
Identification of Rare LRP5 Variants in a Cohort of Males with Impaired Bone Mass

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Genetic Analysis
2.2. Protein Structural Analysis
2.2.1. LRP5 Sequence Conservation and Domain Analysis and Investigation of Variants
2.2.2. Self-Analysis of LRP5 Repeated Domains
2.2.3. Pathogenicity and Stability Predictions
2.2.4. In Silico Classification of LRP5 VUS, Definition of a Prior Odds Ratio
2.2.5. OR for Pathogenicity for In Silico Analysis
2.2.6. Molecular Dynamics Simulation of p.R1036Q and p.R1135C Variants
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. DNA Isolation and Sequencing Analysis
4.3. Variant Filtering and Interpretation of Clinical Relevance
4.4. Bayesian Classificatory Model for LRP5 Pathogenicity Ranking
4.5. Molecular Dynamics Simulation
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kanis, J.A.; Melton, L.J.; Christiansen, C.; Johnston, C.C.; Khaltaev, N. The diagnosis of osteoporosis. J. Bone Miner. Res. 1994, 9, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Eisman, J.A. Genetics of osteoporosis. Endocr. Rev. 1999, 20, 788–804. [Google Scholar] [CrossRef]
- Stewart, T.L.; Ralston, S.H. Role of genetic factors in the pathogenesis of osteoporosis. J. Endocrinol. 2000, 166, 235–245. [Google Scholar] [CrossRef]
- Rivadeneira, F.; Mäkitie, O. Osteoporosis and Bone Mass Disorders: From Gene Pathways to Treatments. Trends Endocrinol. Metab. 2016, 27, 262–281. [Google Scholar] [CrossRef]
- Pouresmaeili, F.; Kamalidehghan, B.; Kamarehei, M.; Goh, Y.M. A comprehensive overview on osteoporosis and its risk factors. Clin. Risk Manag. 2018, 14, 2029–2049. [Google Scholar] [CrossRef]
- Medina-Gomez, C.; Kemp, J.P.; Trajanoska, K.; Luan, J.; Chesi, A.; Ahluwalia, T.S.; Mook-Kanamori, D.O.; Ham, A.; Hartwig, F.P.; Evans, D.S.; et al. Life-Course Genome-wide Association Study Meta-analysis of Total Body BMD and Assessment of Age-Specific Effects. Am. J. Hum. Genet. 2018, 102, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Trajanoska, K.; Rivadeneira, F. The genetic architecture of osteoporosis and fracture risk. Bone 2019, 126, 2–10. [Google Scholar] [CrossRef]
- Rinonapoli, G.; Ruggiero, C.; Meccariello, L.; Bisaccia, M.; Ceccarini, P.; Caraffa, A. Osteoporosis in Men: A Review of an Underestimated Bone Condition. Int. J. Mol. Sci. 2021, 22, 2105. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Semenov, M.; Kato, Y.; Spokony, R.; Liu, C.; Katsuyama, Y.; Hess, F.; Saint-Jeannet, J.P.; He, X. LDL-receptor-related proteins in Wnt signal transduction. Nature 2000, 407, 530–535. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 12, a007880. [Google Scholar] [CrossRef]
- Gong, Y.; Vikkula, M.; Boon, L.; Liu, J.; Beighton, P.; Ramesar, R.; Peltonen, L.; Somer, H.; Hirose, T.; Dallapiccola, B.; et al. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region 11q12-13. Am. J. Hum. Genet. 1996, 59, 146–151. [Google Scholar]
- Johnson, M.L.; Gong, G.; Kimberling, W.; Reckér, S.M.; Kimmel, D.B.; Recker, R.B. Linkage of a gene causing high bone mass to human chromosome 11 (11q12-13). Am. J. Hum. Genet. 1997, 60, 1326–1332. [Google Scholar] [CrossRef]
- Ferrari, S.L.; Deutsch, S.; Choudhury, U.; Chevalley, T.; Bonjour, J.P.; Dermitzakis, E.T.; Rizzoli, R.; Antonarakis, S.E. Polymorphisms in the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with variation in vertebral bone mass, vertebral bone size, and stature in whites. Am. J. Hum. Genet. 2004, 74, 866–875. [Google Scholar] [CrossRef][Green Version]
- Van Meurs, J.B.J.; Rivadeneira, F.; Jhamai, M.; Hugens, W.; Hofman, A.; van Leeuwen, J.P.T.M.; Pols, H.A.P.; Uitterlinden, A.G. Common genetic variation of the low-density lipoprotein receptor-related protein 5 and 6 genes determines fracture risk in elderly white men. J. Bone Miner. Res. 2006, 21, 141–150. [Google Scholar] [CrossRef]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Xiaowei, Z.; Weiyang, B.; Houfeng, Z. Twelve years of GWAS discoveries for osteoporosis and related traits: Advances, challenges and applications. Bone Res. 2021, 9, 23. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Schaper, E.; Kajava, A.V.; Hauser, A.; Anisimova, M. Repeat or not repeat?—Statistical validation of tandem repeat prediction in genomic sequences. Nucleic Acids Res. 2012, 40, 10005–10017. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, L.; Paladin, L.; Piovesan, D.; Tosatto, S.C.E. RepeatsDB-lite: A web server for unit annotation of tandem repeat proteins. Nucleic Acids Res. 2018, 46, 402–407. [Google Scholar] [CrossRef]
- Piovesan, D.; Necci, M.; Escobedo, N.; Monzon, A.M.; Hatos, A.; Mičetić, I.; Quaglia, F.; Paladin, L.; Ramasamy, P.; Dosztányi, Z.; et al. MobiDB: Intrinsically disordered proteins in 2021. Nucleic Acids Res. 2021, 49, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Gonzalez-Sancho, J.M.; Castelo-Soccio, L.A.; Howe, L.R.; Brown, A.M.C. Truncated mutants of the putative Wnt receptor LRP6/Arrow can stabilize beta-catenin independently of Frizzled proteins. Oncogene 2004, 23, 4873–4884. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Acebron, S.P. Wnt Signaling: Multivesicular Bodies Hold GSK3 Captive. Cell 2010, 143, 1044–1066. [Google Scholar] [CrossRef][Green Version]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, 344–347. [Google Scholar] [CrossRef]
- Scaini, M.C.; Minervini, G.; Elefanti, L.; Ghiorzo, P.; Pastorino, L.; Tognazzo, S.; Agata, S.; Quaggio, M.; Zullato, D.; Bianchi-Scarrà, G.; et al. CDKN2A unclassified variants in familial malignant melanoma: Combining functional and computational approaches for their assessment. Hum. Mutat. 2014, 35, 828–840. [Google Scholar] [CrossRef]
- Minervini, G.; Quaglia, F.; Tosatto, S.C.E. Insights into the proline hydroxylase (PHD) family, molecular evolution and its impact on human health. Biochimie 2015, 116, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Body, J.-J.; Bergmann, P.; Boonen, S.; Boutsen, Y.; Bruyere, O.; Devogelaer, J.-P.; Goemaere, S.; Hollevoet, N.; Kaufman, J.-M.; Milisen, K.; et al. Non-pharmacological management of osteoporosis: A consensus of the Belgian Bone Club. Osteoporos. Int. 2011, 22, 2769–2788. [Google Scholar] [CrossRef]
- Peacock, M.; Turner, C.H.; Econs, M.J.; Foroud, T. Genetics of osteoporosis. Endocr. Rev. 2002, 23, 303–326. [Google Scholar] [CrossRef]
- Bek, J.W.; Shochat, C.; De Clercq, A.; De Saffel, H.; Boel, A.; Metz, J.; Rodenburg, F.; Karasik, D.; Willaert, A.; Coucke, P.J. Lrp5 Mutant and Crispant Zebrafish Faithfully Model Human Osteoporosis, Establishing the Zebrafish as a Platform for CRISPR-Based Functional Screening of Osteoporosis Candidate Genes. J. Bone Min. Res. 2021. [Google Scholar] [CrossRef]
- Kato, M.; Patel, M.S.; Levasseur, R.; Lobov, I.; Chang, B.H.J.; Glass, D.A.; Hartmann, C.; Li, L.; Hwang, T.H.; Brayton, C.F.; et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 2002, 157, 303–314. [Google Scholar] [CrossRef]
- Hartikka, H.; Mäkitie, O.; Männikkö, M.; Doria, A.S.; Daneman, A.; Cole, W.G.; Ala-Kokko, L.; Sochett, E.B. Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. Bone Miner. Res. 2005, 20, 783–789. [Google Scholar] [CrossRef]
- Saarinen, A.; Saukkonen, T.; Kivelä, T.; Lahtinen, U.; Laine, C.; Somer, M.; Toiviainen-Salo, S.; Cole, W.G.; Lehesjoki, A.E.; Mäkitie, O. Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and hypercholesterolaemia. Clin. Endocrinol. 2010, 72, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Korvala, J.; Jüppner, H.; Mäkitie, O.; Sochett, E.; Schnabel, D.; Mora, S.; Bartels, C.F.; Warman, M.L.; Deraska, D.; Cole, W.G.; et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med. Genet. 2012, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Biechele, T.; Wei, Z.; Morrone, S.; Moon, R.T.; Wang, L.; Xu, W. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 2011, 18, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Caetano da Silva, C.; Ricquebourg, M.; Orcel, P.; Fabre, S.; Funck-Brentano, T.; Cohen-Solal, M.; Collet, C. More severe phenotype of early-onset osteoporosis associated with recessive form of LRP5 and combination with DKK1 or WNT3A. Mol. Genet. Genom. Med. 2021, 3, e1681. [Google Scholar] [CrossRef]
- Crabbe, P.; Balemans, W.; Willaert, A.; Van Pottelbergh, I.; Cleiren, E.; Coucke, P.J. Missense Mutations in LRP5 Are Not a Common Cause of Idiopathic Osteoporosis in Adult Men. J. Bone Mineral. Res. 2005, 20, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, T.; Maffezzoni, F.; Pezzaioli, L.C.; Delbarba, A.; Cappelli, C.; Ferlin, A. MANAGEMENT OF ENDOCRINE DISEASE: Male osteoporosis: Diagnosis and management—should the treatment and the target be the same as for female osteoporosis? Eur. J. Endocrinol. 2020, 183, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Dessimoz, C.; Gonnet, G.H. OMA Browser--exploring orthologous relations across 352 complete genomes. Bioinformatics 2007, 23, 2180–2182. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 8, 427–432. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 8, 506–515. [Google Scholar]
- Piovesan, D.; Walsh, I.; Minervini, G.; Tosatto, S.C.E. FELLS: Fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Baoshan, G.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, 1062–1067. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G. Ng PC. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, 452–457. [Google Scholar] [CrossRef]
- López-Ferrando, V.; Gazzo, A.; de la Cruz, X.; Orozco, M.; Gelpí, J.L. PMut: A web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017, 45, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qiet, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Piovesan, D.; Minervini, G.; Tosatto, S.C.E. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016, 44, 367–374. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
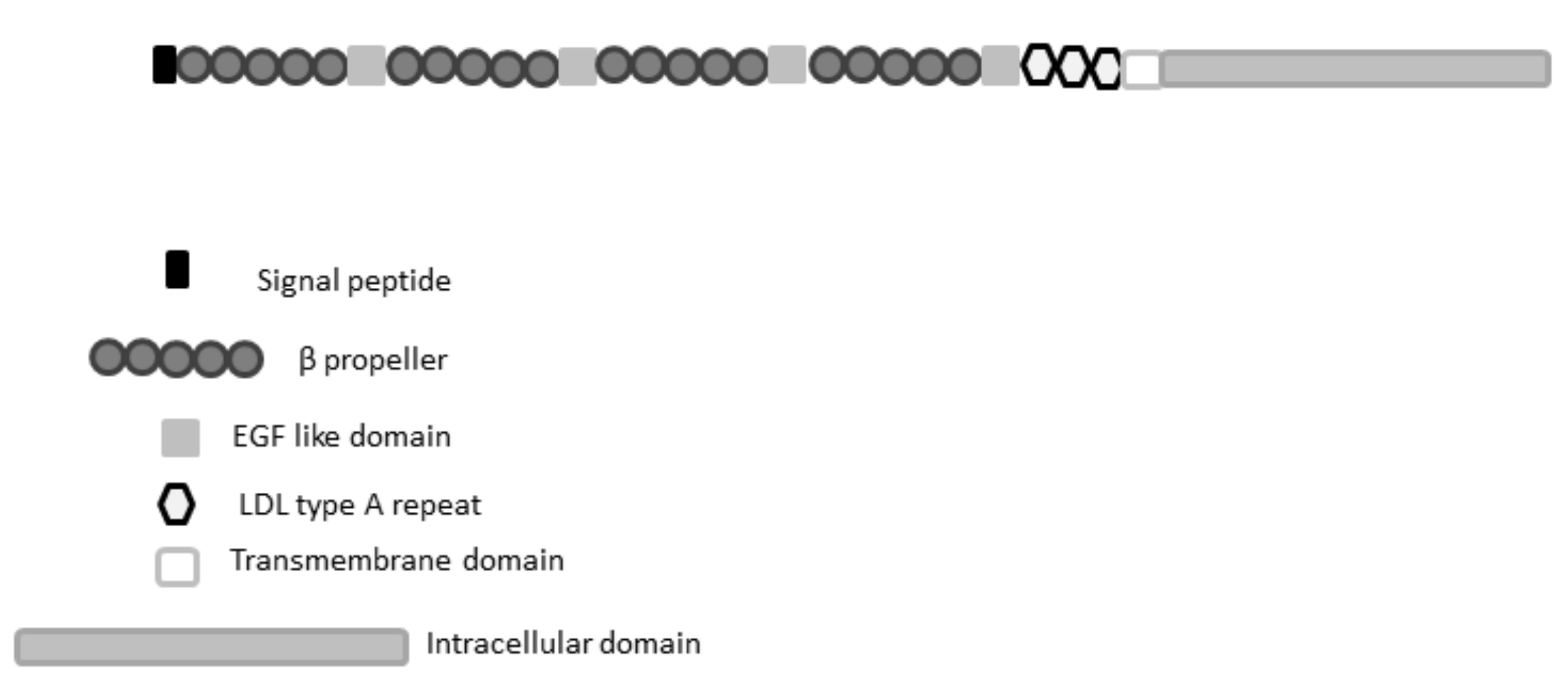{kind=link}
| ID Patients | Protein, Codon | ACMG | Age | BMDL(g/cm2) | TL | ZL | BMDTH(g/cm2) | T TH | Z TH |
|---|---|---|---|---|---|---|---|---|---|
| 11938 | p.V99L, G>T | LP | 55 | 0.90 | −2.6 | −2.3 | 0.86 | −1.6 | −0.9 |
| 9620 | p.G333S, G>A | VUS | 29 | 0.84 | −2.2 | −2.2 | 0.95 | −0.5 | −0.5 |
| 8957 | p.E341K, G>A | VUS | 28 | 0.67 | −3.8 | −3.8 | 0.78 | −1.6 | −1.6 |
| 12639 | p.T443M, C>T | VUS | 45 | 0.85 | −2.2 | −2.1 | 1.10 | 0.5 | 0.6 |
| 7971 | p.R1036Q, G>A | LP | 31 | 0.87 | −2.0 | −2.0 | 0.98 | −0.3 | −0.3 |
| 12141 | p.R1135C, C>T | VUS | 38 | 0.74 | −3.1 | −3.1 | 0.61 | −2.7 | −2.7 |
| 12025 | p.R1342P, C>T | VUS | 43 | 0.86 | −2.0 | −2.0 | 0.94 | −0.6 | −0.5 |
| 11241 | p.A1525V, C>T | B | 43 | 0.86 | −2.1 | −2.0 | 0.81 | −1.4 | −1.2 |
| 3801 | p.A1525V, C>T | B | 34 | 0.81 | −2.5 | −2.5 | 0.91 | −0.8 | −0.7 |
| 10517 | p.A1537V, C>T | VUS | 48 | 0.81 | −2.5 | −2.4 | 1.04 | 0.0 | 0.3 |
| 12,007 | p.S1585L, C>T | VUS | 36 | 0.71 | −3.4 | −3.4 | 0.75 | −1.8 | −1.7 |
| Variant | PolyPhen-2 | SIFT | Pmut | MuPro ΔΔG |
|---|---|---|---|---|
| p.V99L | Benign | Tolerated | Neutral | −0.44734934 (DECREASE stability) |
| p.G333S | Benign | Tolerated | Neutral | −1.3066037 (DECREASE stability) |
| p.E341K | Benign | Tolerated | Disease | −0.82809476 (DECREASE stability) |
| p.T443M | Probably damaging | Tolerated | Neutral | −0.26659723 (DECREASE stability) |
| p.R1036Q | Possibly damaging | Tolerated | Neutral | −1.171288 (DECREASE stability) |
| p.R1135C | Probably damaging | Tolerated | Disease | −0.47636602 (DECREASE stability) |
| p.R1342P | Probably damaging | Tolerated | Disease | −0.94002976 (DECREASE stability) |
| p.A1525V | Benign | Tolerated | Neutral | −0.35033282 (DECREASE stability) |
| p.A1537V | Benign | Not tolerated | Neutral | −0.31014871 (DECREASE stability) |
| p.S1585L | Probably damaging | Not tolerated | Disease | −0.30474209 (DECREASE stability) |
| Variant | Prediction Score | Odd Score | Merged Score In Silico | Prior OR | Posterior OR | Posterior OR% | Meaning | Class |
|---|---|---|---|---|---|---|---|---|
| p.V99L | 1 | 1 | 2 | 1 | 2 | 22.22 | LN | 2 |
| p.G333S | 1 | 1 | 2 | 1 | 2 | 22.22 | LN | 2 |
| p.E341K | 2 | 3 | 5 | 1 | 5 | 55.56 | LP | 4 |
| p.T443M | 2 | 3 | 5 | 1 | 5 | 55.56 | LP | 4 |
| p.R1036Q | 2 | 5 | 7 | 1.25 | 8.745 | 97.22 | P | 5 |
| p.R1135C | 2 | 5 | 8 | 1.25 | 10 | 111.11 | P | 5 |
| p.R1342P | 3 | 3 | 6 | 1 | 6 | 66.67 | LP | 4 |
| p.A1515V | 2 | 3 | 5 | 1.25 | 6.25 | 69.44 | LP | 4 |
| p.A1537V | 2 | 3 | 5 | 1 | 5 | 55.56 | LP | 4 |
| p.S1585L | 4 | 1 | 5 | 1 | 5 | 55.56 | LP | 4 |
| Patient (N = 128) | |
|---|---|
| Age (years) | 41.6 ± 8.7 |
| Height (cm) | 178.1 ± 5.6 |
| Weight (kg) | 77.8 ± 8.4 |
| BMI (kg/m2) | 23.1 ± 2.2 |
| BMDL (g/cm2) | 0.82 ± 0.07 |
| TL | <−2.5 ± 0.55 |
| ZL | <−2.48 ± 0.59 |
| BMDL (g/cm2) | 0.88 ± 0.11 |
| TTH | <−1.07 ± 0.79 |
| ZTH | <−0.99 ± 0.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, M.S.; Minervini, G.; Di Nisio, A.; Merico, M.; Bueno Marinas, M.; De Toni, L.; Pilichou, K.; Garolla, A.; Foresta, C.; Ferlin, A. Identification of Rare LRP5 Variants in a Cohort of Males with Impaired Bone Mass. Int. J. Mol. Sci. 2021, 22, 10834. https://doi.org/10.3390/ijms221910834
Rocca MS, Minervini G, Di Nisio A, Merico M, Bueno Marinas M, De Toni L, Pilichou K, Garolla A, Foresta C, Ferlin A. Identification of Rare LRP5 Variants in a Cohort of Males with Impaired Bone Mass. International Journal of Molecular Sciences. 2021; 22(19):10834. https://doi.org/10.3390/ijms221910834
Chicago/Turabian StyleRocca, Maria Santa, Giovanni Minervini, Andrea Di Nisio, Maurizio Merico, Maria Bueno Marinas, Luca De Toni, Kalliopi Pilichou, Andrea Garolla, Carlo Foresta, and Alberto Ferlin. 2021. "Identification of Rare LRP5 Variants in a Cohort of Males with Impaired Bone Mass" International Journal of Molecular Sciences 22, no. 19: 10834. https://doi.org/10.3390/ijms221910834
APA StyleRocca, M. S., Minervini, G., Di Nisio, A., Merico, M., Bueno Marinas, M., De Toni, L., Pilichou, K., Garolla, A., Foresta, C., & Ferlin, A. (2021). Identification of Rare LRP5 Variants in a Cohort of Males with Impaired Bone Mass. International Journal of Molecular Sciences, 22(19), 10834. https://doi.org/10.3390/ijms221910834