation of Indoles and Acid-Base Properties of the Indolyl Radicals. J. Phys. Chem. 1987, 91, 4403–4406. [Google Scholar] [CrossRef]
- Al-Kazwini, A.T.; O’Neill, P.; Adams, G.E.; Cundall, R.B.; Jacquet, B.; Lang, G.; Junino, A. One-electron Oxidation of Methoxylated and Hydroxylated Indoles by N3.. 1. Characterization of the Primary Indolic Radicals. J. Phys. Chem. 1990, 94, 6666–6670. [Google Scholar] [CrossRef]
- Zhao, R.; Lind, J.; Merenyi, G.; Eriksen, T.E. Kinetics of One-Electron Oxidation of Thiols and Hydrogen Abstraction by Thiyl Radicals from α-Amino C-H Bonds. J. Am. Chem. Soc. 1994, 116, 12010–12015. [Google Scholar] [CrossRef]
- Bors, W.; Michel, C. Antioxidant Capacity of Flavanols and Gallate Esters: Pulse Radiolysis Studies. Free Radic. Biol. Med. 1999, 27, 1413–1426. [Google Scholar] [CrossRef]
- Priyadarsini, K.I.; Khopde, S.M.; Kumar, S.S.; Mohan, H. Free Radical Studies of Ellagic Acid, a Natural Phenoloc Antioxidant. J. Agric. Food Chem. 2002, 50, 2200–2206. [Google Scholar] [CrossRef]
- Skotnicki, K.; Taras-Goslinska, K.; Janik, I.; Bobrowski, K. Radiation-Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions. Molecules 2019, 24, 4402. [Google Scholar] [CrossRef]
- Ram, M.S.; Stanbury, D.M. Electron-Transfer Reactions Involving the Azidyl Radical. J. Phys. Chem. 1986, 90, 3691–3696. [Google Scholar] [CrossRef]
- DeFelippis, M.R.; Faraggi, M.; Klapper, M.H. Redox Potentials of the Azide and Dithiocyanate Radcals. J. Phys. Chem. 1990, 94, 2420–2424. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merenyi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueois solutions: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Prütz, W.A.; Siebert, F.; Butler, J.; Land, E.J.; Menez, A.; Montenay-Garestier, T. Charge transfer in peptides. Intramolecular radical transformation, involving methionine, tryptophan, and tyrosine. Biochim. Biophys. Acta 1982, 705, 139–150. [Google Scholar] [CrossRef]
- Eriksen, T.E.; Fransson, G. Radical-induced oxidation of glutathione in alkaline aqueous solution. Radiat. Phys. Chem. 1988, 32, 163–167. [Google Scholar] [CrossRef]
- Abedinzadeh, Z.; Gardes-Albert, M.; Ferraddini, C. One-electron oxidation of glutathione by azide radicals in neutral medium: A gamma and pulse radiolysis study. Radiat. Phys. Chem. 1991, 38, 1–5. [Google Scholar] [CrossRef]
- Bobrowski, K.; Wierzchowski, K.L.; Holcman, J.; Ciurak, M. Intramolecular electron transfer in peptides containing methionine, tryptophan, and tyrosine: A pulse radiolysis study. Int. J. Radiat. Biol. 1990, 57, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski, K.; Holcman, J.; Poznanski, J.; Ciurak, M.; Wierzchowski, K.L. Pulse radiolysis studies of intramolecular electron transfer in model peptides and proteins. 5. Trp→Tyr radical transformation in H-Trp-(Pro)n-Tyr-OH series of peptides. J. Phys. Chem. 1992, 96, 10036–10043. [Google Scholar] [CrossRef]
- Land, E.J.; Prütz, W.A. Reaction of azide radicals with amino acids and proteins. Int. J. Radiat. Biol. 1979, 36, 75–83. [Google Scholar] [CrossRef]
- Prütz, W.A.; Butler, J.; Land, E.J.; Swallow, A.J. Direct demonstration of electron transfer between tryptophan and tyrosine in proteins. Biochem. Biophys. Res. Commun. 1980, 96, 408. [Google Scholar] [CrossRef]
- Butler, J.; Land, E.J.; Prütz, W.A.; Swallow, A.J. Charge transfer between tryptophan and tyrosine in proteins. Biochim. Biophys. Acta 1982, 705, 150–162. [Google Scholar] [CrossRef]
- Bobrowski, K.; Holcman, J.; Poznanski, J.; Wierzchowski, K.L. Pulse radiolysis of intramolecular electron transfer in model peptides and proteins. 7. Trp→TyrO radical transformation in hen-egg white lysozyme. Effects of pH, temperature, Trp62 oxidation and inhibitor binding. Biophys. Chem. 1997, 63, 153–166. [Google Scholar] [CrossRef]
- Domazou, A.S.; Zhu, H.; Koppenol, W.H. Fast repair of proteins radicals by urate. Free Radic. Biol. Med. 2012, 52, 1929–1936. [Google Scholar] [CrossRef]
- Domazou, A.S.; Koppenol, W.H.; Gebicki, J.M. Efficient repair of protein radicals by ascorbate. Free Radic. Biol. Med. 2009, 46, 1049–1057. [Google Scholar] [CrossRef]
- Jovanovič, S.V.; Harriman, A.; Simic, M.G. Electron-transfer reactions of tryptophan and tyrosine derivatives. J. Phys. Chem. 1986, 90, 1935–1939. [Google Scholar] [CrossRef]
- Prasanthkumar, K.P.; Suresh, C.H.; Aravindakumar, C.T. Dimer radical cation of 4-thiouracil: A pulse radiolysis and theoretical study. J. Phys. Org. Chem. 2013, 26, 510–516. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Janzen, E.G.; Mason, R.P. Spin Trapping of the Azidyl Radical in Azide/Catalase/H2O2 and Various Azide/Peroxidase/H2O2 Peroxidizing Systems. J. Biol. Chem. 1985, 260, 4003–4006. [Google Scholar] [CrossRef]
- Kremers, W.; Singh, A. Electron spin resonance study of spin-trapped azide radicals in aqueous solutions. Can. J. Chem. 1980, 58, 1592–1595. [Google Scholar] [CrossRef]
- Rehorek, D.; Thomas, P.; Hennig, H. Spin Trapping of Azide Radicals in Photolysis of Azido Cobalt(III) Complexes. Inorg. Chim. Acta 1979, 32, L1–L2. [Google Scholar] [CrossRef]
- Chen, Y.-R.; Sturgeon, B.E.; Gunther, M.R.; Mason, R.P. Electron Spin Resonance Investigation of the Cyanyl and Azidyl Radical Formation by Cytochrome c Oxidase. J. Biol. Chem. 1999, 274, 24611–24616. [Google Scholar] [CrossRef] [PubMed]
- Partridge, R.S.; Monroe, S.M.; Parks, J.K.; Johnson, K.; Parker, W.D.; Eaton, G.R.; Eaton, S.S. Spin Trapping of Azidyl and Hydroxyl Radicals in Azide-Inhibited Rat Brain Submitochondrial Particles. Arch. Biochem. Biophys. 1994, 310, 210–217. [Google Scholar] [CrossRef]
- Workentin, M.S.; Schepp, N.P.; Johnston, L.J.; Wayner, D.D.M. Solvation Control of Chemoselectivity in Reactions of Radical Cations. J. Am. Chem. Soc. 1994, 116, 1141–1142. [Google Scholar] [CrossRef]
- Workentin, M.S.; Wagner, B.D.; Lusztyk, J.; Wayner, D.D.M. Azidyl Radical Reactivity. N6− as a Kinetic Probe for the Addition Reactions of Azidyl Radicals with Olefins. J. Am. Chem. Soc. 1995, 117, 119–126. [Google Scholar] [CrossRef]
- Carta, A.; Piras, S.; Loriga, M.; Paglietti, G. Chemistry, biological properties and SAR analysis of quinoxalinones. Mini-Rev. Med. Chem. 2006, 6, 1179–1200. [Google Scholar] [CrossRef]
- Olayiwola, G.; Obafemi, C.A.; Taiwo, F.O. Synthesis and neuropharmacological activity of some quinoxalinone derivatives. Afr. J. Biotechnol. 2007, 6, 777–786. [Google Scholar]
- Tristán-Manzano, M.; Guirado, A.; Martínez-Esparza, M.; Gálvez, J.; García-Peñarrubia, P.; Ruiz-Alcaraz, A.J. Quinoxalines Potential to Target Pathologies. Curr. Med. Chem. 2015, 22, 3075–3108. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, A.C.; Mendonça Nogueira, T.C.; de Souza, M.V.N. Quinoxaline Nucleus: A Promising Scaffold in Anti-cancer Drug Discovery. Anti-Cancer Agents Med. Chem. 2016, 16, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.C.; Cerecetto, H. Quinoxaline derivatives: A patent review (2006—Present). Expert Opin. Ther. Pat. 2012, 22, 1289–1302. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hashem, A.A. Synthesis, Reactions and Biological Activity of Quinoxaline Derivatives. Am. J. Org. Chem. 2015, 5, 14–56. [Google Scholar]
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.D.S.; Fernandes, R.; Prudencio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, its derivatives and applications: A State of the Art review. Eur. J. Med. Chem 2015, 97, 664–672. [Google Scholar] [CrossRef]
- Srivastava, S.; Banerjee, J.; Srestha, N. Quinoxaline as a potent heterocyclic moiety. IOSR J. Pharm. 2014, 4, 17–27. [Google Scholar] [CrossRef]
- Skotnicki, K.; De la Fuente, J.R.; Canñete, A.; Berrios, E.; Bobrowski, K. Radical Ions of 3-Styryl-quinoxalin-2-one Derivatives Studied by Pulse Radiolysis in Organic Solvents. J. Phys. Chem. B 2018, 122, 4051–4066. [Google Scholar] [CrossRef]
- Skotnicki, K.; De la Fuente, J.R.; Canñete, A.; Bobrowski, K. Radiation-induced reduction of quinoxalin-2one derivatives in aqueous solution. Radiat. Phys. Chem. 2016, 124, 91–98. [Google Scholar] [CrossRef]
- Skotnicki, K.; De la Fuente, J.R.; Canñete, A.; Bobrowski, K. Spectral and Kinetic Properties of Radicals Derived from Oxidation of Quinoxalin-2-ones and Its Methyl Derivative. Molecules 2014, 19, 19152–19171. [Google Scholar] [CrossRef]
- Kulkarni, M.S.; Rao, B.S.M. Redox chemistry of copper complexes of 6,7-dicyanodipyridoquinoxaline: A pulse radiolysis study. Inorg. Chim. Acta 2010, 363, 1813–1817. [Google Scholar] [CrossRef]
- Kulkarni, M.S.; Kumbhar, A.S.; Mohan, H.; Rao, B.S.M. Pulse radiolysis study of pyridine substituted quinoxalines. Res. Chem. Intermed. 2005, 31, 63–72. [Google Scholar] [CrossRef]
- Díaz-Hernandez, D.; Canñete, A.; Pavez, L.; Perez-Sanhueza, A.; Gunther, G.; Szreder, T.; De la Fuente, J.R. Spectral and Kinetic Study of 3-Styrylquinoxalin-2(1H)-ones Photoreduced by N-Phenylglycine and Amines. J. Phys. Chem. B 2019, 123, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, J.R.; Canñete, A.; Carathanassis, N.; Bernazar, L.; Saitz, C.; Díaz-Hernandez, D. Spectral and Kinetic Study of 3-Methylquinoxalin-2-ones Photoreduced by Amino Acids: N-Phenylglycine Radical Chain Reactions and N-Acetyltryptophan Decarboxylation. J. Phys. Chem. A 2016, 120, 2787–2807. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, J.R.; Canñete, A.; Jullian, C.; Saitz, C.; Aliaga, C. Unexpected Imidazoquinoxalinone Annulation Products in the Photoinitiated Reaction of Substituted-3-Methyl-Quinoxalin-2-Ones with N-Phenylglycine. Photochem. Photobiol. 2013, 89, 1335–1345. [Google Scholar] [CrossRef]
- De la Fuente, J.R.; Canñete, A.; Saitz, C.; Jullian, C. Photoreduction of 3-phenylquinoxalin-2-ones by amines. Transient-absorption and semiempirical quantum-chemical studies. J. Phys. Chem. A 2002, 106, 7113–7120. [Google Scholar] [CrossRef]
- De la Fuente, J.R.; Canñete, A.; Zanocco, A.L.; Saitz, C.; Jullian, C. Formal hydride transfer mechanism for photoreduction of 3-phenylquinoxalin-2-ones by amines. Association of 3-phenylquinoxalin-2-one with aliphatic amines. J. Org. Chem. 2000, 65, 7949–7958. [Google Scholar] [CrossRef]
- Zimpl, M.; Skopalova, J.; Jirovsky, D.; Bartak, P.; Navratil, T.; Sedonikova, J.; Kotoucek, M. Electrochemical Behavior of Quinoxalin-2-one Derivatives at Mercury Electrodes and Its Analytical Use. Sci. World J. 2012, 409378. [Google Scholar] [CrossRef]
- Bodini, M.E.; del Valle, M.A.; Copia, G.E.; Soto, C. Voltammetric study of the redox chemistry of 2,3-dihydroxy-quinoxaline and its zinc complexes in non-aqueous medium. Polyhedron 1998, 17, 2109–2114. [Google Scholar] [CrossRef]
- Babu, N.S.; Tedesse, S.; Lelisho, T.A. Computational and electrochemical studies on the redox reaction of for quinoxalin-2(H)-one and its derivatives in aqueous solution. J. Chem. Pharm. Res. 2013, 5, 61–69. [Google Scholar]
- Udilova, N.; Kozlov, A.V.; Bieberschulte, W.; Frei, K.; Ehrenberger, K.; Nohl, K. The antioxidant activity of caroverine. Biochem. Pharmacol. 2003, 65, 59–65. [Google Scholar] [CrossRef]
- Jonsson, M.; Lind, J.; Reitberger, T.; Eriksen, T.E. Redox Chemistry of Substituted Benzenes. The One-Electron Reduction Potentials of Methoxy-Substituted Benzene Radical Cations. J. Phys. Chem. 1993, 97, 11278–11282. [Google Scholar] [CrossRef]
- Jonsson, M.; Lind, J.; Eriksen, T.E.; Merenyi, G. Redox and Acidity Properties of 4-Substituted Aniline Radical Cations in Water. J. Am. Chem. Soc. 1994, 116, 1423–1427. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Assary, R.S.; Brushett, F.R.; Curtiss, L.A. Reduction potential predictions of some aromatic nitrogen-containing molecules. RSC Adv. 2014, 4, 57442–57451. [Google Scholar] [CrossRef]
- Hirai, H.; Takahashi-Suziki, I.; Shimomura, T.; Fukusawa, K.; Machida, T.; Takaki, T.; Kobayashi, M.; Eguchi, T.; Oki, H.; Arai, T.; et al. Potent anti-tumor activity of a macrocycle-quinoxalinone class pan-Cdk inhibitor in vitro and in vivo. Investig. New Drugs 2011, 29, 534–543. [Google Scholar] [CrossRef]
- Mori, Y.; Hirokawa, T.; Aoki, K.; Satomi, H.; Takeda, S.; Aburada, M.; Miyamoto, K. Structure Activity Relationships of Quinoxalin-2-one Derivatives as Platelet-Derived Growth Factor-β Receptor (PDGFβ R) Inhibitors, Derived from Molecular Modeling. Chem. Pharm. Bull. 2008, 56, 682–687. [Google Scholar] [CrossRef]
- Bobrowski, K. Radiation induced radical reactions. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; John Wiley&Sons Ltd.: Hoboken, NJ, USA, 2012; Volume 1, pp. 395–432. [Google Scholar]
- Houée-Levin, C.; Bobrowski, K. The use of methods of radiolysis to explore the mechanisms of free radical modifications in proteins. J. Proteom. 2013, 92, 51–62. [Google Scholar] [CrossRef]
- Augusto, O.; Bonini, M.G.; Amanso, A.M.; Linares, E.; Santos, C.C.X.; De Menezes, S.L. Nitrogen dioxide and carbonate radical anion: Two emerging radicals in biology. Free Radic. Biol. Med. 2002, 32, 841–859. [Google Scholar] [CrossRef]
- Wojnarovits, L.; Toth, T.; Takacs, E. Rate constants of carbonate radical anion reactions with molecules of environmental interestin aqueous solution: A review. Sci. Total Environ. 2020, 717, 137219. [Google Scholar] [CrossRef]
- Medinas, D.B.; Cerchiaro, G.; Trindade, D.F.; Augusto, O. The Carbonate Radical and Related Oxidants Derived from Bicarbonate Buffer. IUBMB Life 2007, 59, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Hoffman, M.Z. Rate constants for the reaction of the carbonate radical with compounds of biochemical interest in neutral aqueous solution. Radiat. Res. 1973, 56, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Eibenberger, H.; Steenken, S.; O’Neill, P.; Schulte-Frohlinde, D. Pulse radiolysis and electron spin resonance studies concerning the reaction of SO4●− with alcohols and ethers in aqueous solution. J. Phys. Chem. 1978, 82, 749–750. [Google Scholar] [CrossRef]
- Dogan, I.; Steenken, S.; Schulte-Frohlinde, D.; Icli, S. Electron spin resonance and pulse radiolysis studies on the reaction of hydroxyl radical and sulfate (1−) radical with five-membered heterocyclic compounds in aqueous solution. J. Phys. Chem. 1990, 94, 1887–1894. [Google Scholar] [CrossRef]
- O’Neill, P.; Steenken, S.; Schulte-Frohlinde, D. Formation of radical cations of methoxylated benzenes by reaction with OH radicals, Tl2+, Ag2+, and SO4●− in aqueous solution. An optical and conductometric pulse radiolysis and in situ radiolysis electron spin resonance study. J. Phys. Chem. 1975, 79, 2773–2779. [Google Scholar] [CrossRef]
- Wojnarovits, L.; Takacs, E. Rate constants of sulfate radical anion reactions with organic molecules: A review. Chemosphere 2019, 220, 1014–1032. [Google Scholar] [CrossRef]
- Dixon, W.T.; Murphy, D. Determination of the Acidity Constants of some Phenol Radical Cations by means of Electron Spin Resonance. J. Chem. Soc. Faraday Trans. 1976, 72, 1221–1230. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Structure and Acid-Base Properties of One-Electron-Oxidized Deoxyguanosine, Guanosine and 1-Methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Jovanovič, S.V.; Simic, M.G. Repair of tryptophan radicals by antioxidants. J. Free Radic. Biol. Med. 1985, 1, 125–129. [Google Scholar] [CrossRef]
- Newkome, G.R.; Hager, D.C. Indoles, Part 1. Chemistry of Heterocyclic Compounds; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Posener, M.L.; Adams, G.E.; Wardman, P.; Cundall, R.B. Mechanism of Tryptophan Oxidation by some Inorganic Radical-Anions: A Pulse Radiolysis Study. J. Chem. Soc. Faraday Trans. 1976, 72, 2231–2239. [Google Scholar] [CrossRef]
- Balkas, T.I.; Fendler, J.H.; Schuler, R.H. Radiolysis of Aqueous Solutions of Methyl Chloride. The Concentration Dependence for Scavenging Electrons within Spurs. J. Phys. Chem. 1970, 74, 4497–4505. [Google Scholar]
- Schuler, R.H.; Hartzell, A.L.; Behar, B. Track effects in radiation chemistry. Concentration dependence for the scavenging of OH by ferrocyanide in N2O-saturated aqueous solutions. J. Phys. Chem. 1981, 85, 192–199. [Google Scholar] [CrossRef]
- Wardman, P. The reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1753. [Google Scholar] [CrossRef]
- Bobrowski, K. Free radicals in chemistry, biology, and medicine: Contribution of radiation chemistry. Nukleonika 2005, 50 (Suppl. 3), S67–S76. [Google Scholar]
- Mirkowski, J.; Wisniowski, P.; Bobrowski, K. INCT Annual Report 2000; INCT: Warsaw, Poland, 2001. [Google Scholar]
- Schuler, R.H.; Patterson, L.K.; Janata, E. Yield for the scavenging of hydroxyl radicals in the radiolysis of nitrous oxide-saturated aqueous solutions. J. Phys. Chem. 1980, 84, 2088–2090. [Google Scholar] [CrossRef]
- Buxton, G.V. An overview of the radiation chemistry of liquids. In Radiation Chemistry: From Basics to Applications in Material and Life Sciences; Spotheim-Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Les Ulis, France, 2008; pp. 3–16. [Google Scholar]
- Janata, E.; Schuler, R.H. Rate constant for scavenging eaq- in N2O-saturated solutions. J. Phys. Chem. 1982, 86, 2078–2084. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (●OH/O●−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Milosavljevič, B.H.; LaVerne, J.A. Pulse Radiolysis of Aqueous Thiocyanate Solutions. J. Phys. Chem. A 2005, 109, 165–168. [Google Scholar] [CrossRef][Green Version]
- Hohenberg, P.C.; Kohn, W.; Sham, L.J. The beginning and some thoughts on the future. In Density Functional Theory of Many-Fermion Systems; Trickey, S.B., Ed.; Academic Press: San Diego, CA, USA, 1990; pp. 7–26. [Google Scholar]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA—Type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning Jr, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Thanthiriwatte, K.S.; Hohenstein, E.G.; Burns, L.A.; Sherrill, C.D. Assessment of the Performance of DFT and DFT-D Methods for Describing Distance Dependence of Hydrogen-Bonded Interactions. J. Chem. Theory Comput. 2011, 7, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Chen, H. Comparative Assessment of DFT Performances in Ru- and Rh-Promoted σ-Bond Activations. J. Chem. Theory Comput. 2015, 11, 1428–1438. [Google Scholar] [CrossRef]
- Dobrowolski, J.C.; Lipiński, P.F.; Karpińska, G. Substituent Effect in the First Excited Singlet State of Monosubstituted Benzenes. J. Phys. Chem. A 2018, 122, 4609–4621. [Google Scholar] [CrossRef]
- Graef, E.L.; Martins, J.B.L. Analysis of lowest energy transitions at TD-DFT of pyrene in vacuum and solvent. J. Mol. Model. 2019, 25, 183. [Google Scholar] [CrossRef]
- Larsson, J.; Tong, L.; Chen, T.; Nolan, M.; Greer, J.C. A basis set study for the calculation of electronic excitations using Monte Carlo configuration interaction. J. Chem. Phys. 2001, 114, 15. [Google Scholar] [CrossRef]
- Hickey, A.L.; Rowley, C.N. Benchmarking Quantum Chemical Methods for the Calculation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A 2014, 118, 3678–3687. [Google Scholar] [CrossRef]
- Witte, J.; Neaton, J.B.; Head-Gordon, M. Push it to the limit: Characterizing the convergence of common sequences of basis sets for intermolecular interactions as described by density functional theory. J. Chem. Phys. A 2016, 144, 194306. [Google Scholar] [CrossRef]
- Kovacs, A.; Dobrowolski, J.C.; Ostrowski, S.; Rode, J.E. Benchmarking density functionals in conjunction withGrimme’s dispersion correction for noble gas dimers(Ne2,Ar2,Kr2,Xe2,Rn2). Int. J. Quantum Chem. 2017, 117, e25358. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- SigmaPlot Version 13; Systat Software, Inc.: San Jose, CA, USA, 2017.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 7-R-3-MeQ R | kforward M−1 s−1 | kbackward s−1 | K M−1 |
|---|---|---|---|
| −F | (6.1 ± 0.9) × 109 | (6.4 ± 0.9) × 105 | 9.5 × 103 |
| −H | (8.3 ± 1.1) × 109 | (1.3 ± 0.2) × 106 | 6.4 × 103 |
| −CF3 | (7.8 ± 1.4) × 109 | (6.7 ± 0.5) × 105 | 1.2 × 104 |
| −CN | (9.8 ± 1.0) × 109 | (3.7 ± 0.3) × 106 | 2.9 × 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skotnicki, K.; Ostrowski, S.; Dobrowolski, J.C.; De la Fuente, J.R.; Cañete, A.; Bobrowski, K. Spectral Probe for Electron Transfer and Addition Reactions of Azide Radicals with Substituted Quinoxalin-2-Ones in Aqueous Solutions. Int. J. Mol. Sci. 2021, 22, 633. https://doi.org/10.3390/ijms22020633
Skotnicki K, Ostrowski S, Dobrowolski JC, De la Fuente JR, Cañete A, Bobrowski K. Spectral Probe for Electron Transfer and Addition Reactions of Azide Radicals with Substituted Quinoxalin-2-Ones in Aqueous Solutions. International Journal of Molecular Sciences. 2021; 22(2):633. https://doi.org/10.3390/ijms22020633
Chicago/Turabian StyleSkotnicki, Konrad, Slawomir Ostrowski, Jan Cz. Dobrowolski, Julio R. De la Fuente, Alvaro Cañete, and Krzysztof Bobrowski. 2021. "Spectral Probe for Electron Transfer and Addition Reactions of Azide Radicals with Substituted Quinoxalin-2-Ones in Aqueous Solutions" International Journal of Molecular Sciences 22, no. 2: 633. https://doi.org/10.3390/ijms22020633
APA StyleSkotnicki, K., Ostrowski, S., Dobrowolski, J. C., De la Fuente, J. R., Cañete, A., & Bobrowski, K. (2021). Spectral Probe for Electron Transfer and Addition Reactions of Azide Radicals with Substituted Quinoxalin-2-Ones in Aqueous Solutions. International Journal of Molecular Sciences, 22(2), 633. https://doi.org/10.3390/ijms22020633