
Evaluating Molecular Docking Software for Small Molecule Binding to G-Quadruplex DNA

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
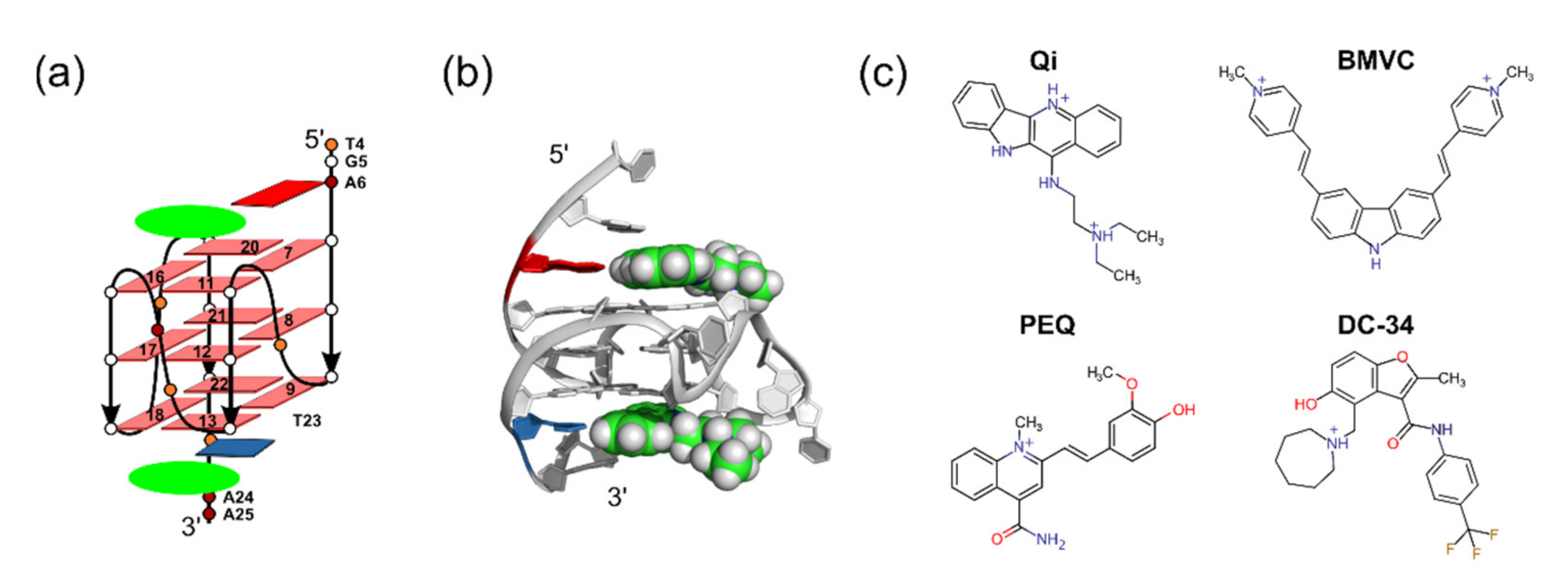
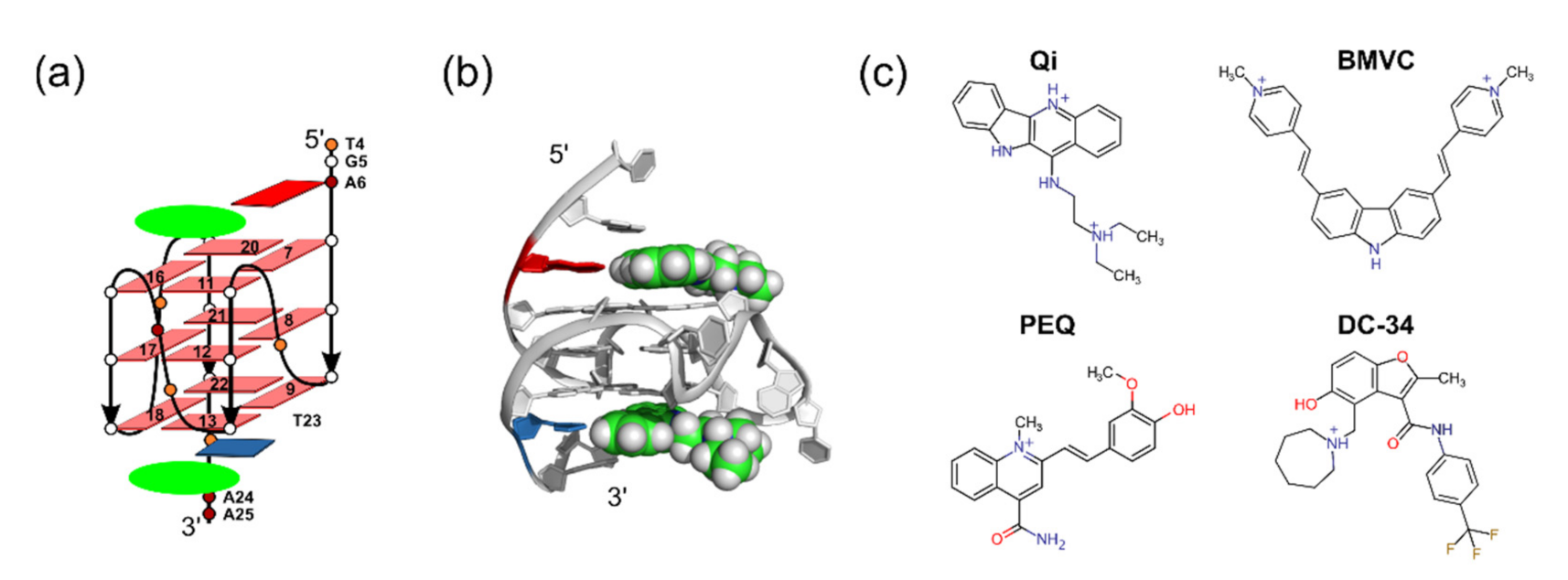
2.1. Using MycG4 as a Model System for Docking Evaluation
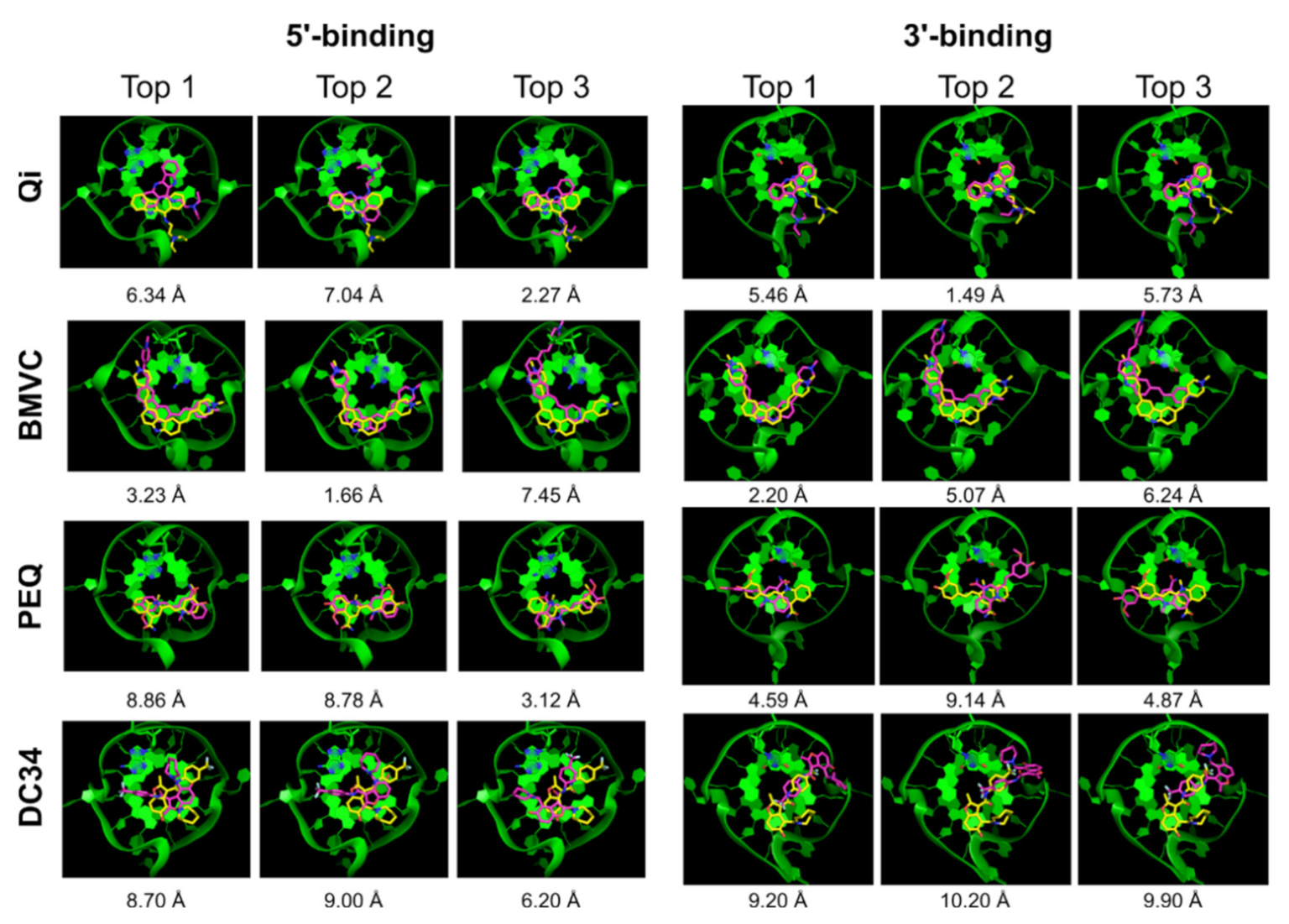
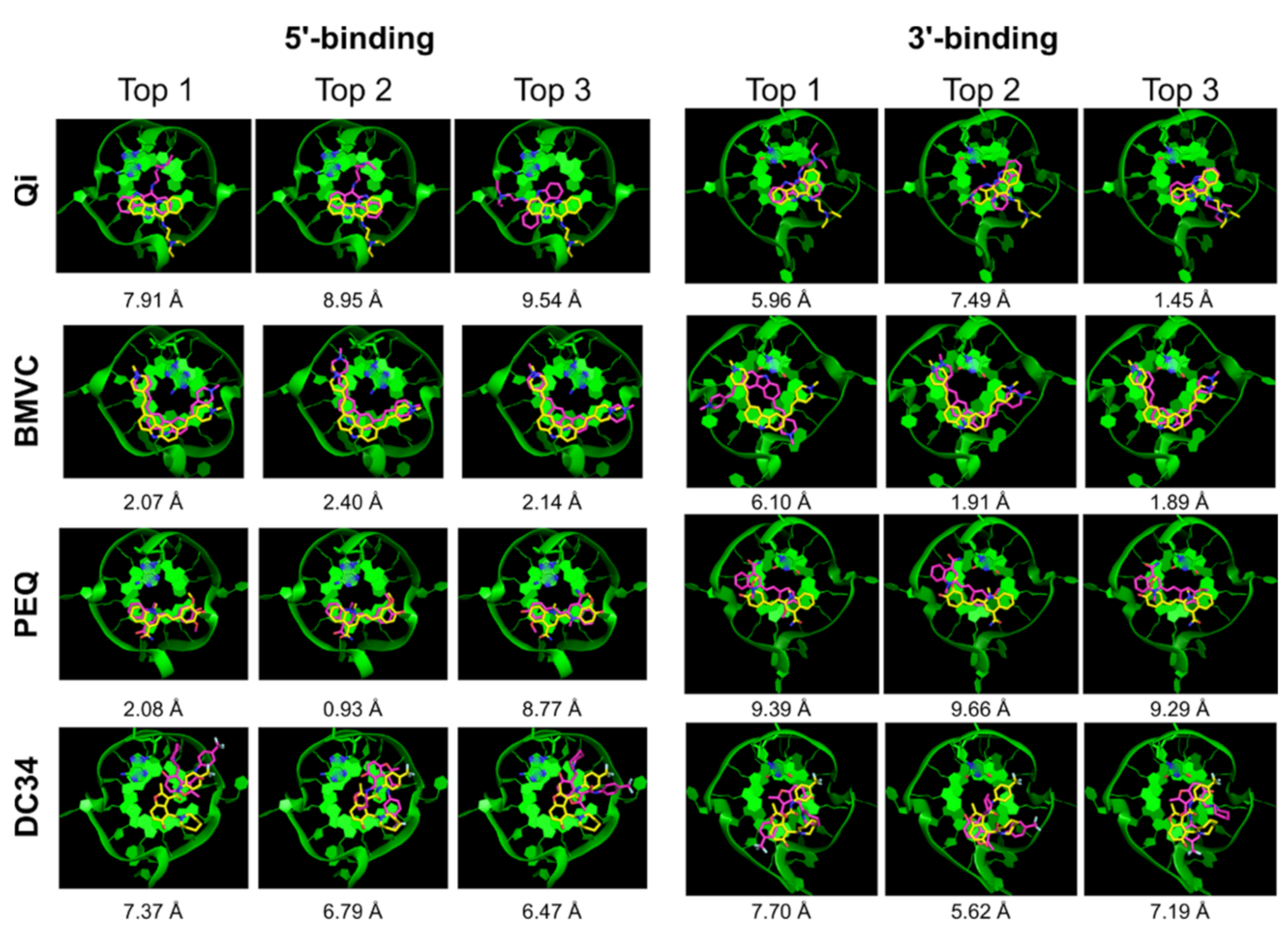
2.2. Docking Study Using Four Commonly Used Docking Programs
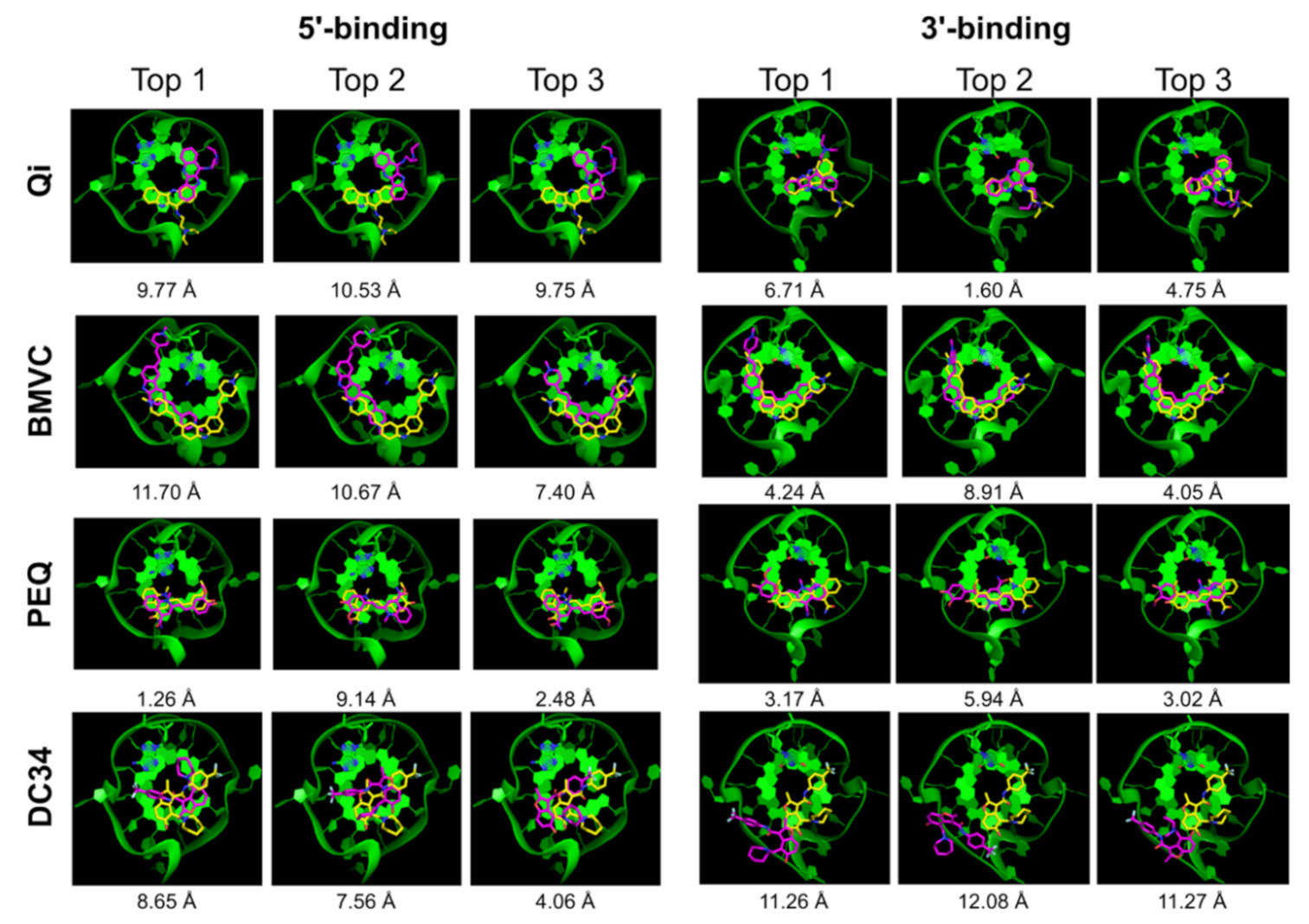
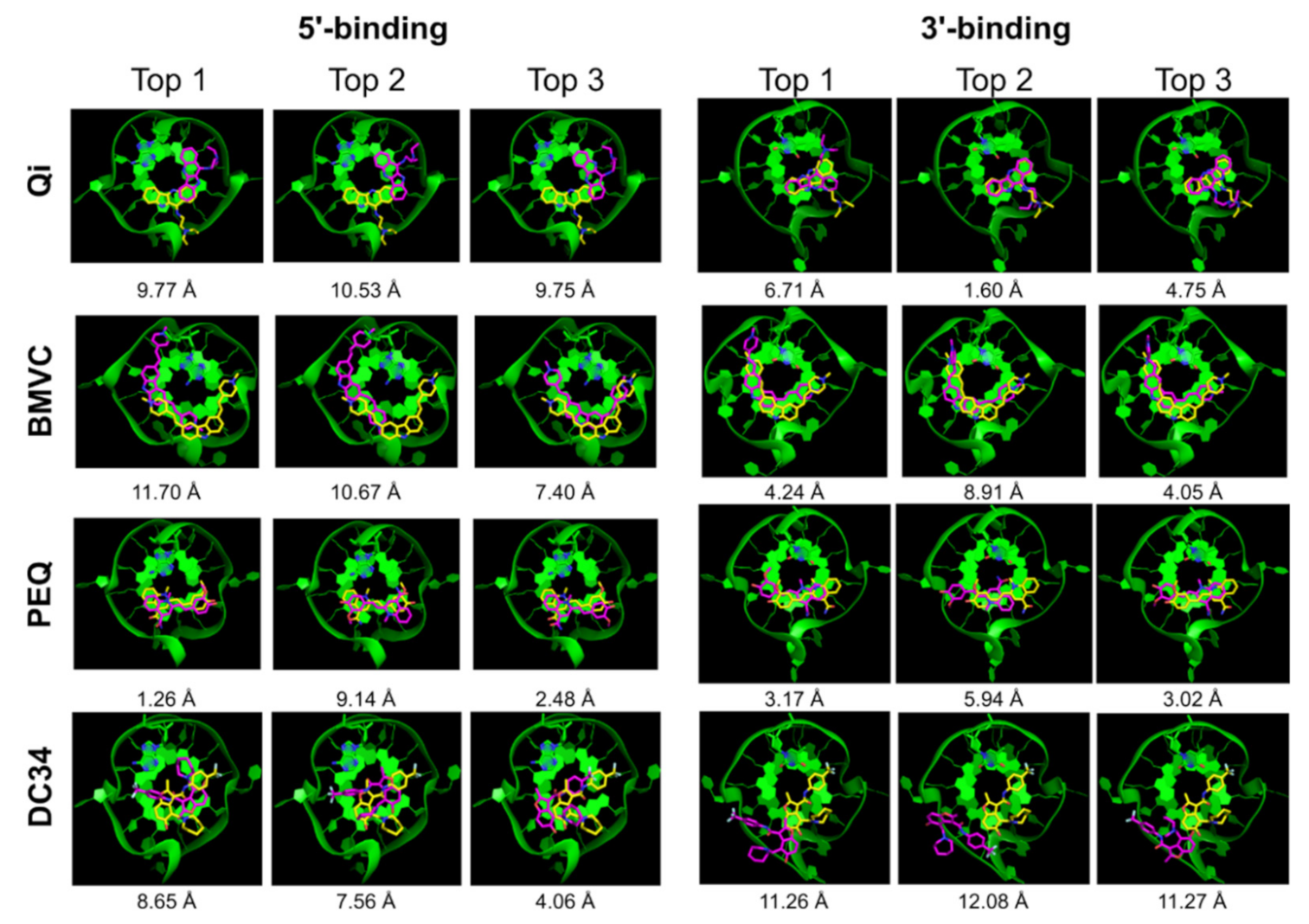
2.2.1. AutoDock Vina
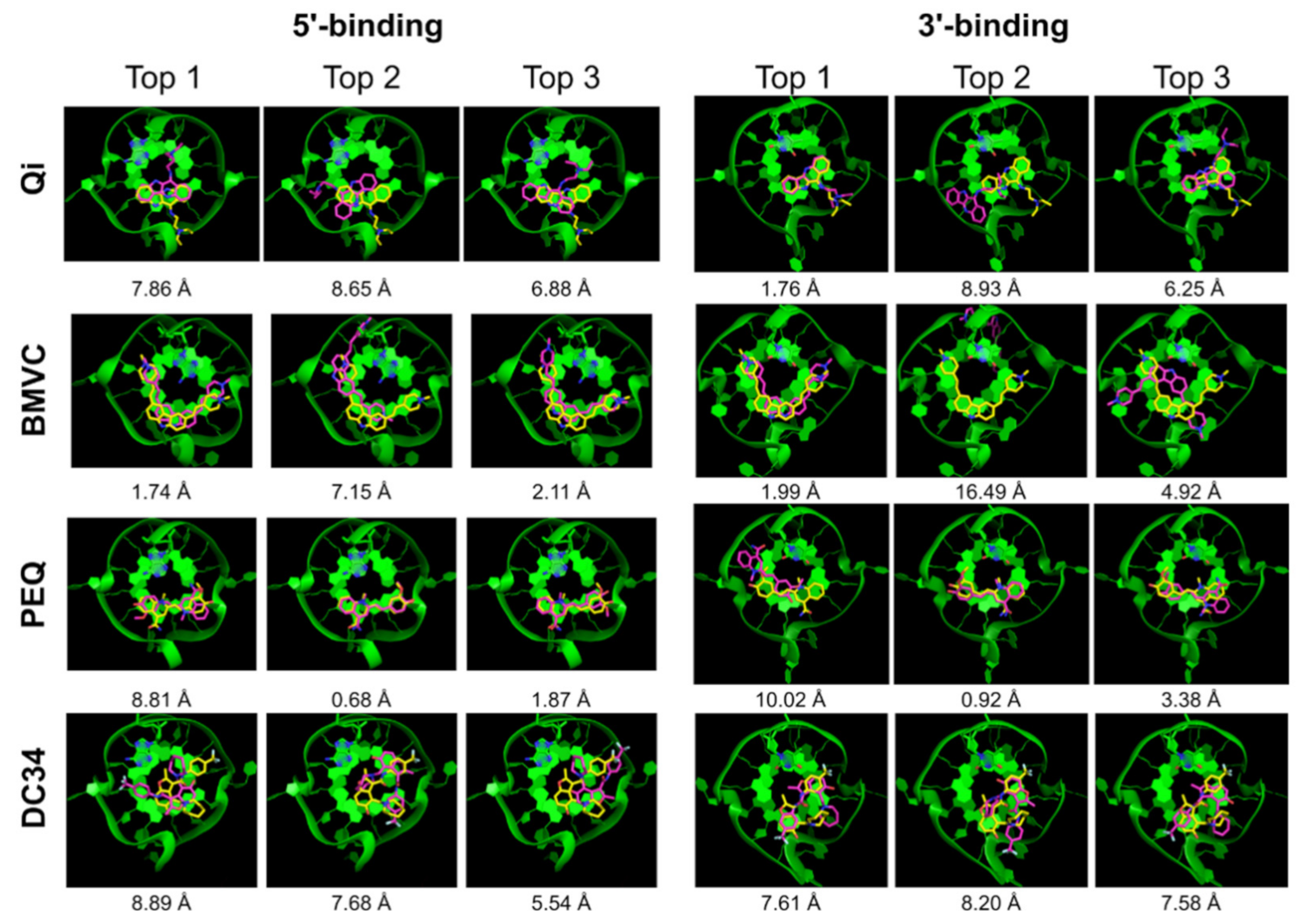
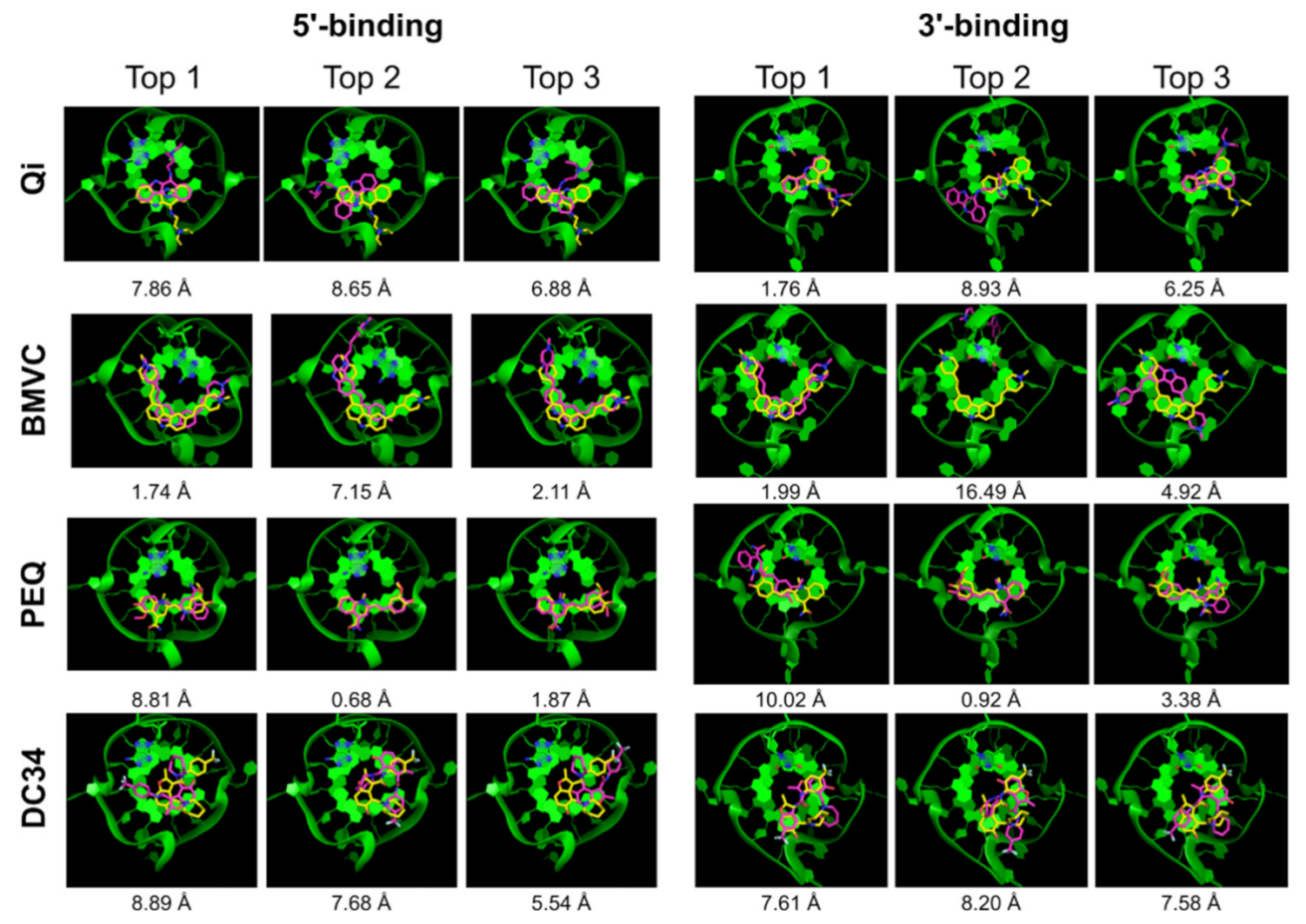
2.2.2. DOCK 6
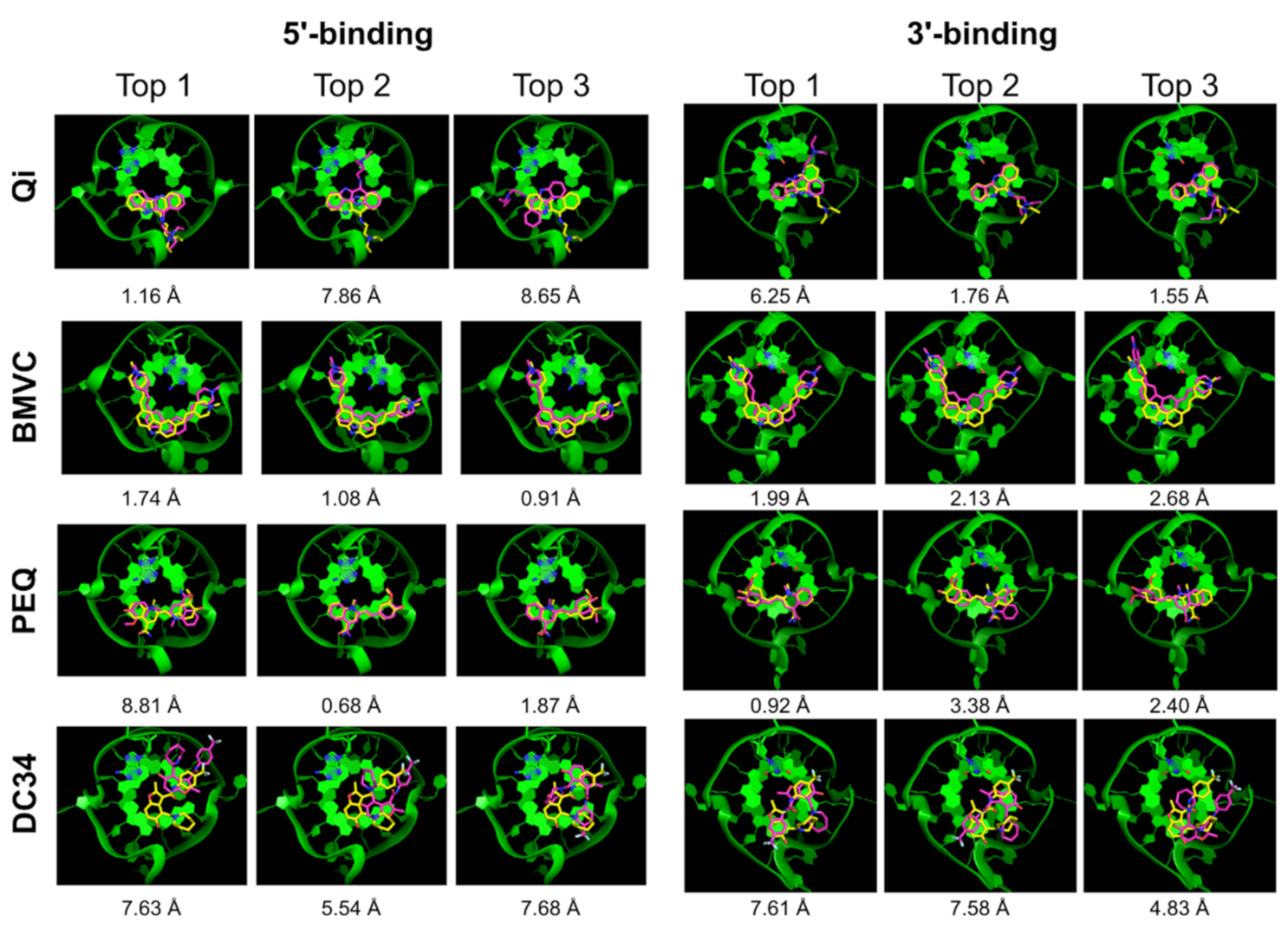
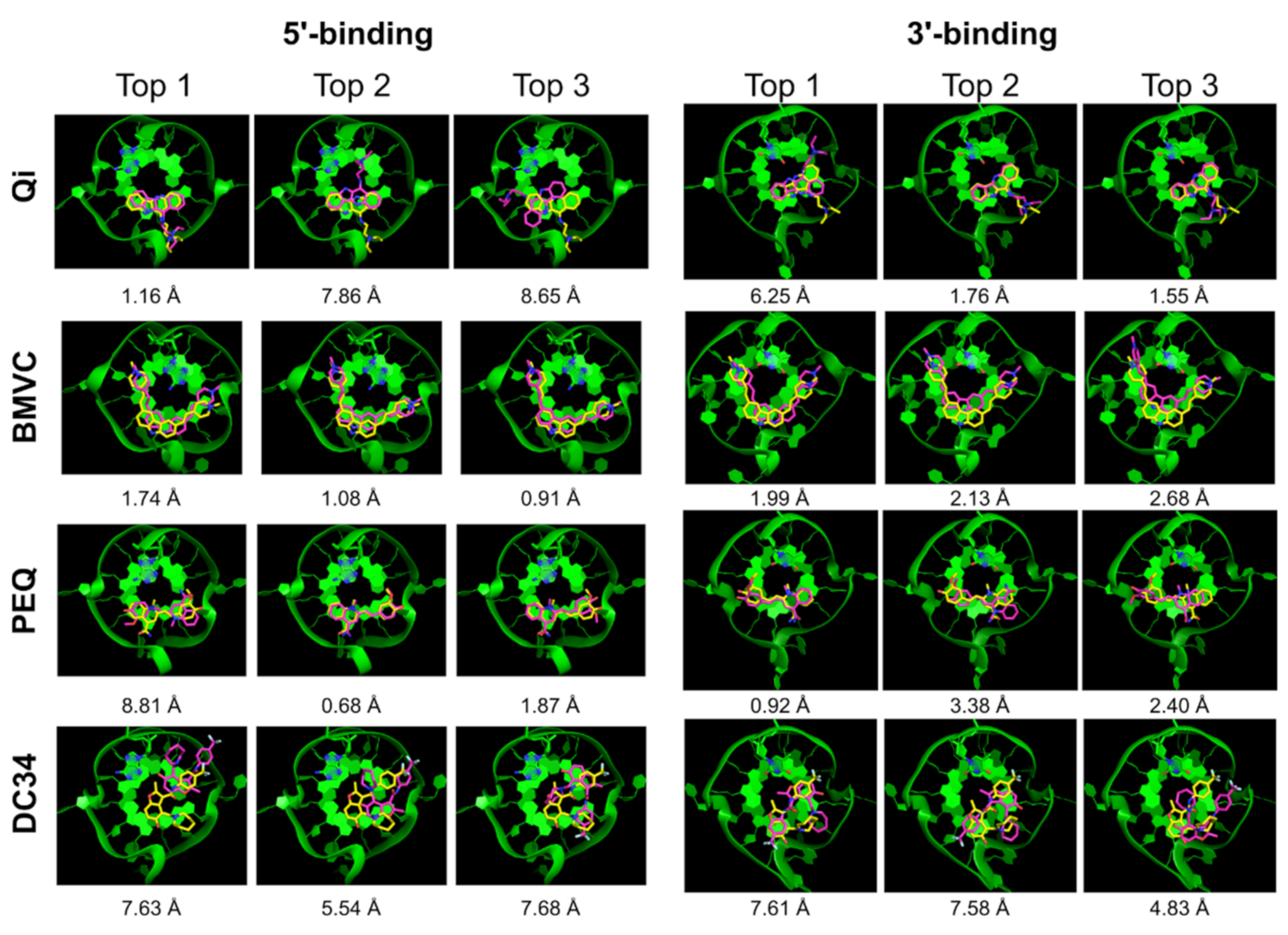
2.2.3. Glide SP
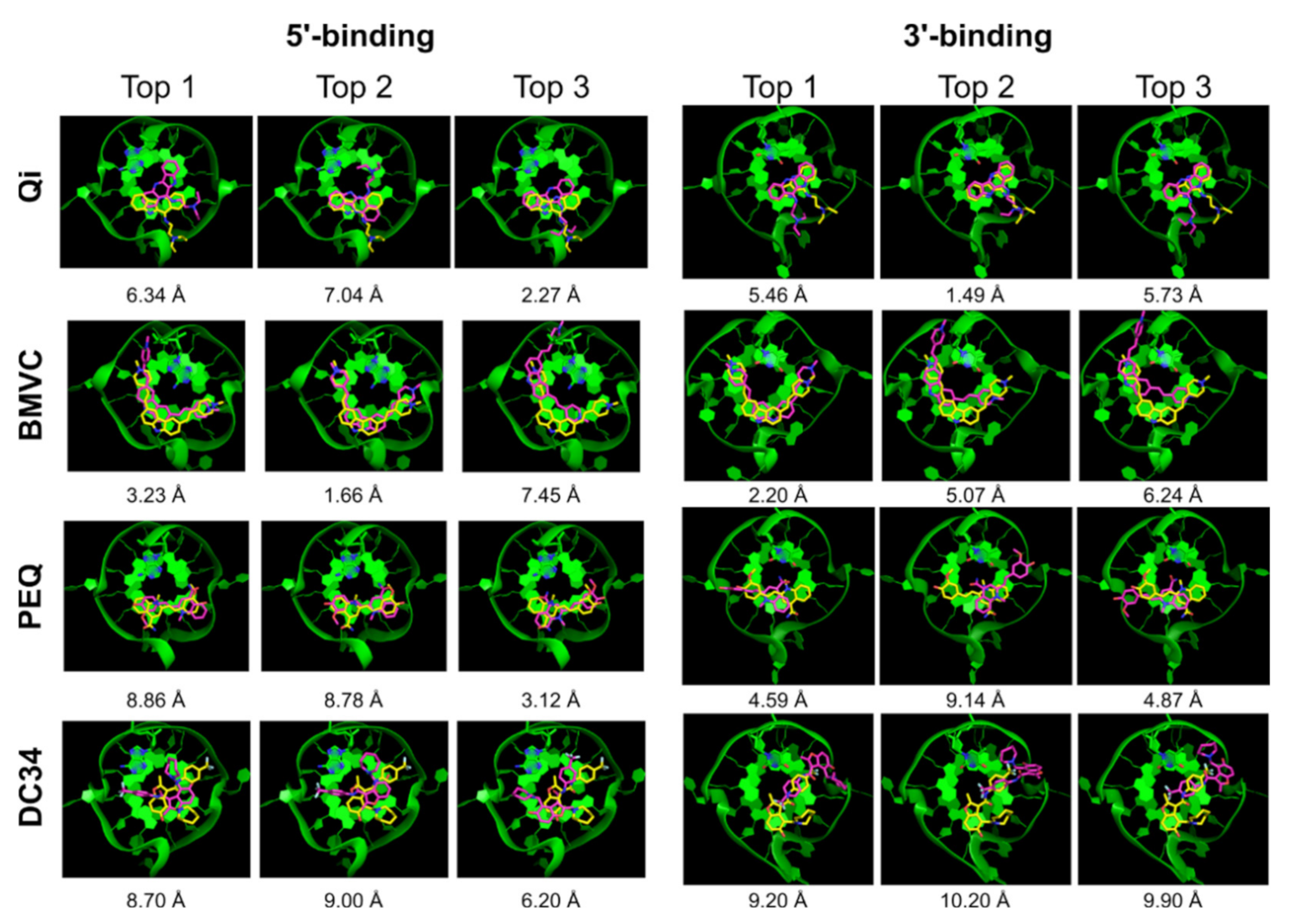
2.2.4. RxDock
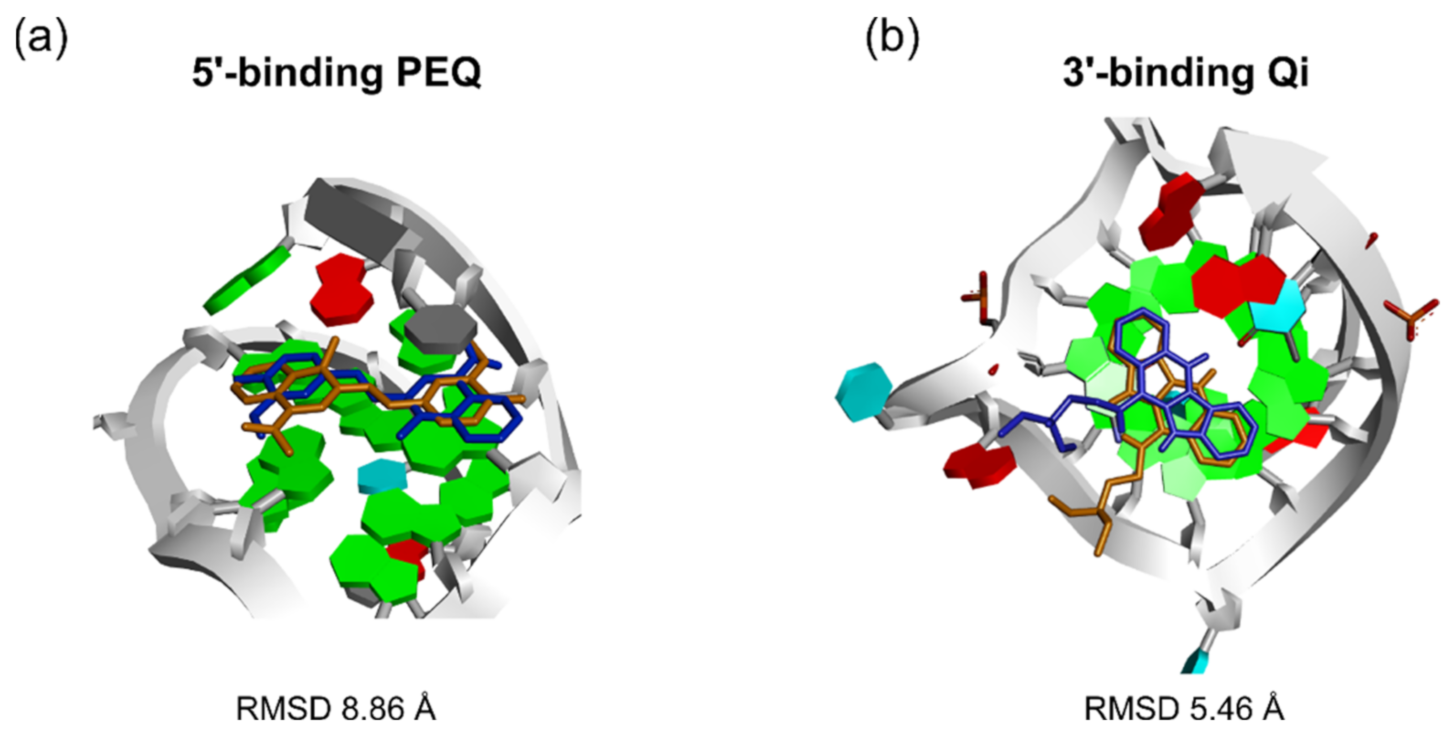
2.3. Comparison of the Four Docking Programs
3. Materials and Methods
3.1. Structure Files
3.2. AutoDock Vina
3.3. DOCK 6
3.4. Glide
3.5. RxDock
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Futur. Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef] [Green Version]
- Haensel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, D. Sequence, stability, and structure of G-quadruplexes and their interactions with drugs. Curr. Protoc. Nucleic Acid Chem. 2012, 50, 17.5.1–17.5.17. [Google Scholar] [CrossRef]
- Yang, D. G-quadruplex DNA and RNA. In G-Quadruplex Nucleic Acids; Yang, D., Lin, C., Eds.; Springer: New York, NY, USA, 2019; Volume 2035, pp. 1–24. ISBN 978-1-4939-9665-0. [Google Scholar]
- Neidle, S. Quadruplex nucleic acids as novel therapeutic targets. J. Med. Chem. 2016, 59, 5987–6011. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, E.; Richter, S.N. Survey and summary G-quadruplexes and G-quadruplex ligands: Targets and tools in antiviral therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef]
- Lightfoot, H.L.; Hagen, T.; Tatum, N.; Hall, J. The diverse structural landscape of quadruplexes. FEBS Lett. 2019, 593, 2083–2102. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, E.P.; Holmes, A.; Verga, D.; Teulade-Fichou, M.-P.; Nicolas, A.; Londoño-Vallejo, A. Thermodynamically stable and genetically unstable G-quadruplexes are depleted in genomes across species. Nucleic Acids Res. 2019, 47, 6098–6113. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic MYC as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Hurley, L.H. Targeting MYC expression through G-quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.-B.; Elsayed, M.S.A.; Wu, G.; Deng, N.; Cushman, M.; Yang, D. Indenoisoquinoline topoisomerase inhibitors strongly bind and stabilize the MYC promoter g-quadruplex and downregulate MYC. J. Am. Chem. Soc. 2019, 141, 11059–11070. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking screens for novel ligands conferring new biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Wang, S.; Balius, T.; Singh, I.; Levit, A.; Moroz, Y.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nat. Cell Biol. 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Sullivan, H.-J.; Chen, B.; Wu, C. Molecular dynamics study on the binding of an anticancer DNA G-quadruplex stabilizer, CX-5461, to human telomeric, c-KIT1, and c-Myc G-quadruplexes and a DNA duplex. J. Chem. Inf. Model. 2020, 60, 5203–5224. [Google Scholar] [CrossRef]
- Deng, N.; Xia, J.; Wickstrom, L.; Lin, C.; Wang, K.; He, P.; Yin, Y.; Yang, D. Ligand selectivity in the recognition of protoberberine alkaloids by hybrid-2 human telomeric G-quadruplex: Binding free energy calculation, fluorescence binding, and NMR experiments. Molecules 2019, 24, 1574. [Google Scholar] [CrossRef] [Green Version]
- Deng, N.; Wickstrom, L.; Cieplak, P.; Lin, C.; Yang, D. Resolving the ligand-binding specificity in c-MYC G-quadruplex DNA: Absolute binding free energy calculations and SPR experiment. J. Phys. Chem. B 2017, 121, 10484–10497. [Google Scholar] [CrossRef]
- Rocca, R.; Costa, G.; Artese, A.; Parrotta, L.; Ortuso, F.; Maccioni, E.; Pinato, O.; Greco, M.L.; Sissi, C.; Alcaro, S.; et al. Hit identification of a novel dual binder forh-telo/c-myc G-quadruplex by a combination of pharmacophore structure-based virtual screening and docking refinement. Chem. Med. Chem. 2016, 11, 1721–1733. [Google Scholar] [CrossRef]
- Kaserer, T.; Rigo, R.; Schuster, P.; Alcaro, S.; Sissi, C.; Schuster, D. Optimized virtual screening workflow for the identification of novel G-quadruplex ligands. J. Chem. Inf. Model. 2016, 56, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, S.; Musetti, C.; Distinto, S.; Casatti, M.; Zagotto, G.; Artese, A.; Parrotta, L.; Moraca, F.; Costa, G.; Ortuso, F.; et al. Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure-based virtual screening approaches. J. Med. Chem. 2013, 56, 843–855. [Google Scholar] [CrossRef]
- Hou, J.-Q.; Chen, S.-B.; Zan, L.-P.; Ou, T.-M.; Tan, J.-H.; Luyt, L.G.; Huang, Z.-S. Identification of a selective G-quadruplex DNA binder using a multistep virtual screening approach. Chem. Commun. 2014, 51, 198–201. [Google Scholar] [CrossRef]
- Monsen, R.C.; Trent, J.O. G-quadruplex virtual drug screening: A review. Biochimie 2018, 152, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Ang, D.L.; Kelso, C.; Beck, J.L.; Ralph, S.F.; Harman, D.G.; Aldrich-Wright, J.R. A study of Pt (II)–phenanthroline complex interactions with double-stranded and G-quadruplex DNA by ESI–MS, circular dichroism, and computational docking. Nat. Prod. Res. 2021, 35, 2583–2587. [Google Scholar] [CrossRef]
- Ribaudo, G.; Ongaro, A.; Zagotto, G.; Memo, M.; Gianoncelli, A. Evidence on selective binding to G-quadruplex DNA of isoflavones from Maclura pomifera by mass spectrometry and molecular docking. Nat. Prod. Res. 2019, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tomar, J.S. In-Silico modeling studies of G-quadruplex with soy isoflavones having anticancerous activity. J. Mol. Model. 2015, 21, 193. [Google Scholar] [CrossRef]
- Salem, A.A.; El Haty, I.A.; Abdou, I.M.; Mu, Y. Interaction of human telomeric G-quadruplex DNA with thymoquinone: A possible mechanism for thymoquinone anticancer effect. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.; Rocca, R.; Moraca, F.; Talarico, C.; Romeo, I.; Ortuso, F.; Alcaro, S.; Artese, A. A Comparative docking strategy to identify polyphenolic derivatives as promising antineoplastic binders of G-quadruplex DNAc-mycandbcl-2 sequences. Mol. Inform. 2016, 35, 391–402. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Talarico, C.; Ortuso, F.; Da Ros, S.; Nicoletto, G.; Sissi, C.; Alcaro, S.; Artese, A. In Silico identification of piperidinyl-amine derivatives as novel dual binders of oncogene c-myc/c-kit G-quadruplexes. ACS Med. Chem. Lett. 2018, 9, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Saxena, S.; Srivastava, P.; Sharma, T.; Kundu, N.; Kaur, S.; Shankaraswamy, J. Screening the binding potential of quercetin with parallel, antiparallel and mixed G-quadruplexes of human telomere and cancer protooncogenes using molecular docking approach. SN Appl. Sci. 2020, 2, 490. [Google Scholar] [CrossRef] [Green Version]
- Bessi, I.; Bazzicalupi, C.; Richter, C.; Jonker, H.R.A.; Saxena, K.; Sissi, C.; Chioccioli, M.; Bianco, S.; Bilia, A.R.; Schwalbe, H.; et al. Spectroscopic, molecular modeling, and NMR-spectroscopic investigation of the binding mode of the natural alkaloids berberine and sanguinarine to human telomeric G-quadruplex DNA. ACS Chem. Biol. 2012, 7, 1109–1119. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Cross, J.B.; Thompson, D.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yatsunyk, L.; Neidle, S. Water spines and networks in G-quadruplex structures. Nucleic Acids Res. 2021, 49, 519–528. [Google Scholar] [CrossRef]
- Luo, J.; Wei, W.; Waldispühl, J.; Moitessier, N. Challenges and current status of computational methods for docking small molecules to nucleic acids. Eur. J. Med. Chem. 2019, 168, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Hurley, L.H.; Yang, D. Solution structure of a 2:1 quindoline–c-MYC G-quadruplex: Insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011, 133, 17673–17680. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Lin, C.; Wu, G.; Dai, J.; Chang, T.-C.; Yang, D. Structures of 1:1 and 2:1 complexes of BMVC and MYC promoter G-quadruplex reveal a mechanism of ligand conformation adjustment for G4-recognition. Nucleic Acids Res. 2019, 47, 11931–11942. [Google Scholar] [CrossRef] [PubMed]
- Dickerhoff, J.; Dai, J.; Yang, D. Structural recognition of the MYC promoter G-quadruplex by a quinoline derivative: Insights into molecular targeting of parallel G-quadruplexes. Nucleic Acids Res. 2021, 49, 5905–5915. [Google Scholar] [CrossRef]
- Calabrese, D.; Chen, X.; Leon, E.C.; Gaikwad, S.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.R.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Fochtman, B.C.; Balius, T.E.; Rizzo, R.C. Customizable de novo design strategies for dock: Application to HIVgp41 and other therapeutic targets. J. Comput. Chem. 2017, 38, 2641–2663. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Wu, G.; Wang, K.; Onel, B.; Sakai, S.; Shao, Y.; Yang, D. Molecular recognition of the hybrid-2 human telomeric G-quadruplex by epiberberine: Insights into conversion of telomeric G-quadruplex structures. Angew. Chem. Int. Ed. 2018, 57, 10888–10893. [Google Scholar] [CrossRef]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and accurate solution to the induced fit docking problem for protein-ligand binding. J. Chem. Theory Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein−ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, G.D.; Cramer, C.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.; Truhlar, D.G. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision making in structure-based drug discovery: Visual inspection of docking results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.1; Schrödinger LLC: New York, NY, USA, 2018.
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock vina for virtual screening. J. Comput. Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, H.W. The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 1955, 2, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Munkres, J. Algorithms for the assignment and transportation problems. J. Soc. Ind. Appl. Math. 1957, 5, 32–38. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | 5′-Complex | 3′-Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Correct Pose | Pose RMSD | Correct Pose | Pose RMSD | |||||||
| All | Top 1 | Top 5 | Top | Best | All | Top 1 | Top 5 | Top | Best | |
| Qi | x | x | x | 9.77 | 5.34 | √ | x | √ | 6.71 | 1.60 |
| BMVC | √ | x | x | 11.70 | 1.98 | x | x | x | 4.24 | 3.23 |
| PEQ | √ | √ | √ | 1.26 | 1.26 | x | x | x | 3.17 | 2.64 |
| DC34 | √ | x | x | 8.65 | 2.41 | x | x | x | 11.26 | 11.26 |
| Ligand | 5′-Complex | 3′-Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Correct Pose | Pose RMSD | Correct Pose | Pose RMSD | |||||||
| All | Top 1 | Top 5 | Top | Best | All | Top 1 | Top 5 | Top | Best | |
| Qi | √ | x | √ | 7.86 | 1.16 | √ | √ | √ | 1.76 | 1.55 |
| GB/SA | - | √ | √ | 1.16 | 1.16 | - | x | √ | 6.25 | 1.55 |
| BMVC | √ | √ | √ | 1.74 | 0.91 | √ | √ | √ | 1.99 | 1.30 |
| GB/SA | - | √ | √ | 1.74 | 0.91 | - | √ | √ | 1.99 | 1.30 |
| PEQ | √ | x | √ | 8.81 | 0.68 | √ | x | √ | 10.02 | 0.92 |
| GB/SA | - | x | √ | 8.81 | 0.68 | - | √ | √ | 0.92 | 0.92 |
| DC34 | x | x | x | 8.89 | 3.63 | √ | x | x | 7.61 | 1.85 |
| GB/SA | - | x | x | 7.63 | 3.63 | - | x | x | 7.61 | 1.85 |
| Ligand | 5′-Complex | 3′-Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Correct Pose | Pose RMSD | Correct Pose | Pose RMSD | |||||||
| All | Top 1 | Top 5 | Top | Best | All | Top 1 | Top 5 | Top | Best | |
| Qi | √ | x | √ | 6.34 | 2.27 | √ | x | √ | 5.46 | 1.49 |
| BMVC | √ | x | √ | 3.23 | 1.16 | √ | √ | √ | 2.20 | 2.20 |
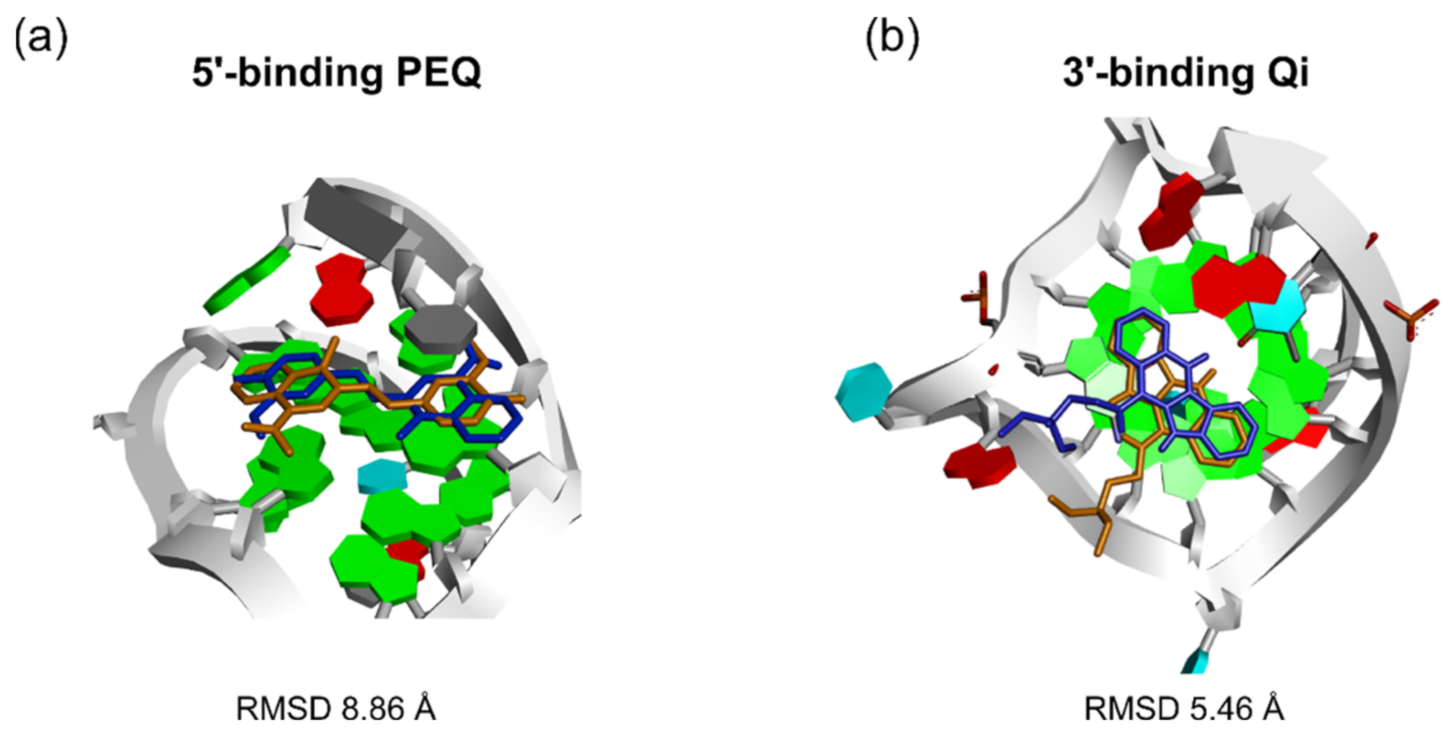
| PEQ | √ | x | √ | 8.86 | 0.40 | x | x | x | 4.59 | 4.11 |
| DC34 | x | x | x | 8.70 | 6.20 | √ | x | x | 9.20 | 1.98 |
| Ligand | 5′-Complex | 3′-Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Correct Pose | Pose RMSD | Correct Pose | Pose RMSD | |||||||
| All | Top 1 | Top 5 | Top | Best | All | Top 1 | Top 5 | Top | Best | |
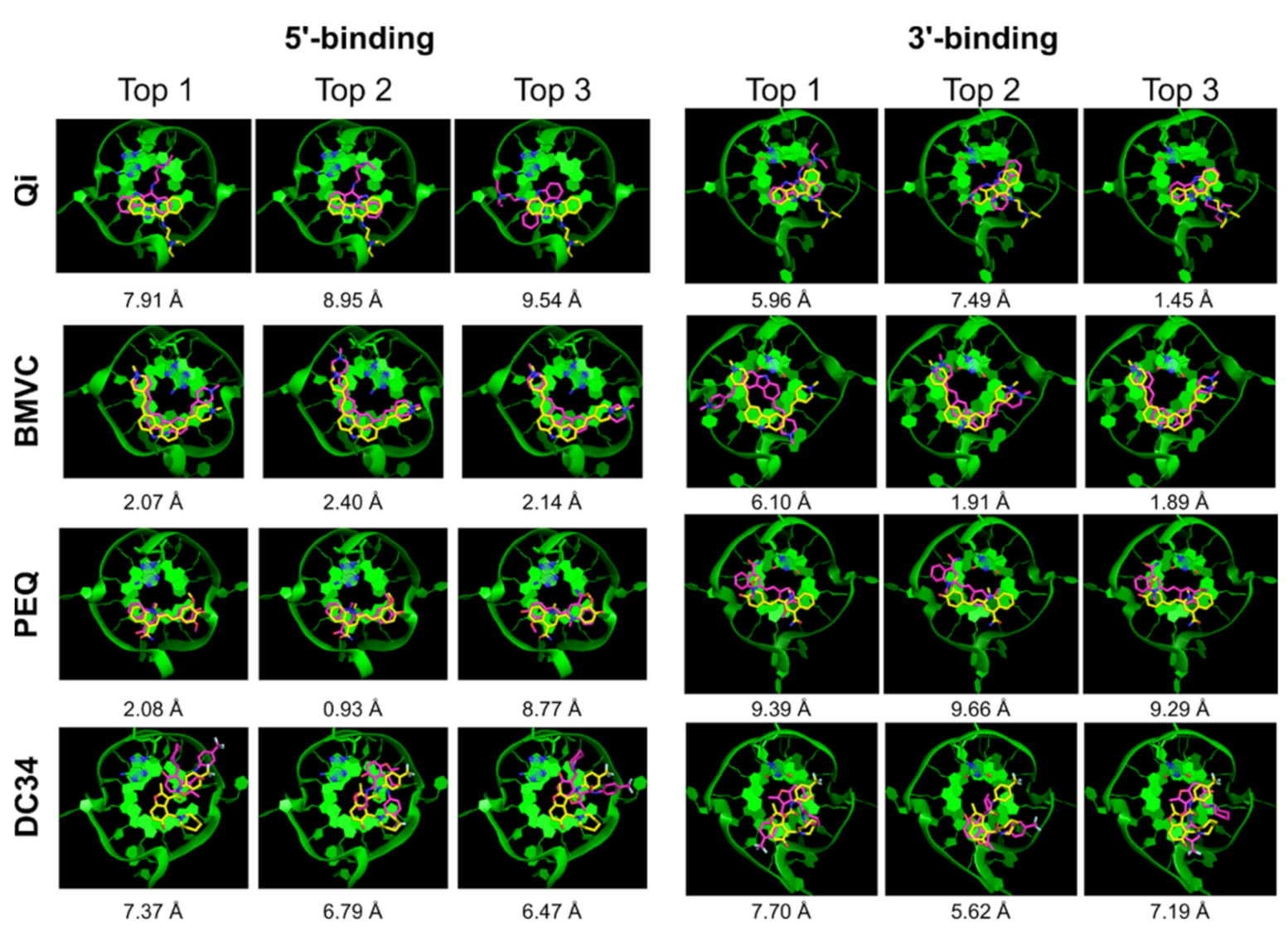
| Qi | √ | x | x | 7.91 | 1.52 | √ | x | √ | 5.96 | 0.85 |
| BMVC | √ | √ | √ | 2.07 | 1.42 | √ | x | √ | 6.10 | 1.16 |
| PEQ | √ | √ | √ | 2.08 | 0.93 | √ | x | x | 9.39 | 0.38 |
| DC34 | x | x | x | 7.37 | 3.54 | √ | x | √ | 7.70 | 1.48 |
| Ligand | Correct Pose | Correct Pose Top1 | Correct Pose Top5 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Auto Dock Vina | Dock 6 | Glide | RxDock | Auto Dock Vina | Dock 6 | Dock 6 GB/SA | Glide | RxDock | Auto Dock Vina | Dock 6 | Dock 6 GB/SA | Glide | RxDock | ||
| Qi | 5′ | x | √ | √ | √ | x | x | √ | x | x | x | √ | √ | √ | x |
| 3′ | √ | √ | √ | √ | x | √ | x | x | x | √ | √ | √ | √ | √ | |
| BMVC | 5′ | √ | √ | √ | √ | x | √ | √ | x | √ | x | √ | √ | √ | √ |
| 3′ | x | √ | √ | √ | x | √ | √ | √ | x | x | √ | √ | √ | √ | |
| PEQ | 5′ | √ | √ | √ | √ | √ | x | x | x | √ | √ | √ | √ | √ | √ |
| 3′ | x | √ | x | √ | x | x | √ | x | x | x | √ | √ | x | x | |
| DC34 | 5′ | √ | x | x | x | x | x | x | x | x | x | x | x | x | x |
| 3′ | x | √ | √ | √ | x | x | x | x | x | x | x | x | x | √ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickerhoff, J.; Warnecke, K.R.; Wang, K.; Deng, N.; Yang, D. Evaluating Molecular Docking Software for Small Molecule Binding to G-Quadruplex DNA. Int. J. Mol. Sci. 2021, 22, 10801. https://doi.org/10.3390/ijms221910801
Dickerhoff J, Warnecke KR, Wang K, Deng N, Yang D. Evaluating Molecular Docking Software for Small Molecule Binding to G-Quadruplex DNA. International Journal of Molecular Sciences. 2021; 22(19):10801. https://doi.org/10.3390/ijms221910801
Chicago/Turabian StyleDickerhoff, Jonathan, Kassandra R. Warnecke, Kaibo Wang, Nanjie Deng, and Danzhou Yang. 2021. "Evaluating Molecular Docking Software for Small Molecule Binding to G-Quadruplex DNA" International Journal of Molecular Sciences 22, no. 19: 10801. https://doi.org/10.3390/ijms221910801
APA StyleDickerhoff, J., Warnecke, K. R., Wang, K., Deng, N., & Yang, D. (2021). Evaluating Molecular Docking Software for Small Molecule Binding to G-Quadruplex DNA. International Journal of Molecular Sciences, 22(19), 10801. https://doi.org/10.3390/ijms221910801