
Differential Transcriptional Regulation of Polymorphic p53 Codon 72 in Metabolic Pathways

Abstract
1. Introduction
2. Results
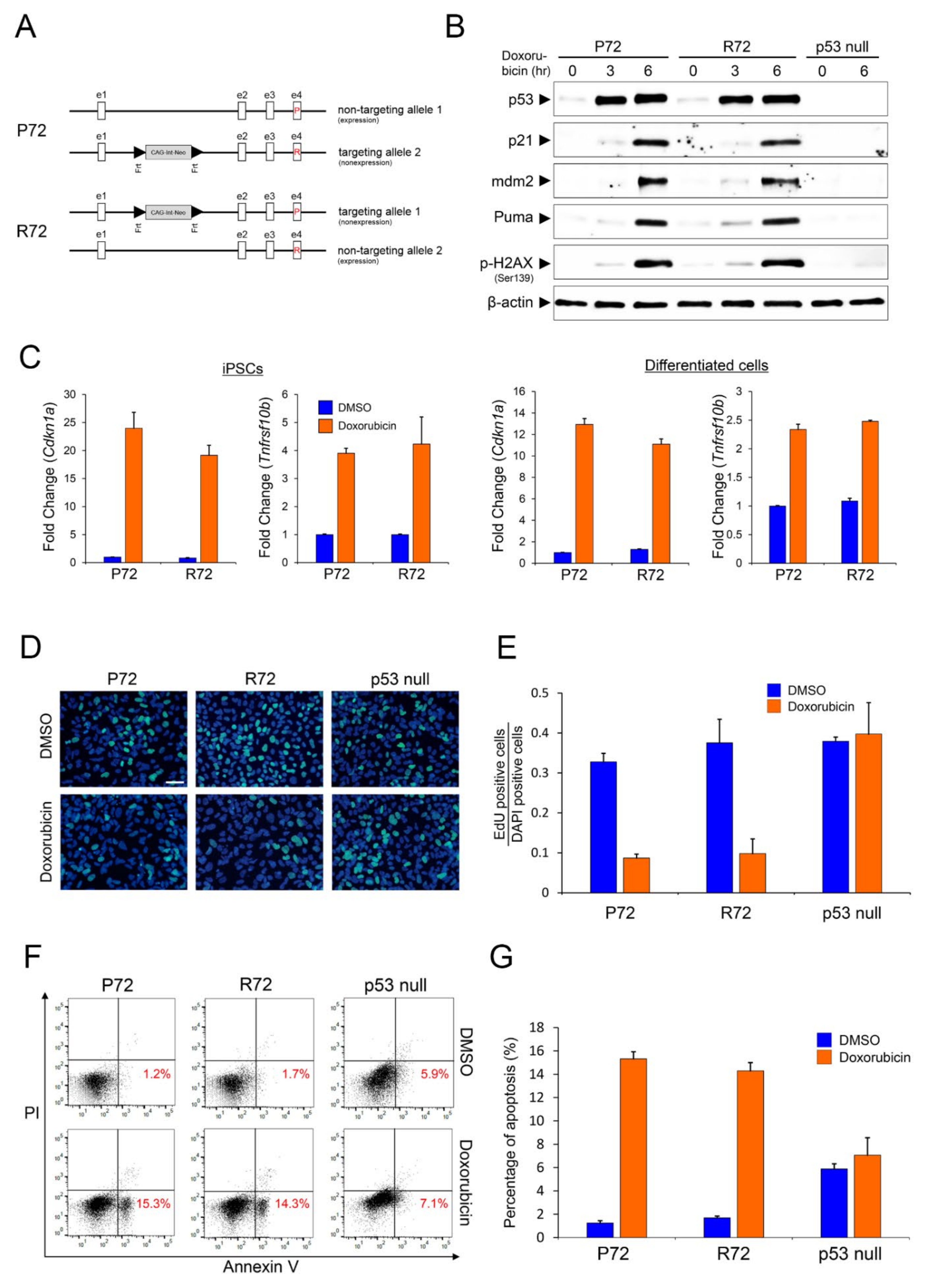
2.1. Expression Pattern of Representative p53 Target Genes
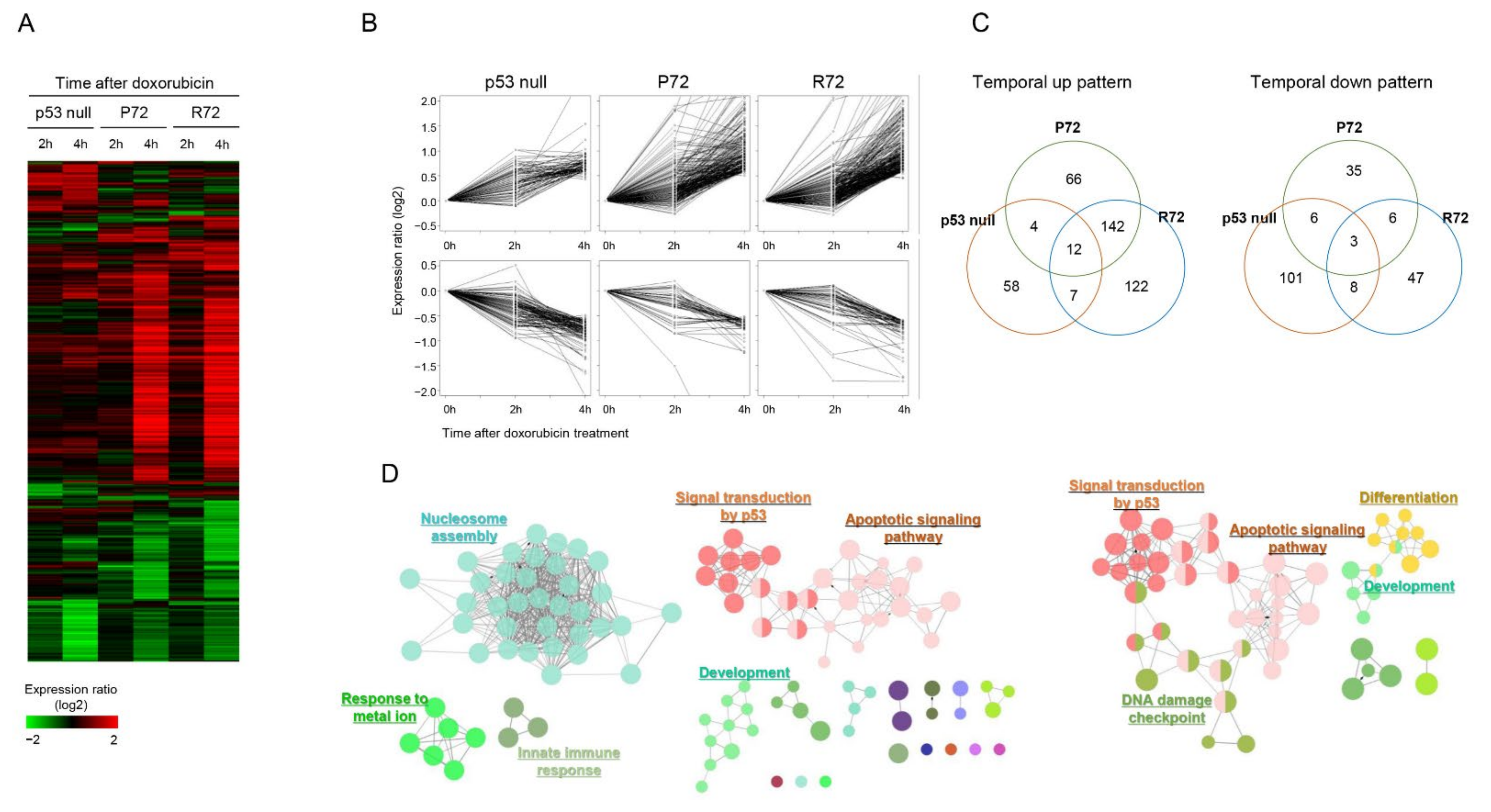
2.2. Similar Global Expression Pattern between the Polymorphic p53 under DNA Damage Stress
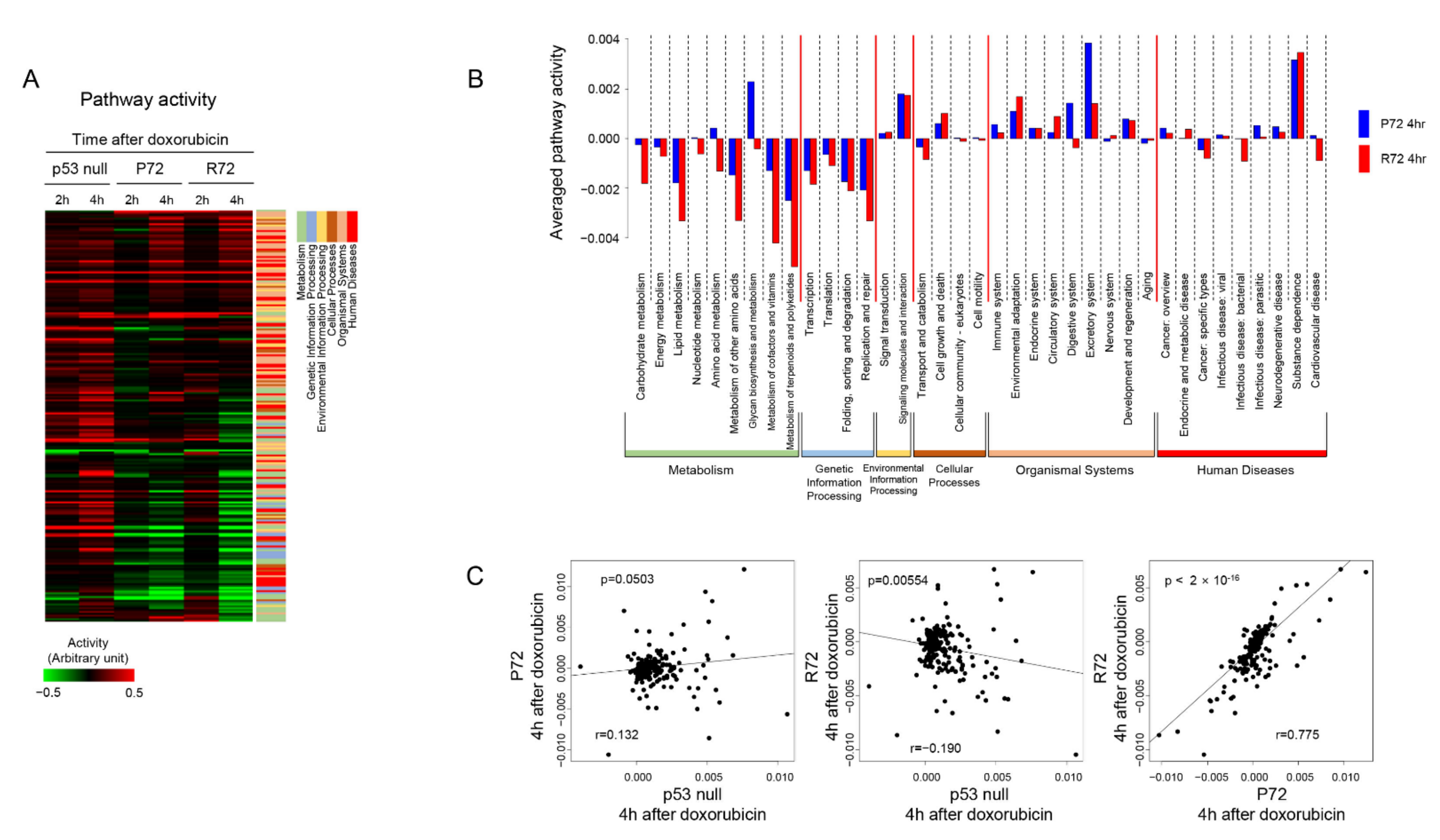
2.3. Patterns of Cellular Signaling Pathways between the Polymorphic p53 Codon 72
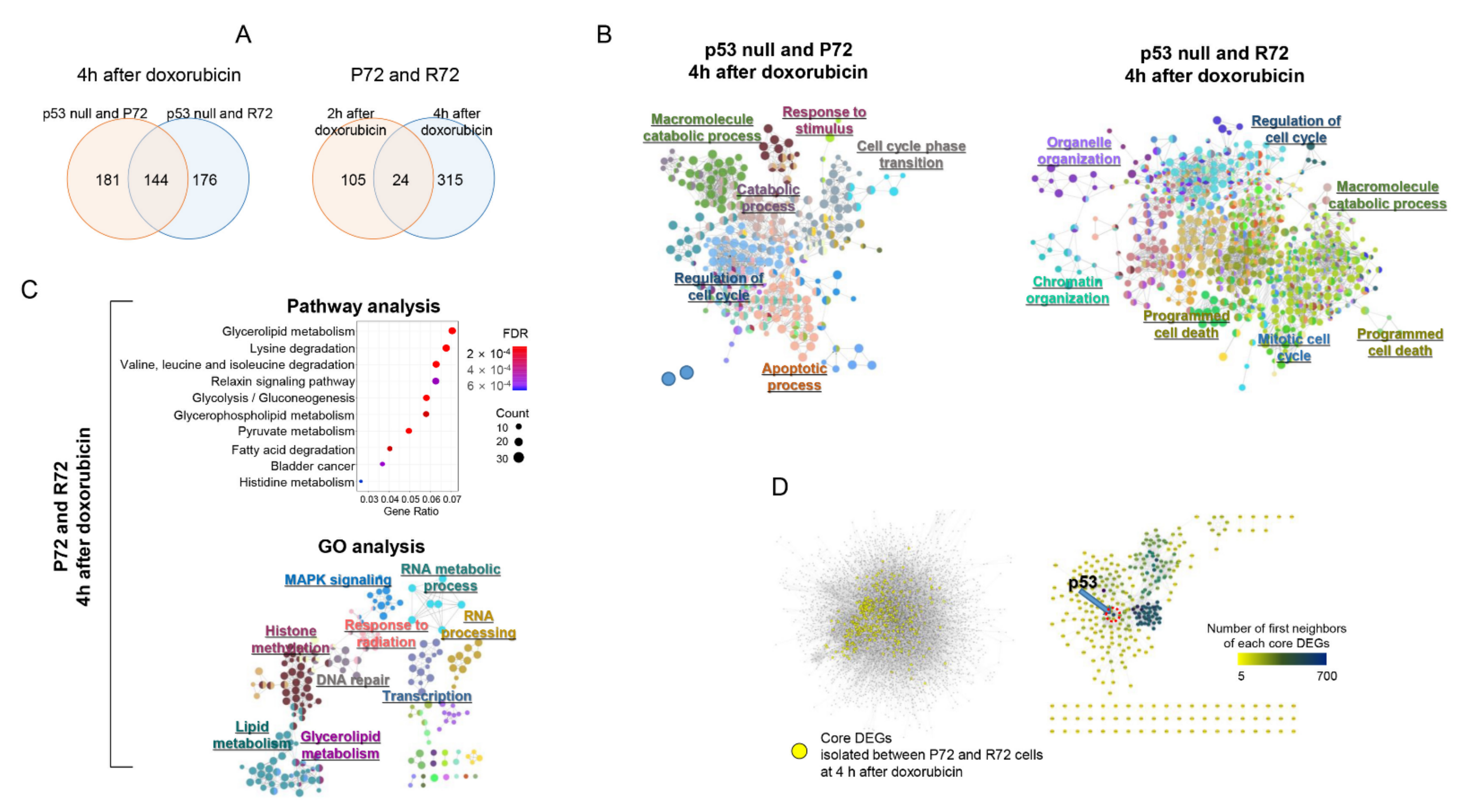
2.4. Neighbor-Based Isolation of Doxorubicin Responsive Genes
2.5. Distances in Network between Metabolism, p53, and DNA Damage
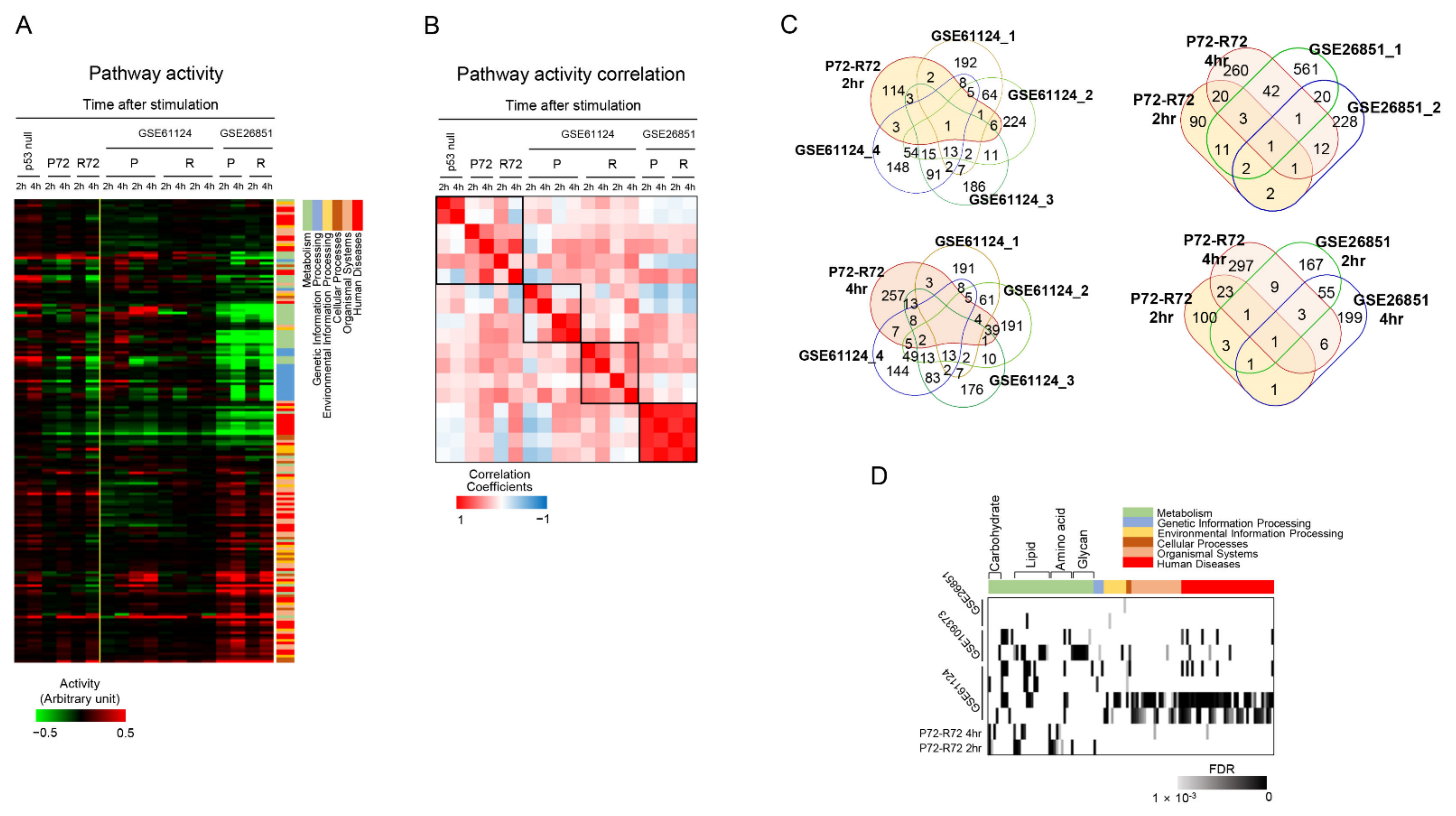
2.6. Analysis of External Public Datasets
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Protein Extraction and Western Blotting
4.3. RNA Extraction and Quantitative Real-Time PCR
4.4. Cell Growth Arrest Assay
4.5. Annexin V Analysis
4.6. Statistical Analysis
4.7. RNA Isolation, Library Preparation, and Sequencing for QuantSeq Analysis
4.8. QuantSeq Data Analysis
4.9. Clustering of Genes and Isolation of Temporally Regulated Genes
4.10. Construction of Interaction Network
4.11. Isolation of Core DEGs
4.12. Functional Enrichemtbn of GO-Terms
4.13. Pathway Activity
4.14. Measurement of Distance between Genes in the Network
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Song, H.; Chung, S.K.; Xu, Y. Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell 2010, 6, 80–89. [Google Scholar] [CrossRef]
- Bieging, K.T.; Attardi, L.D. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012, 22, 97–106. [Google Scholar] [CrossRef]
- Pavlakis, E.; Stiewe, T. p53’s Extended Reach: The Mutant p53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef]
- Garritano, S.; Inga, A.; Gemignani, F.; Landi, S. More targets, more pathways and more clues for mutant p53. Oncogenesis 2013, 2, e54. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Andera, L.; Opattova, A.; Vodickova, L. The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction. Cancers 2021, 13, 479. [Google Scholar] [CrossRef]
- Mackay, H.L.; Moore, D.; Hall, C.; Birkbak, N.J.; Jamal-Hanjani, M.; Karim, S.A.; Phatak, V.M.; Pinon, L.; Morton, J.P.; Swanton, C.; et al. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat. Commun. 2018, 9, 3070. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Dolle, M.E.; Berton, T.R.; Kuiper, R.V.; Capps, C.; Espejo, A.; McArthur, M.J.; Bedford, M.T.; van Steeg, H.; de Vries, A.; et al. Mouse models for the p53 R72P polymorphism mimic human phenotypes. Cancer Res. 2010, 70, 5851–5859. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Kalita, A.; Labrecque, S.; Pim, D.; Banks, L.; Matlashewski, G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol. Cell. Biol. 1999, 19, 1092–1100. [Google Scholar] [CrossRef]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C., 3rd; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Lin, H.Y.; Huang, C.H.; Wu, W.J.; Chang, L.C.; Lung, F.W. TP53 codon 72 Gene Polymorphism Paradox in Associated with Various Carcinoma Incidences, Invasiveness and Chemotherapy Responses. Int. J. Biomed. Sci. IJBS 2008, 4, 248–254. [Google Scholar]
- Pim, D.; Banks, L. p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int. J. Cancer 2004, 108, 196–199. [Google Scholar] [CrossRef]
- Bonafe, M.; Salvioli, S.; Barbi, C.; Trapassi, C.; Tocco, F.; Storci, G.; Invidia, L.; Vannini, I.; Rossi, M.; Marzi, E.; et al. The different apoptotic potential of the p53 codon 72 alleles increases with age and modulates in vivo ischaemia-induced cell death. Cell Death Differ. 2004, 11, 962–973. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jeong, B.S.; Hu, W.; Belyi, V.; Rabadan, R.; Levine, A.J. Differential levels of transcription of p53-regulated genes by the arginine/proline polymorphism: p53 with arginine at codon 72 favors apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, J.H.; Jeong, S.; Kim, B.Y.; Kang, Y.K.; Xu, Y.; Chung, S.K. K120R mutation inactivates p53 by creating an aberrant splice site leading to nonsense-mediated mRNA decay. Oncogene 2019, 38, 1597–1610. [Google Scholar] [CrossRef]
- Zehbe, I.; Voglino, G.; Wilander, E.; Genta, F.; Tommasino, M. Codon 72 polymorphism of p53 and its association with cervical cancer. Lancet 1999, 354, 218–219. [Google Scholar] [CrossRef]
- Dokianakis, D.N.; Spandidos, D.A. P53 codon 72 polymorphism as a risk factor in the development of HPV-associated cervical cancer. Mol. Cell. Biol. Res. Commun. MCBRC 2000, 3, 111–114. [Google Scholar] [CrossRef]
- Dokianakis, D.N.; Koumantaki, E.; Billiri, K.; Spandidos, D.A. P53 codon 72 polymorphism as a risk factor in the development of HPV-associated non-melanoma skin cancers in immunocompetent hosts. Int. J. Mol. Med. 2000, 5, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, Z.; Strom, S.; Spitz, M.; Lee, J.; Gershenwald, J.; Ross, M.; Mansfield, P.; Duvic, M.; Ananthaswamy, H.; et al. p53 Codon 72 Arg Homozygotes are associated with an increased risk of cutaneous melanoma. J. Investig. Dermatol. 2003, 121, 1510–1514. [Google Scholar] [CrossRef]
- Katkoori, V.; Manne, U.; Chaturvedi, L.; Bassin, M.; Haan, P.; Coffey, D.; Bumpers, H. Functional consequence of the p53 codon 72 polymorphism in colorectal cancer. Oncotarget 2017, 8, 76574–76586. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.N.; Ryan, A.; Al-Jehani, R.M.; Storey, A.; Harwood, C.A.; Jacobs, I.J. p53 codon 72 polymorphism and risk of cervical cancer in UK. Lancet 1998, 352, 871–872. [Google Scholar] [CrossRef]
- Minaguchi, T.; Kanamori, Y.; Matsushima, M.; Yoshikawa, H.; Taketani, Y.; Nakamura, Y. No evidence of correlation between polymorphism at codon 72 of p53 and risk of cervical cancer in Japanese patients with human papillomavirus 16/18 infection. Cancer Res. 1998, 58, 4585–4586. [Google Scholar] [PubMed]
- Hamel, N.; Black, M.J.; Ghadirian, P.; Foulkes, W.D. No association between P53 codon 72 polymorphism and risk of squamous cell carcinoma of the head and neck. Br. J. Cancer 2000, 82, 757–759. [Google Scholar] [CrossRef]
- Ma, Y.L.; Yang, J.J.; Liu, Z.H.; Zhang, P.; Yang, Z.; Wang, Y.; Qin, H.L. No significant association between the TP53 codon 72 polymorphism and breast cancer risk: A meta-analysis of 21 studies involving 24,063 subjects. Breast Cancer Res. Treat. 2011, 125, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Chandirasekar, R.; Kumar, B.L.; Venkatesan, R.; Sasikala, K. No Association Between the Trp53 Codon 72 Polymorphism and Head and Neck Cancer: A Case-Control Study in a South Indian Population. Asian Pac. J. Cancer P 2010, 11, 1749–1753. [Google Scholar]
- Jiang, N.; Pan, J.; Wang, L.; Duan, Y.Z. No significant association between p53 codon 72 Arg/Pro polymorphism and risk of oral cancer. Tumour Biol. 2013, 34, 587–596. [Google Scholar] [CrossRef]
- Li, M.S.; Liu, J.L.; Wu, Y.; Wang, P.; Teng, H. Meta-analysis demonstrates no association between p53 codon 72 polymorphism and prostate cancer risk. Genet. Mol. Res. 2011, 10, 2924–2933. [Google Scholar] [CrossRef]
- Kim, B.Y.; Lim, H.S.; Kim, Y.; Kim, Y.J.; Koo, I.; Jeong, S.J. Evaluation of Animal Models by Comparison with Human Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 9234–9250. [Google Scholar] [CrossRef]
- Kung, C.P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol. Cancer Res. 2015, 13, 250–262. [Google Scholar] [CrossRef]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef]
- Frank, A.; Leu, J.I.; Zhou, Y.; Devarajan, K.; Nedleko, T.; Klein-Szanto, A.; Hollstein, M.; Murphy, M.E. The codon 72 polymorphism of p53 regulates interaction with NFkB and transactivation of genes involved in immunity and inflammation. Mol. Cell. Biol. 2011, 31, 1201–1213. [Google Scholar] [CrossRef]
- Kung, C.P.; Leu, J.I.; Basu, S.; Khaku, S.; Anokye-Danso, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep. 2016, 14, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, K.S.; Grarup, N.; Justesen, J.M.; Harder, M.N.; Witte, D.R.; Jorgensen, T.; Sandbaek, A.; Lauritzen, T.; Madsbad, S.; Hansen, T.; et al. Studies of the association of Arg72Pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 Europeans. PLoS ONE 2011, 6, e15813. [Google Scholar] [CrossRef] [PubMed]
- Bonfigli, A.R.; Sirolla, C.; Testa, R.; Cucchi, M.; Spazzafumo, L.; Salvioli, S.; Ceriello, A.; Olivieri, F.; Festa, R.; Procopio, A.D.; et al. The p53 codon 72 (Arg72Pro) polymorphism is associated with the degree of insulin resistance in type 2 diabetic subjects: A cross-sectional study. Acta Diabetol. 2013, 50, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- De Hoon, M.J.; Imoto, S.; Nolan, J.; Miyano, S. Open source clustering software. Bioinformatics 2004, 20, 1453–1454. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.Y.; Lim, H.S.; Kim, Y.J.; Sohn, E.; Kim, Y.H.; Koo, I.; Jeong, S.J. Similarity of therapeutic networks induced by a multi-component herbal remedy, Ukgansan, in neurovascular unit cells. Sci. Rep. 2020, 10, 2658. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53 Null-P72 4 h after Doxorubicin | p53 Null-R72 4 h after Doxorubicin | P72-R72 4 h after Doxorubicin | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Symbol | # of First Neighbors | Empirical p-Value | Common Gene | Symbol | # of First Neighbors | Empirical p-Value | Common Gene | Symbol | # of First Neighbors | Empirical p-Value | Common Gene |
| TP53 | 684 | 0.001 | null-R P-R | MYC | 908 | <0.001 | TP53 | 684 | <0.001 | null-P null-R | |
| UBC | 383 | 0.003 | null-R | TP53 | 684 | <0.001 | null-P P-R | POLR2A | 291 | <0.001 | |
| MDM2 | 347 | <0.001 | null-R | UBC | 383 | <0.001 | null-P | U2AF2 | 249 | 0.007 | null-R |
| AR | 327 | <0.001 | null-R | MDM2 | 347 | <0.001 | null-P | H2AX | 212 | <0.001 | null-R |
| SP1 | 272 | <0.001 | null-R | AR | 327 | <0.001 | null-P | SIRT1 | 204 | 0.001 | null-R |
| CREBBP | 268 | <0.001 | null-R | HDAC1 | 299 | 0.003 | ACOT8 | 193 | <0.001 | ||
| RELA | 243 | <0.001 | null-R | SP1 | 272 | <0.001 | null-P | HSPA4 | 192 | <0.001 | |
| JUN | 232 | 0.004 | null-R | CREBBP | 268 | 0.003 | null-P | DLD | 190 | <0.001 | |
| HIF1A | 228 | 0.001 | null-R | U2AF2 | 249 | 0.001 | P-R | ACAA2 | 187 | <0.001 | |
| NFKB1 | 201 | 0.001 | null-R | RELA | 243 | 0.001 | null-P | KMT2A | 171 | <0.001 | |
| SMAD3 | 179 | 0.001 | null-R | JUN | 232 | 0.001 | null-P | STUB1 | 160 | <0.001 | null-R |
| SQSTM1 | 178 | 0.001 | null-R | HIF1A | 228 | 0.001 | null-P | HSPA8 | 152 | <0.001 | |
| BTRC | 172 | 0.001 | null-R | UBE2I | 228 | 0.001 | PRMT5 | 144 | <0.001 | ||
| NR3C1 | 165 | 0.001 | null-R | H2AX | 212 | 0.001 | P-R | HRAS | 137 | <0.001 | |
| RHOA | 159 | 0.001 | null-R | SIRT1 | 204 | 0.001 | P-R | CREB1 | 135 | <0.001 | |
| STAT3 | 158 | 0.002 | null-R | H3C1 | 203 | 0.001 | LPCAT1 | 129 | <0.001 | ||
| UBE2D1 | 155 | 0.004 | null-R | NFKB1 | 201 | 0.002 | null-P | STAT1 | 119 | <0.001 | |
| CUL3 | 152 | 0.002 | null-R | PCNA | 182 | 0.002 | KMT2D | 119 | <0.001 | ||
| ITCH | 148 | 0.002 | null-R | SMAD3 | 179 | 0.002 | null-P | POLR1A | 119 | <0.001 | |
| UBE2D3 | 146 | 0.003 | null-R | SQSTM1 | 178 | 0.002 | null-P | HADHB | 118 | <0.001 | |
| CBX5 | 135 | 0.003 | null-R | BTRC | 172 | 0.002 | null-P | ACAA1 | 115 | <0.001 | |
| CBL | 132 | 0.01 | null-R | RB1 | 170 | 0.004 | SF3A2 | 114 | <0.001 | null-R | |
| RBX1 | 130 | 0.003 | null-R | VCP | 169 | 0.003 | NCOA1 | 114 | <0.001 | ||
| ABL1 | 128 | 0.003 | null-R | E2F2 | 167 | 0.003 | SETD1A | 114 | <0.001 | ||
| RARA | 125 | 0.003 | null-R | NR3C1 | 165 | 0.042 | null-P | TERF1 | 112 | <0.001 | |
| TRAF2 | 122 | 0.007 | null-R | STUB1 | 160 | 0.042 | P-R | KDM2A | 111 | <0.001 | |
| CDK9 | 114 | 0.003 | null-R | RHOA | 159 | 0.004 | null-P | PCK2 | 108 | <0.001 | |
| ETS1 | 107 | 0.003 | STAT3 | 158 | 0.004 | null-P | KMT2B | 106 | <0.001 | ||
| FZR1 | 105 | 0.001 | null-R | UBE2D1 | 155 | 0.004 | null-P | KMT5C | 106 | <0.001 | |
| CHEK2 | 104 | 0.004 | null-R | CUL3 | 152 | 0.004 | null-P | PLOD1 | 106 | <0.001 | |
| XIAP | 93 | 0.004 | PML | 149 | 0.006 | LPCAT4 | 105 | <0.001 | |||
| AURKA | 92 | 0.004 | null-R | ITCH | 148 | 0.004 | null-P | SMYD3 | 105 | <0.001 | |
| UBE2N | 88 | 0.004 | null-R | UBE2D3 | 146 | 0.005 | null-P | KRAS | 104 | <0.001 | null-R |
| TSG101 | 88 | 0.004 | null-R | UBE2D2 | 139 | 0.005 | PLOD2 | 104 | <0.001 | ||
| SPRTN | 84 | 0.002 | null-R | CBX5 | 135 | 0.005 | null-P | CDC20 | 103 | <0.001 | null-R |
| FBXW11 | 77 | 0.002 | null-R | CBL | 132 | 0.005 | null-P | RIOX1 | 103 | 0.002 | |
| PTK2 | 76 | 0.005 | null-R | RBX1 | 130 | 0.005 | null-P | ALDH3A2 | 103 | 0.001 | |
| BCL2L1 | 75 | 0.005 | ABL1 | 128 | 0.002 | null-P | ADSS2 | 101 | 0.003 | ||
| NBN | 74 | 0.005 | null-R | ATM | 127 | 0.002 | ALDH2 | 101 | 0.001 | ||
| UBE2E1 | 72 | 0.005 | null-R | RARA | 125 | 0.002 | null-P | PARK7 | 100 | 0.001 | |
| CENPA | 68 | 0.005 | null-R | TRAF2 | 122 | 0.006 | null-P | ALDH7A1 | 100 | 0.001 | |
| SUMO2 | 68 | 0.005 | null-R | SF3A2 | 114 | 0.006 | P-R | PIN1 | 99 | 0.009 | null-R |
| EEF1A1 | 68 | 0.004 | null-R | SMAD4 | 114 | 0.002 | KMT5B | 99 | 0.001 | ||
| SCP2 | 68 | 0.001 | P-R | TBP | 112 | 0.007 | ALDH9A1 | 99 | 0.003 | ||
| FANCD2 | 67 | 0.005 | null-R | ETS1 | 107 | 0.007 | null-P | ALDH1B1 | 98 | 0.001 | |
| CCNA2 | 64 | 0.008 | null-R | FZR1 | 105 | 0.003 | null-P | SETD5 | 97 | 0.004 | |
| NOTCH1 | 60 | 0.006 | null-R | CHEK2 | 104 | 0.002 | null-P | WDR5 | 91 | 0.001 | |
| KEAP1 | 57 | 0.006 | KRAS | 104 | 0.008 | P-R | HSPA1A | 89 | 0.001 | ||
| CERS4 | 56 | 0.006 | null-R | CDC20 | 103 | 0.009 | P-R | PDHA1 | 87 | 0.004 | |
| CERS5 | 56 | 0.002 | null-R | RXRA | 99 | 0.003 | APP | 86 | 0.002 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-Y.; Lee, S.-Y.; Chung, S.-K. Differential Transcriptional Regulation of Polymorphic p53 Codon 72 in Metabolic Pathways. Int. J. Mol. Sci. 2021, 22, 10793. https://doi.org/10.3390/ijms221910793
Kim B-Y, Lee S-Y, Chung S-K. Differential Transcriptional Regulation of Polymorphic p53 Codon 72 in Metabolic Pathways. International Journal of Molecular Sciences. 2021; 22(19):10793. https://doi.org/10.3390/ijms221910793
Chicago/Turabian StyleKim, Bu-Yeo, Seo-Young Lee, and Sun-Ku Chung. 2021. "Differential Transcriptional Regulation of Polymorphic p53 Codon 72 in Metabolic Pathways" International Journal of Molecular Sciences 22, no. 19: 10793. https://doi.org/10.3390/ijms221910793
APA StyleKim, B.-Y., Lee, S.-Y., & Chung, S.-K. (2021). Differential Transcriptional Regulation of Polymorphic p53 Codon 72 in Metabolic Pathways. International Journal of Molecular Sciences, 22(19), 10793. https://doi.org/10.3390/ijms221910793