
Single-Tear Proteomics: A Feasible Approach to Precision Medicine

 ,
, 
 ,
,  ,
,  ,
,  , , , and
, , , and 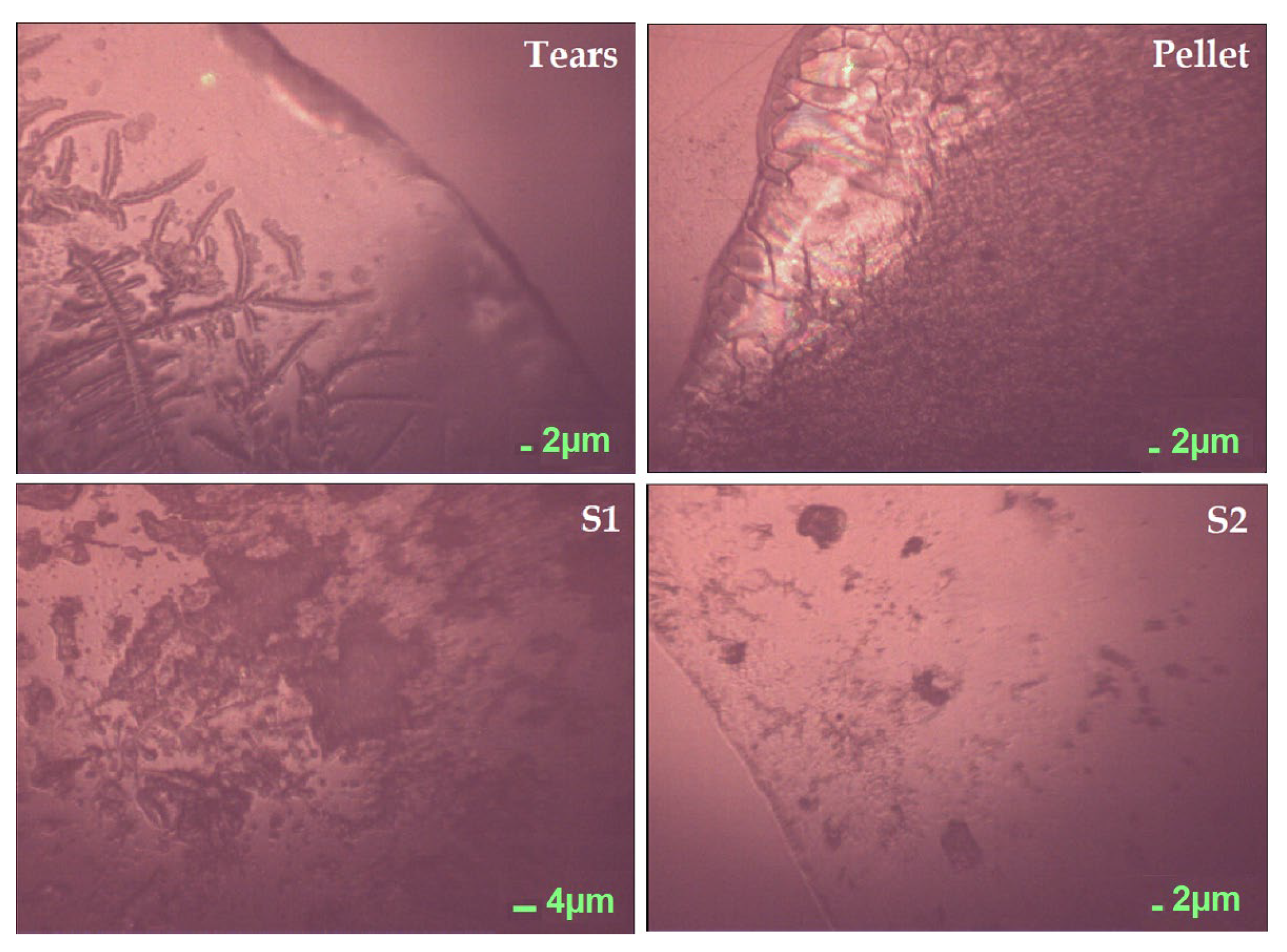{kind=link}
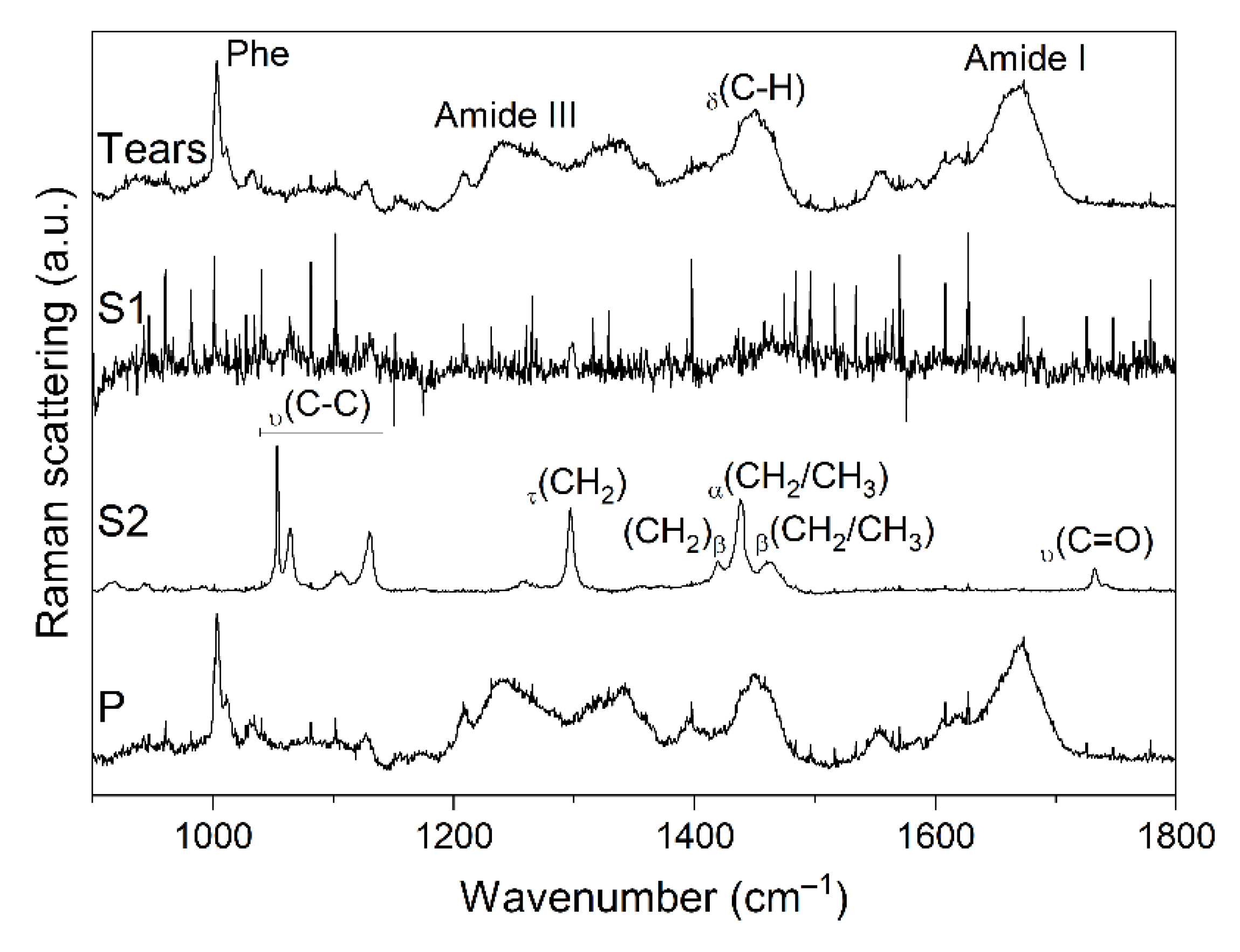{kind=link}
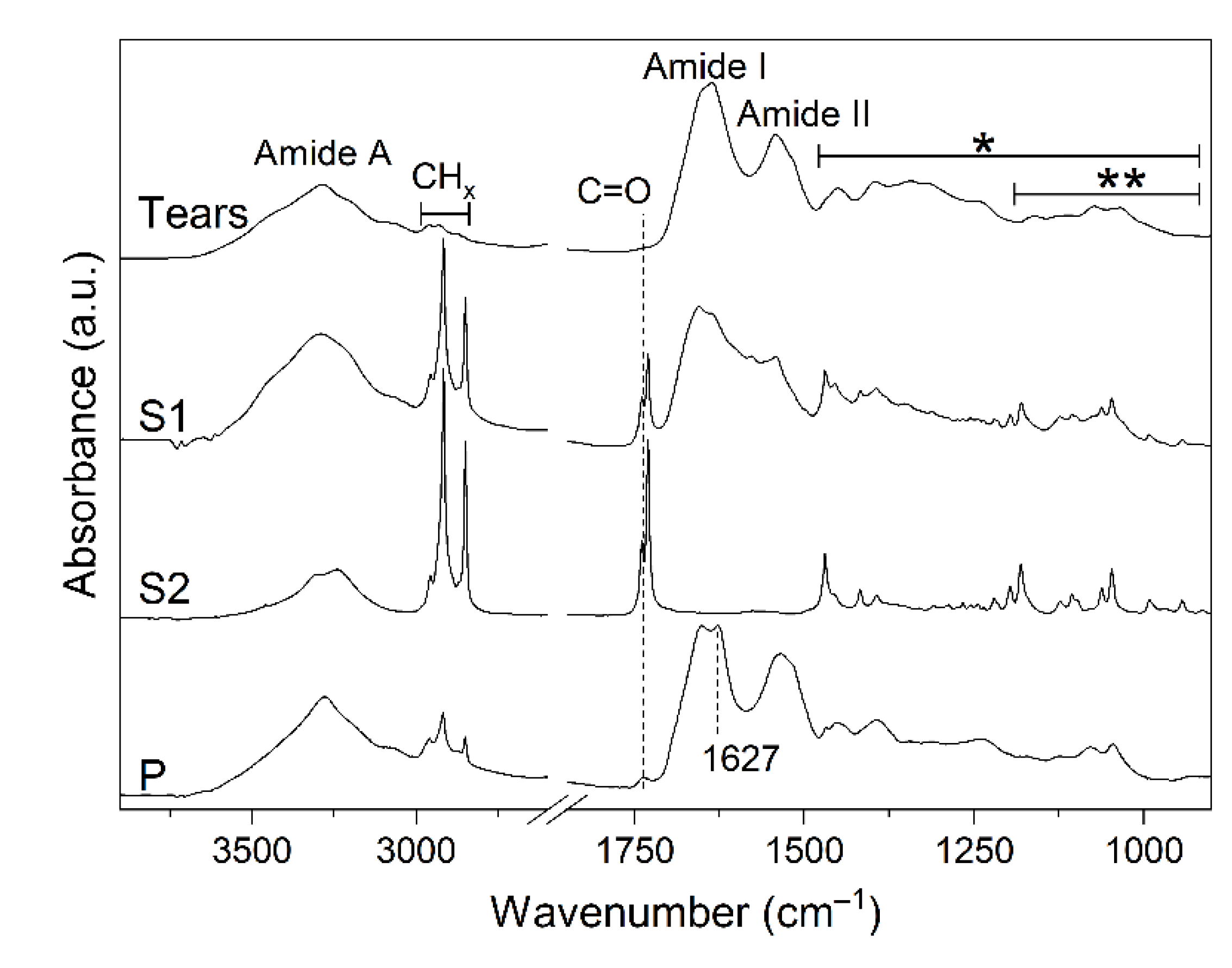{kind=link}
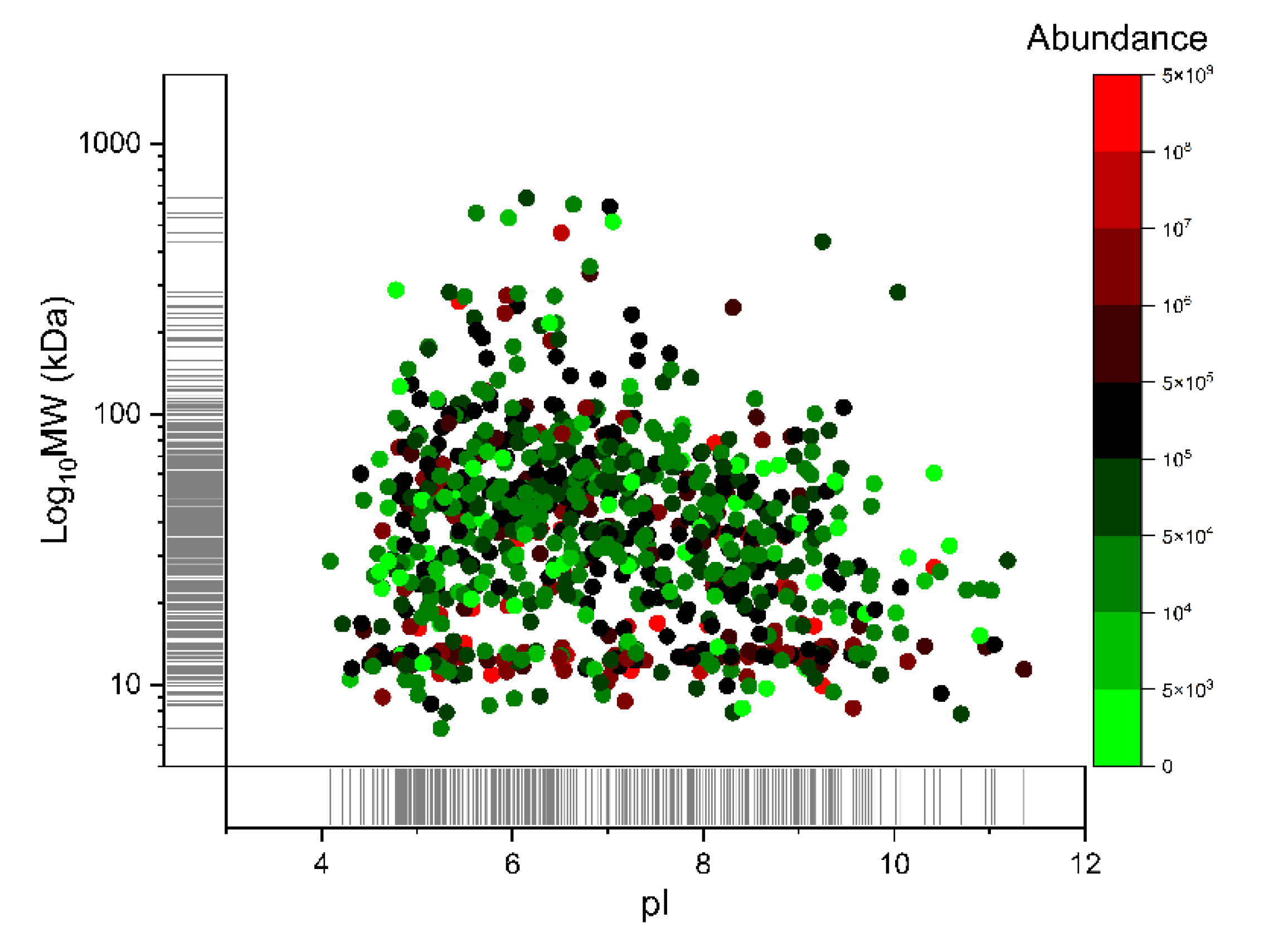{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
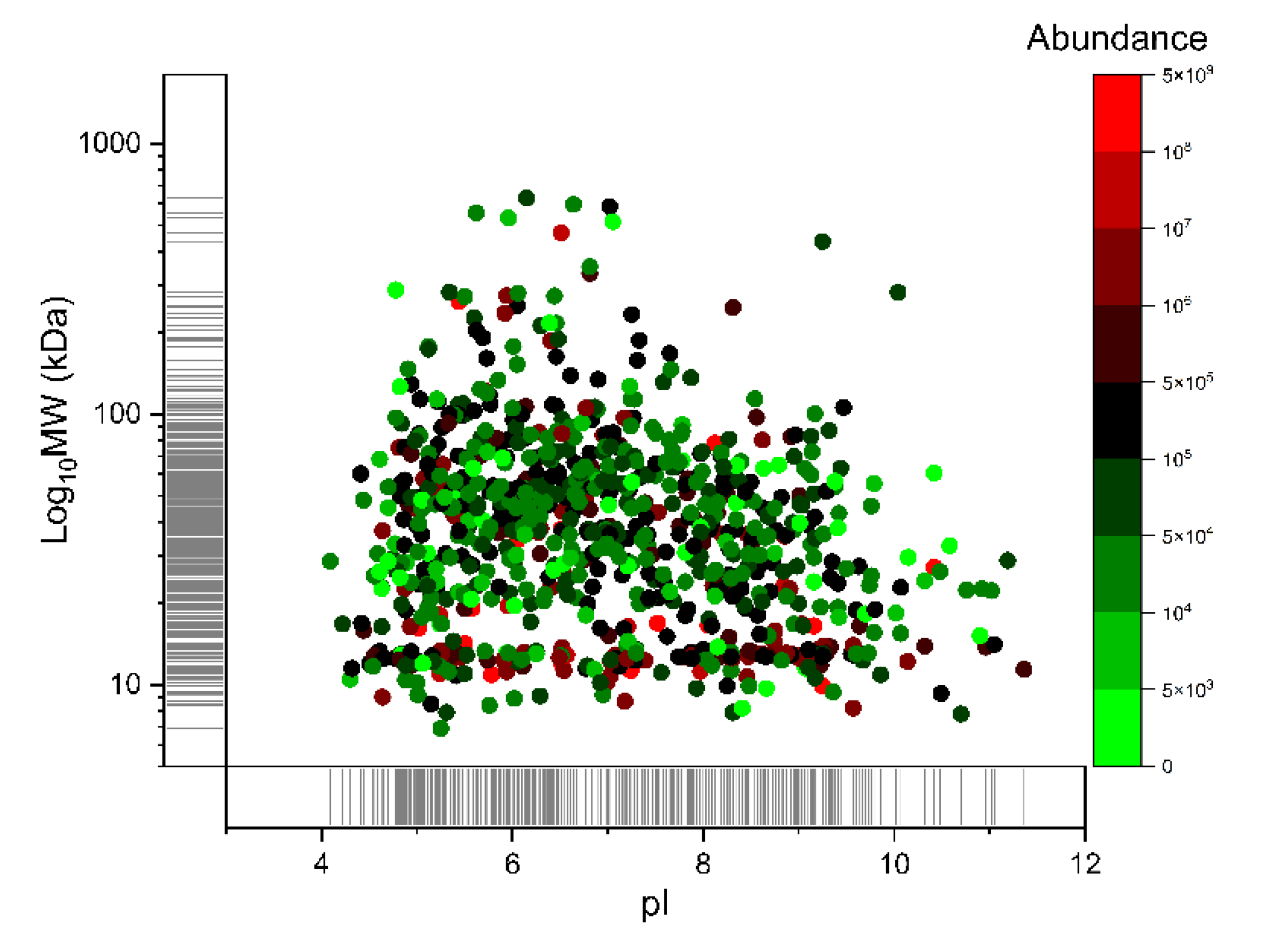
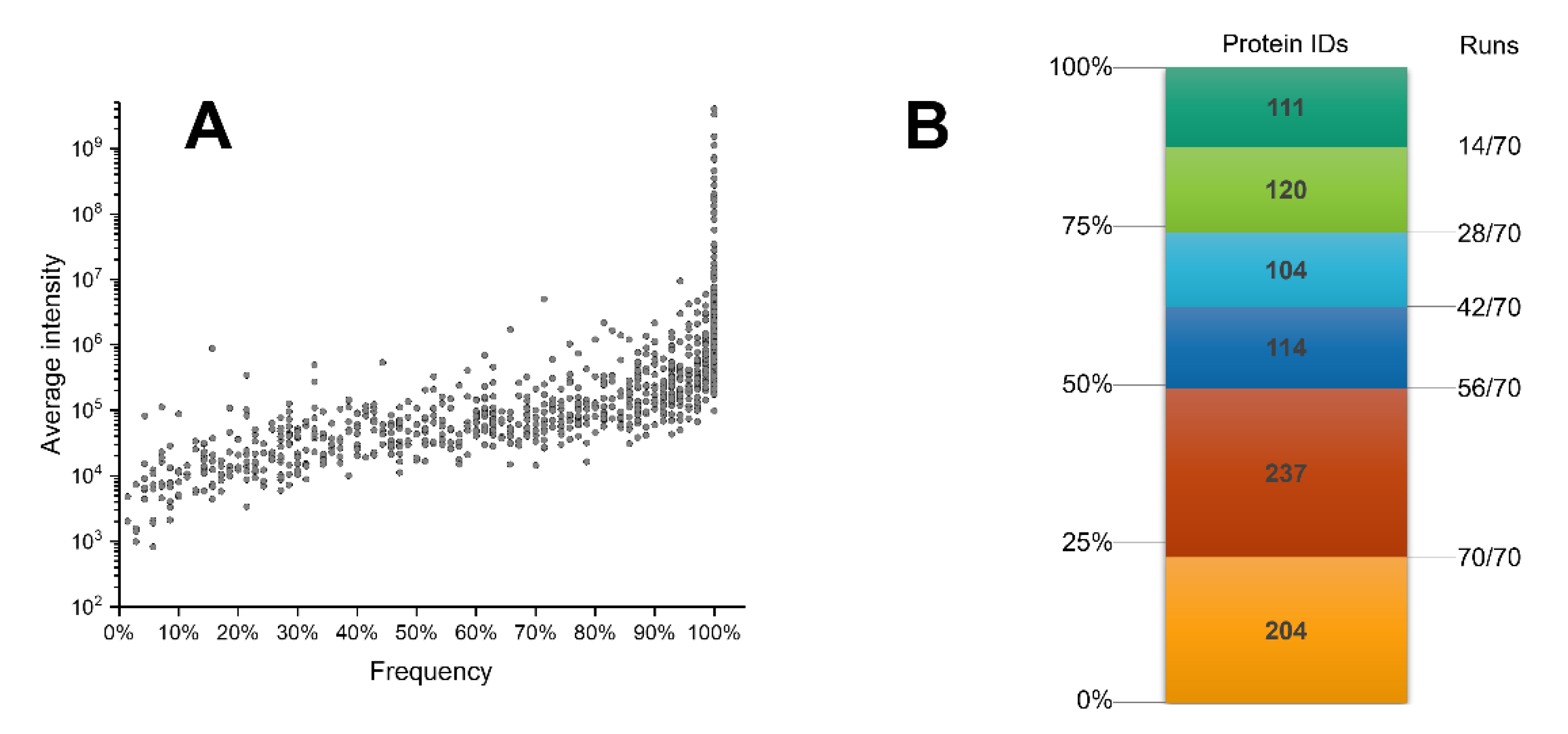
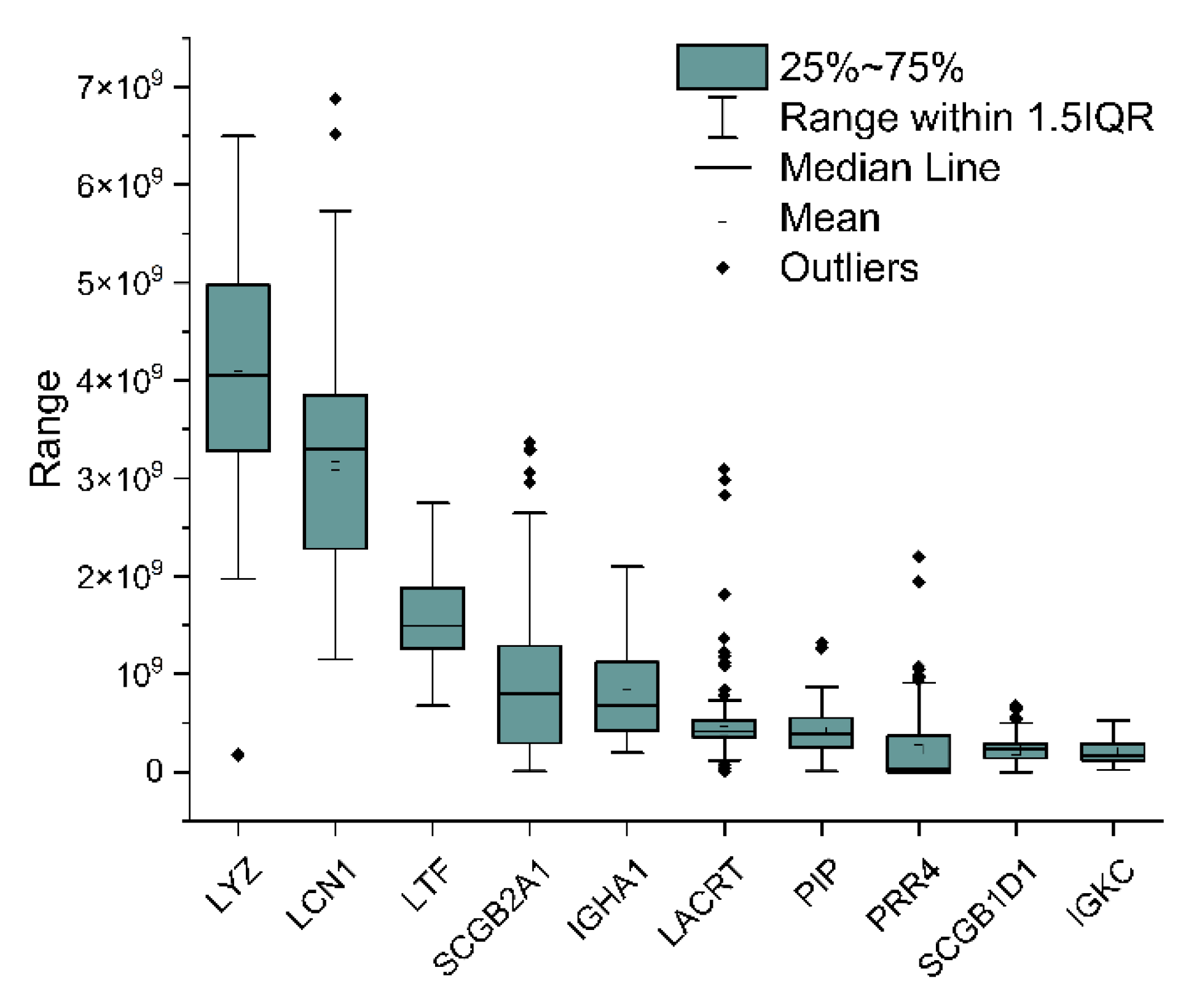
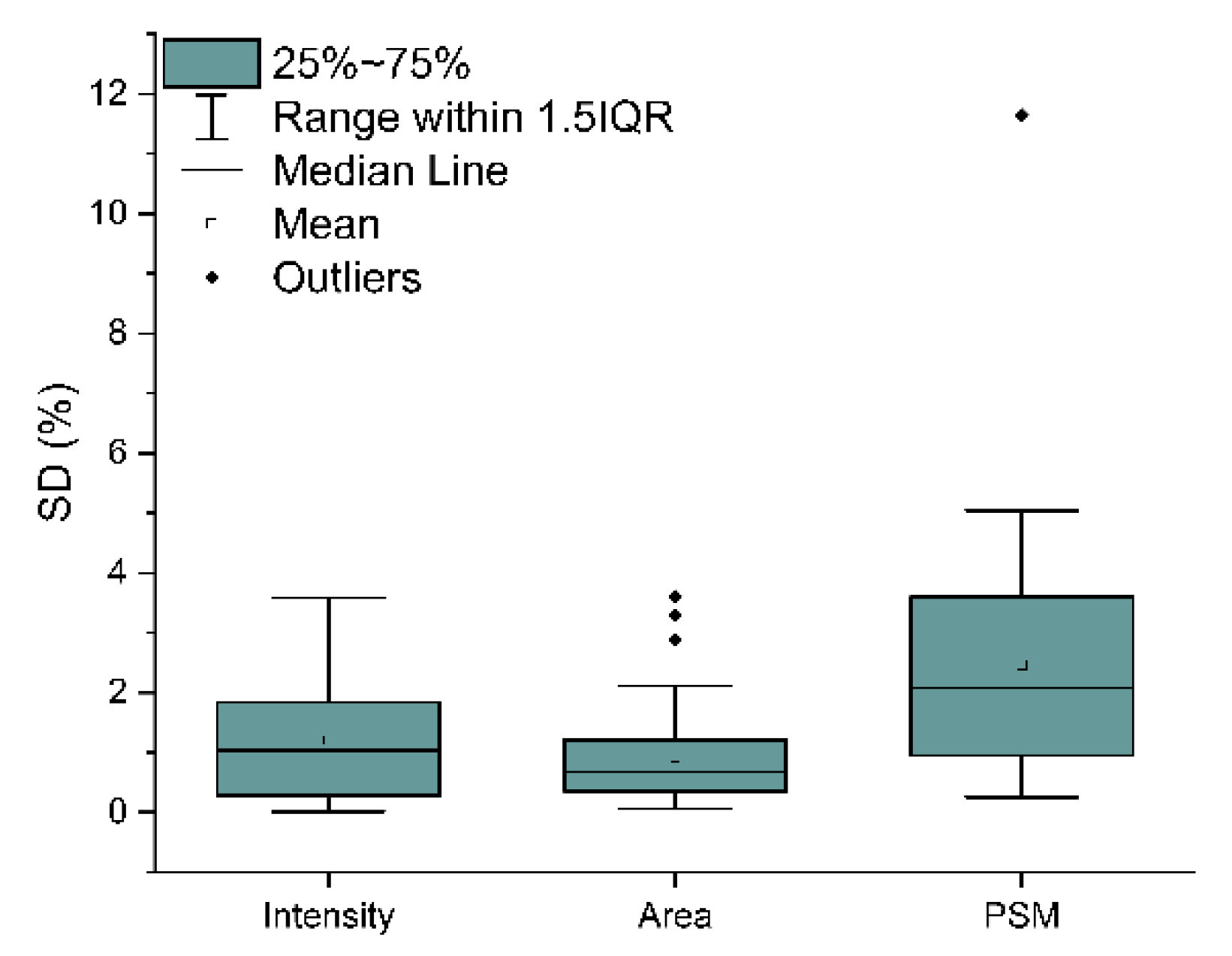
2. Results
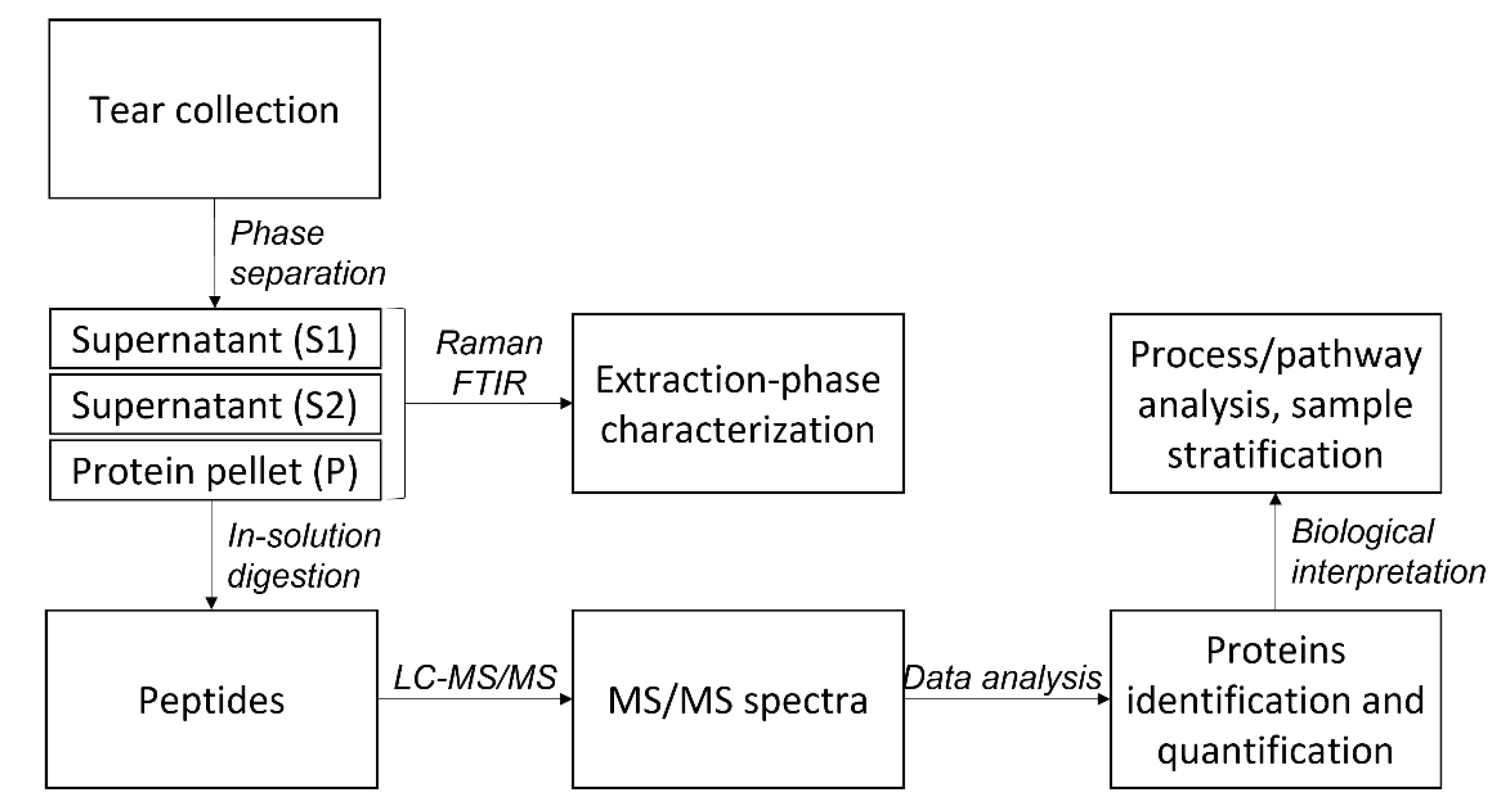
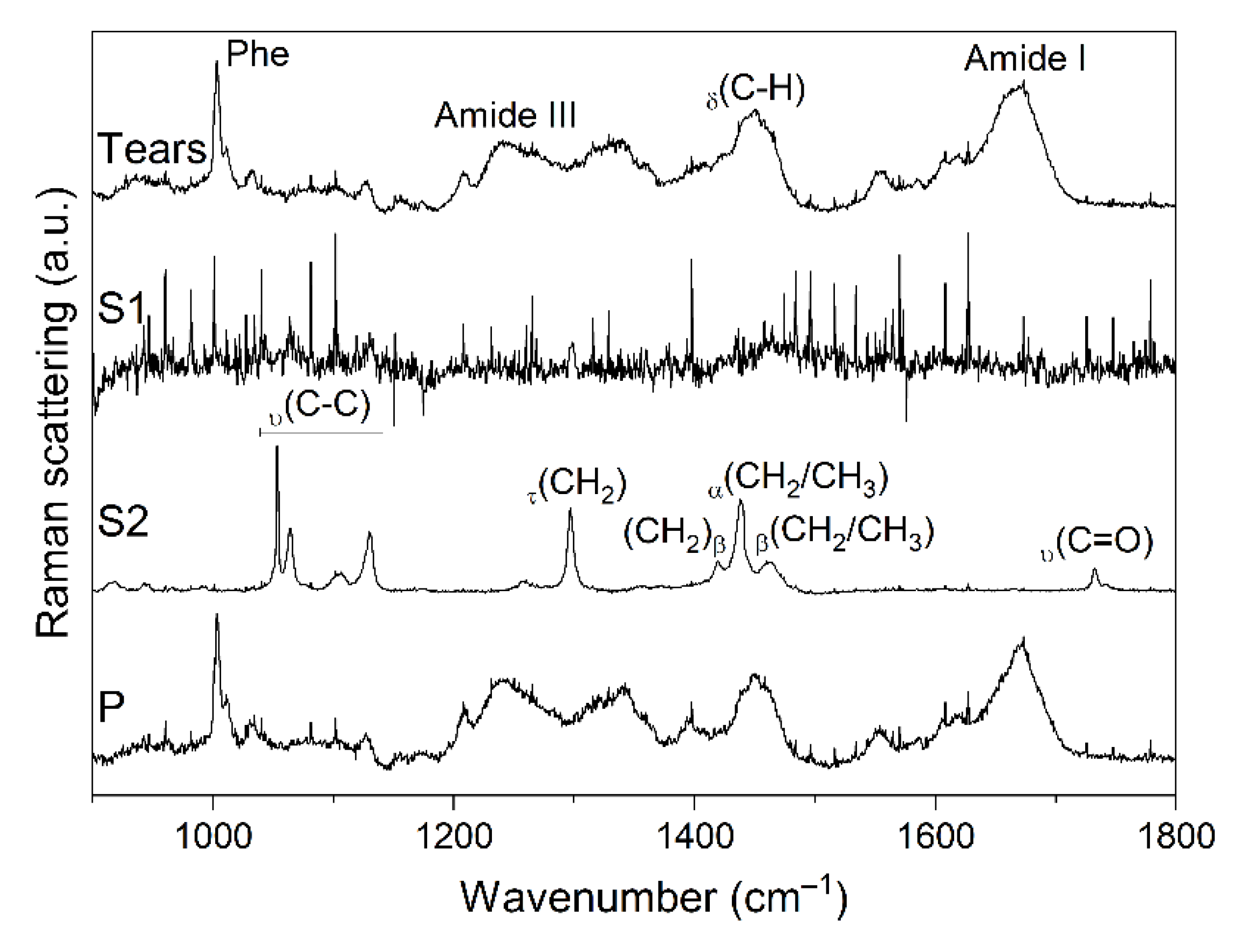
2.1. Protein Extraction
2.2. LC-MS/MS Analysis
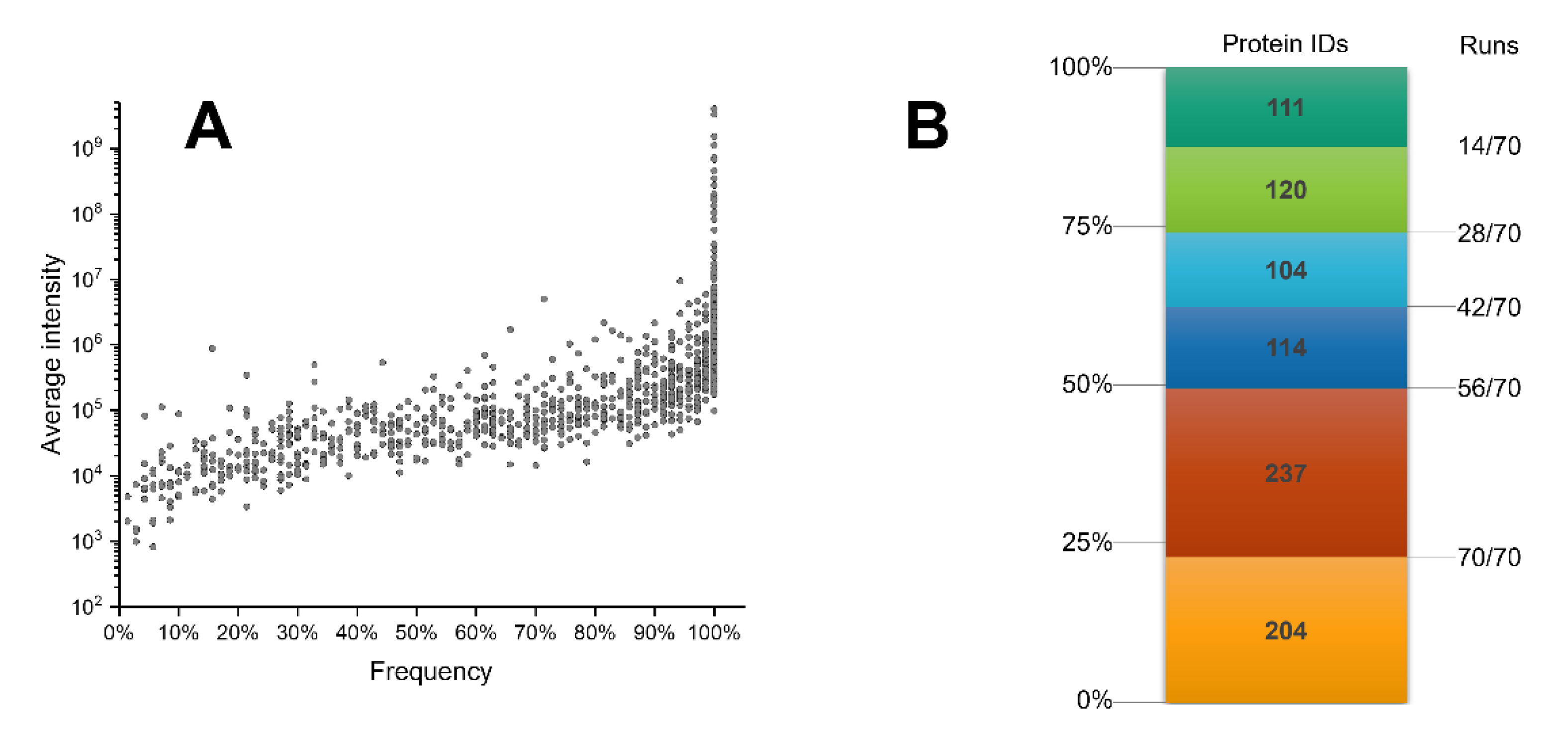
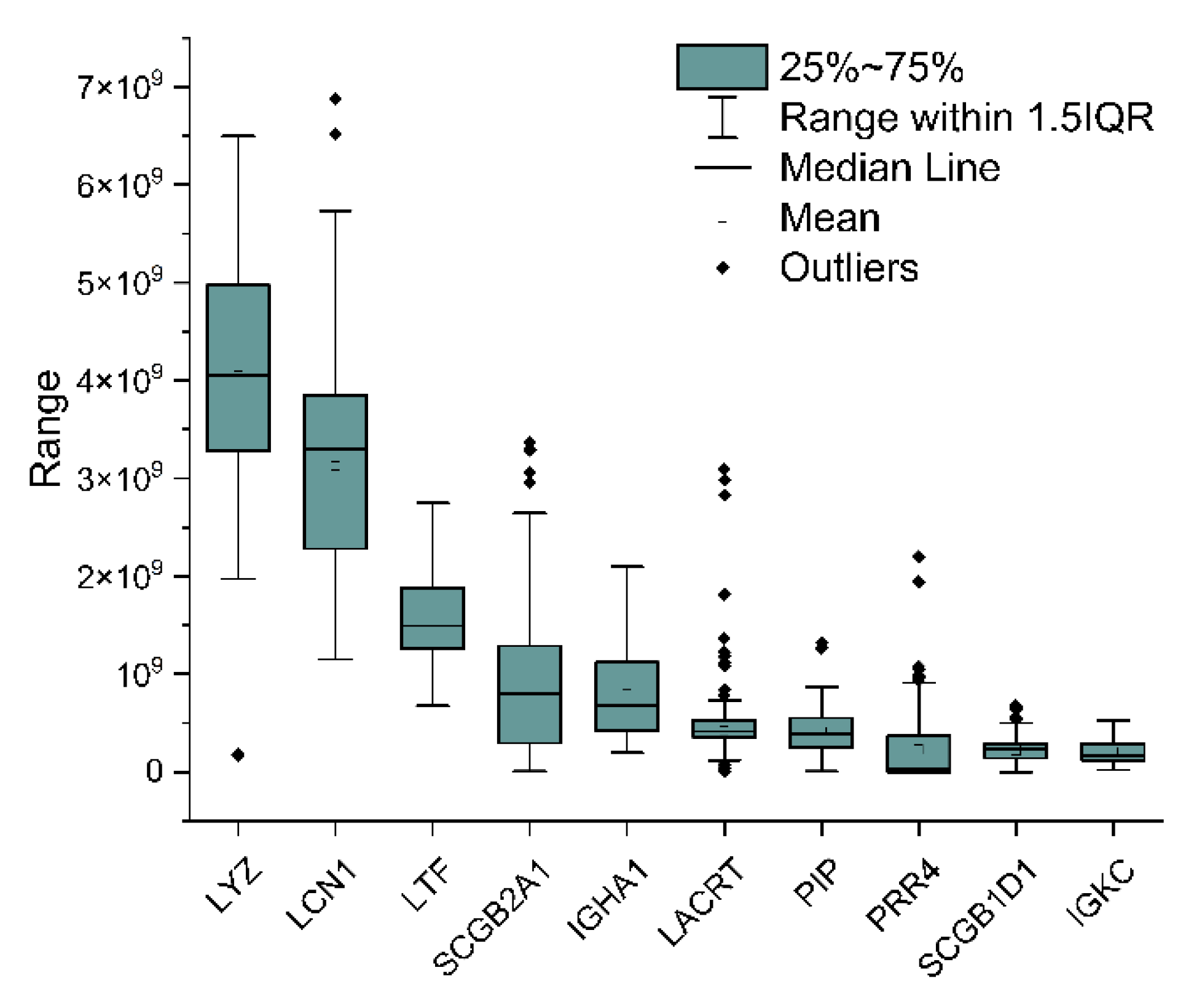
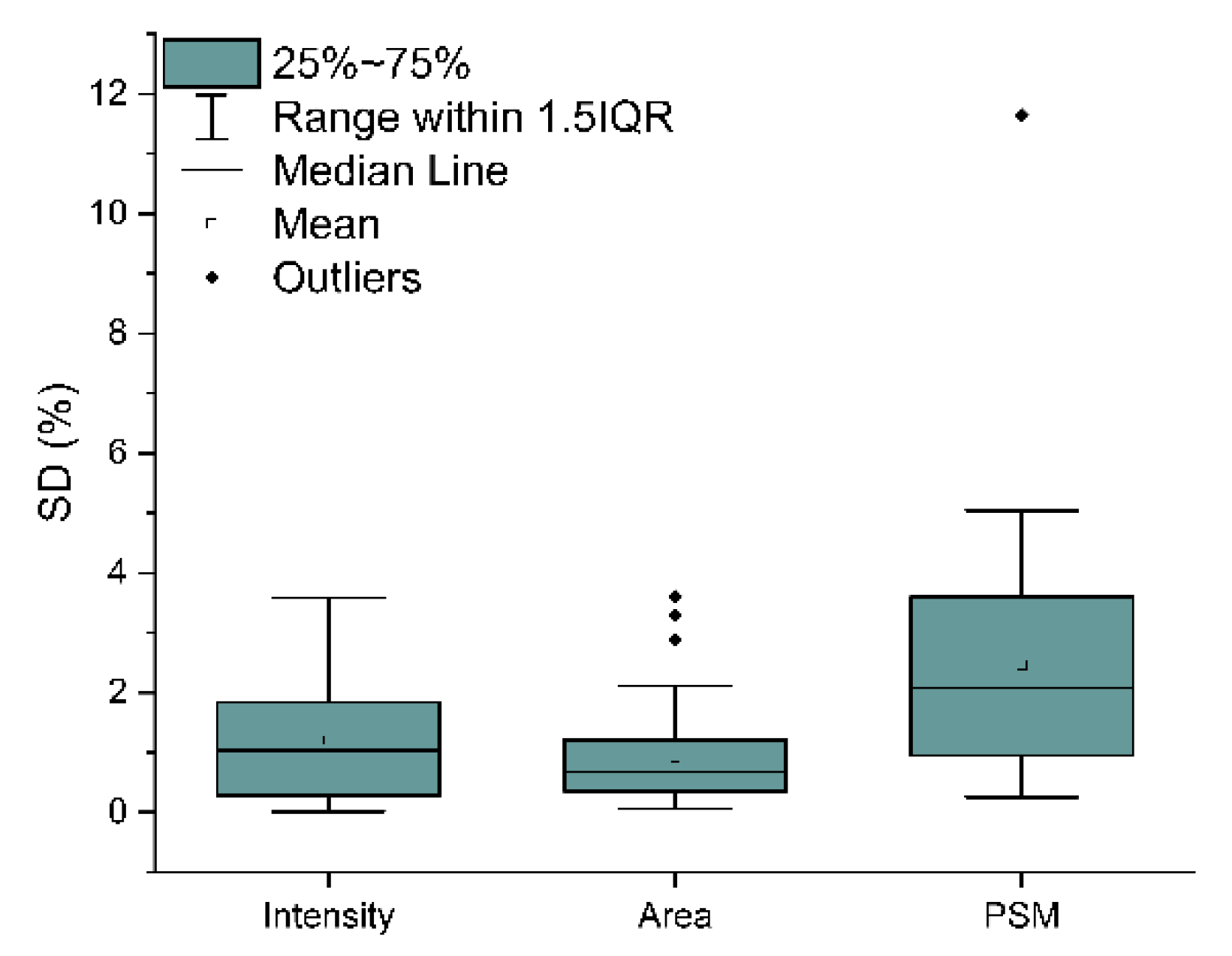
2.3. Analytical Power Evaluation
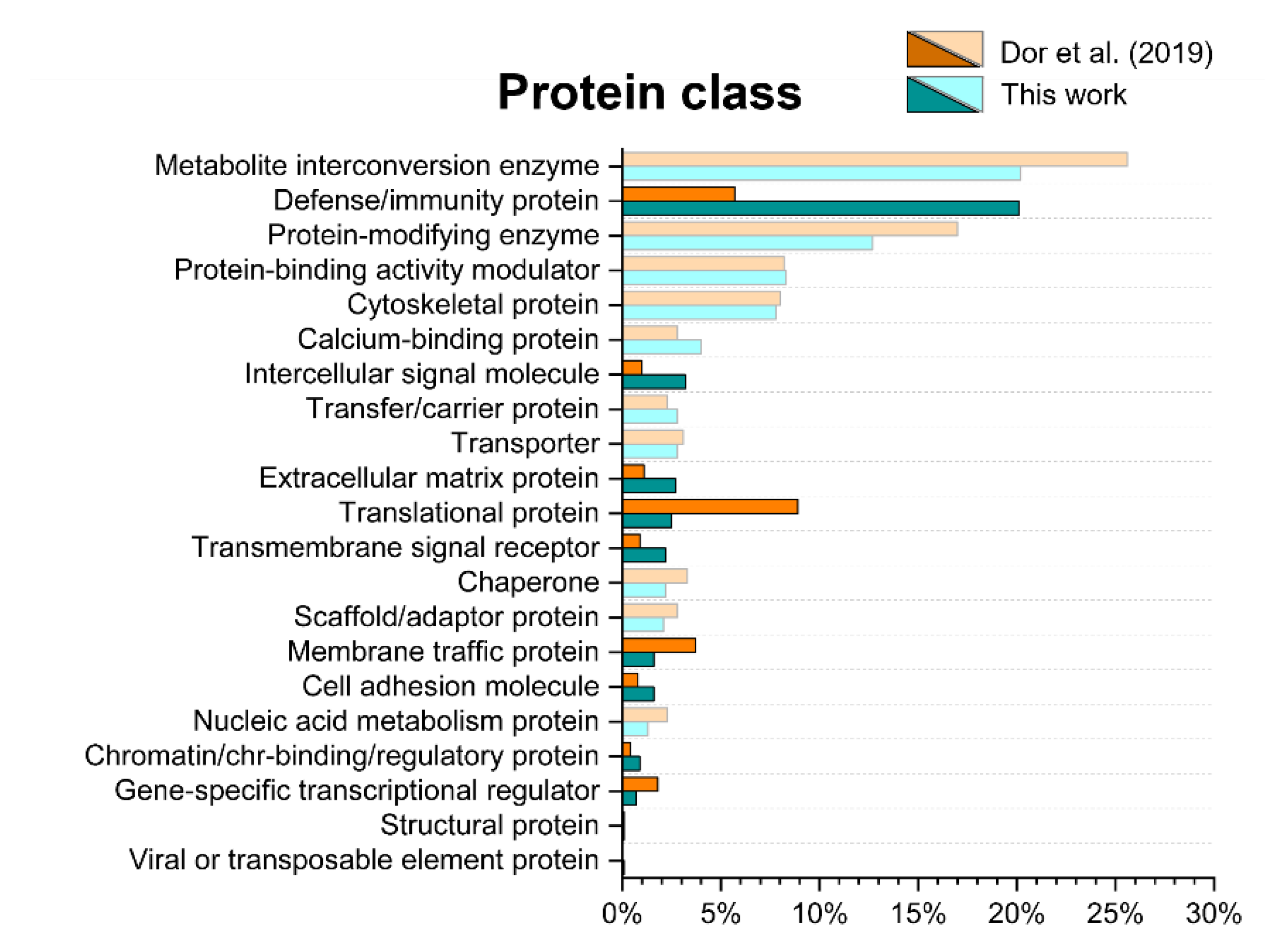
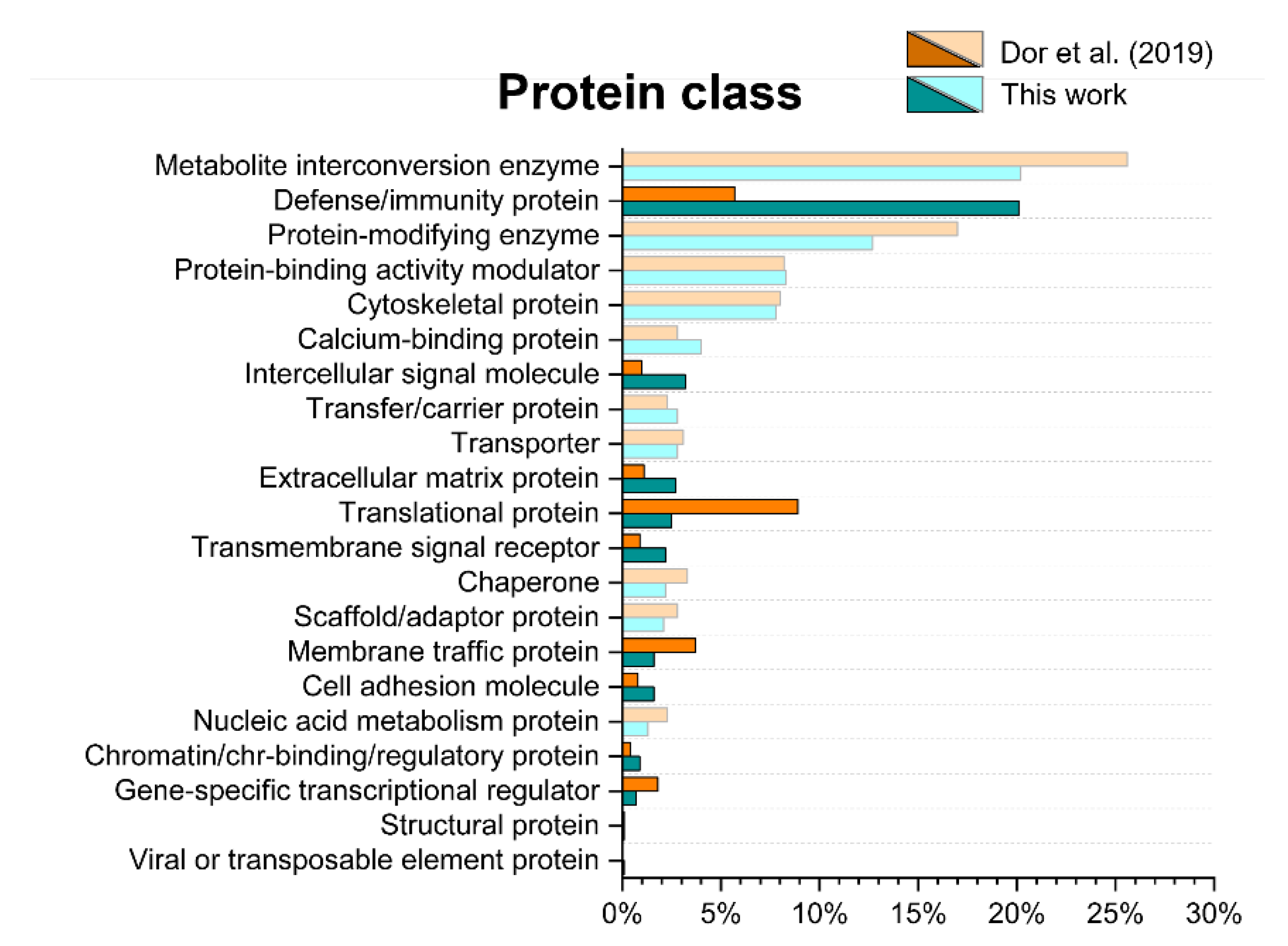
2.4. Gene ontology Analysis
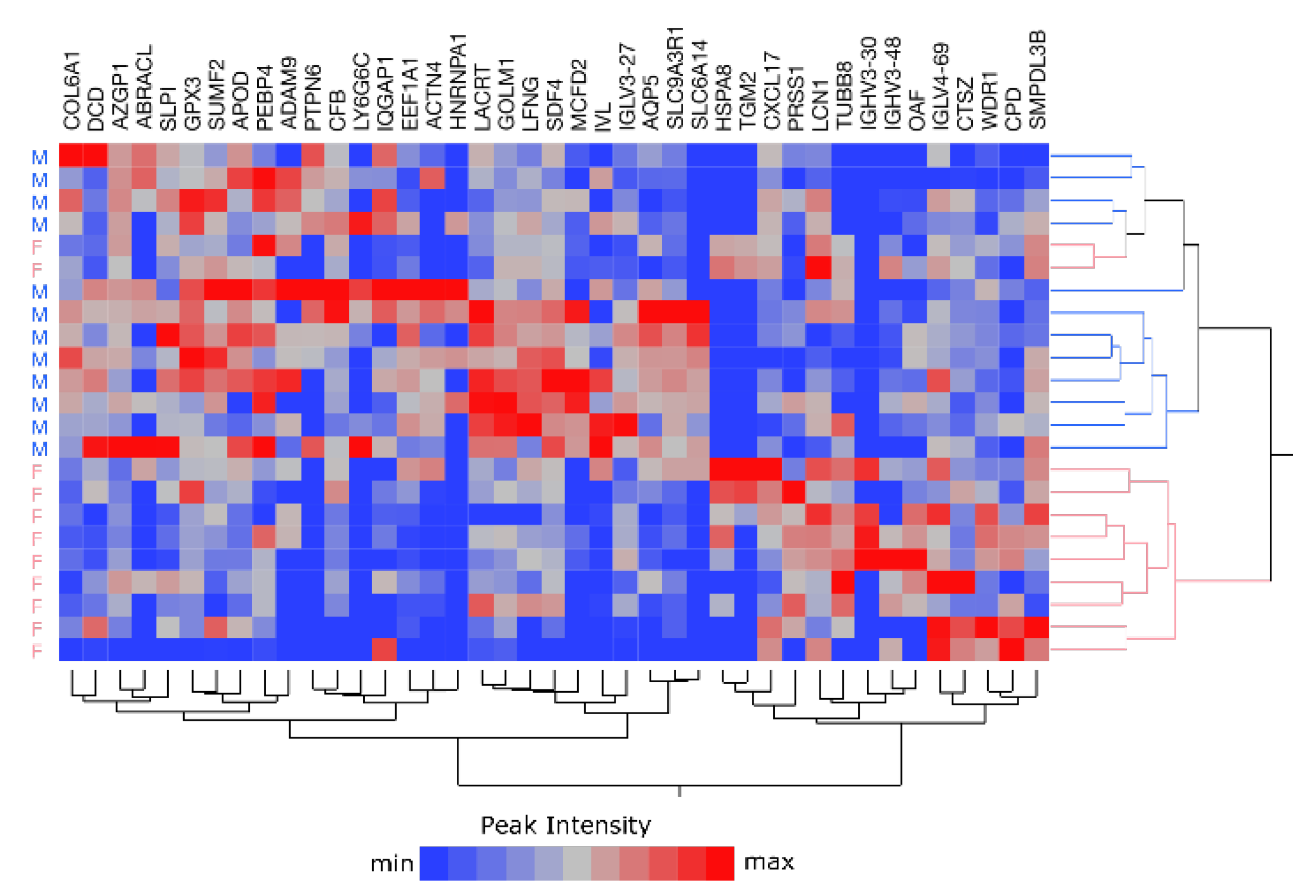
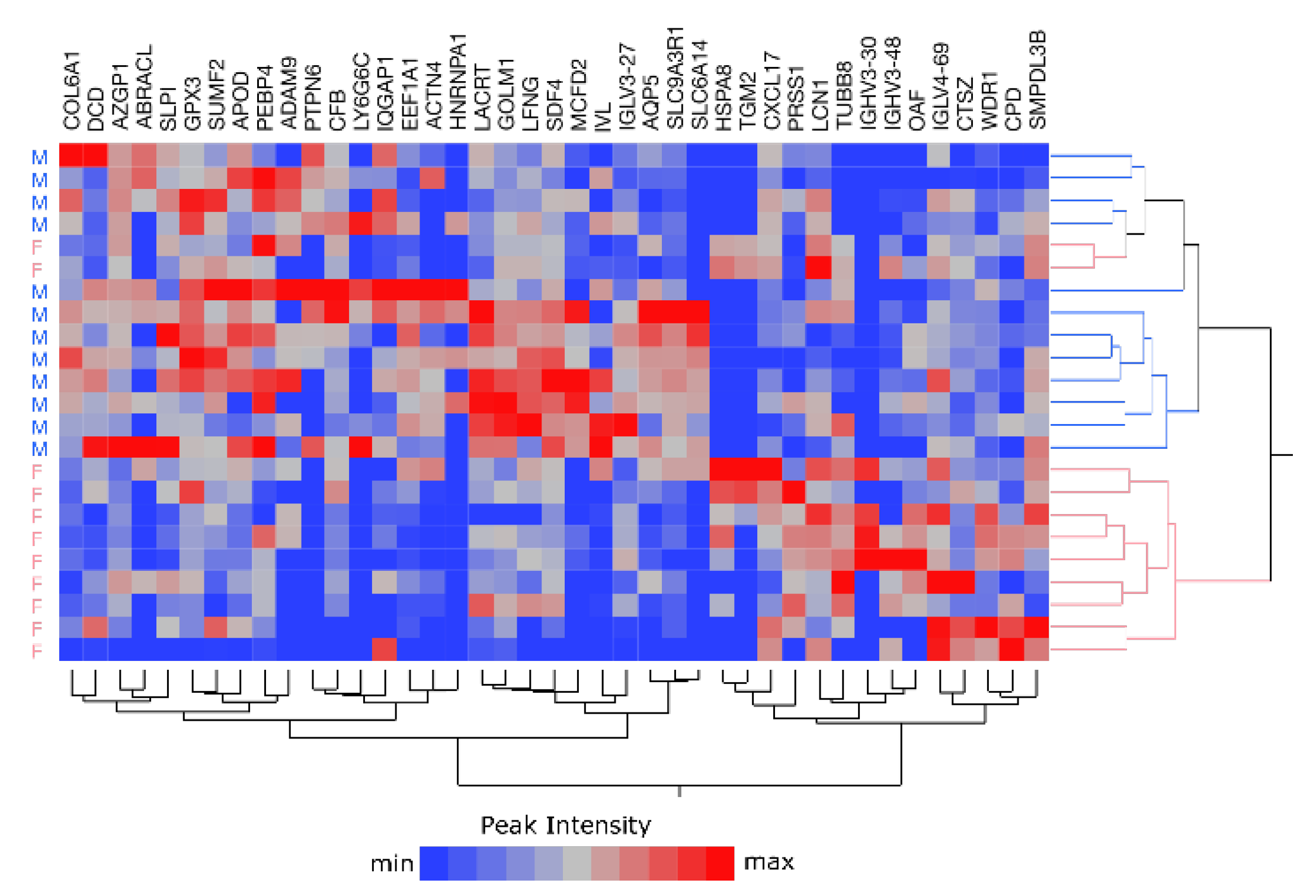
2.5. Sample Stratification
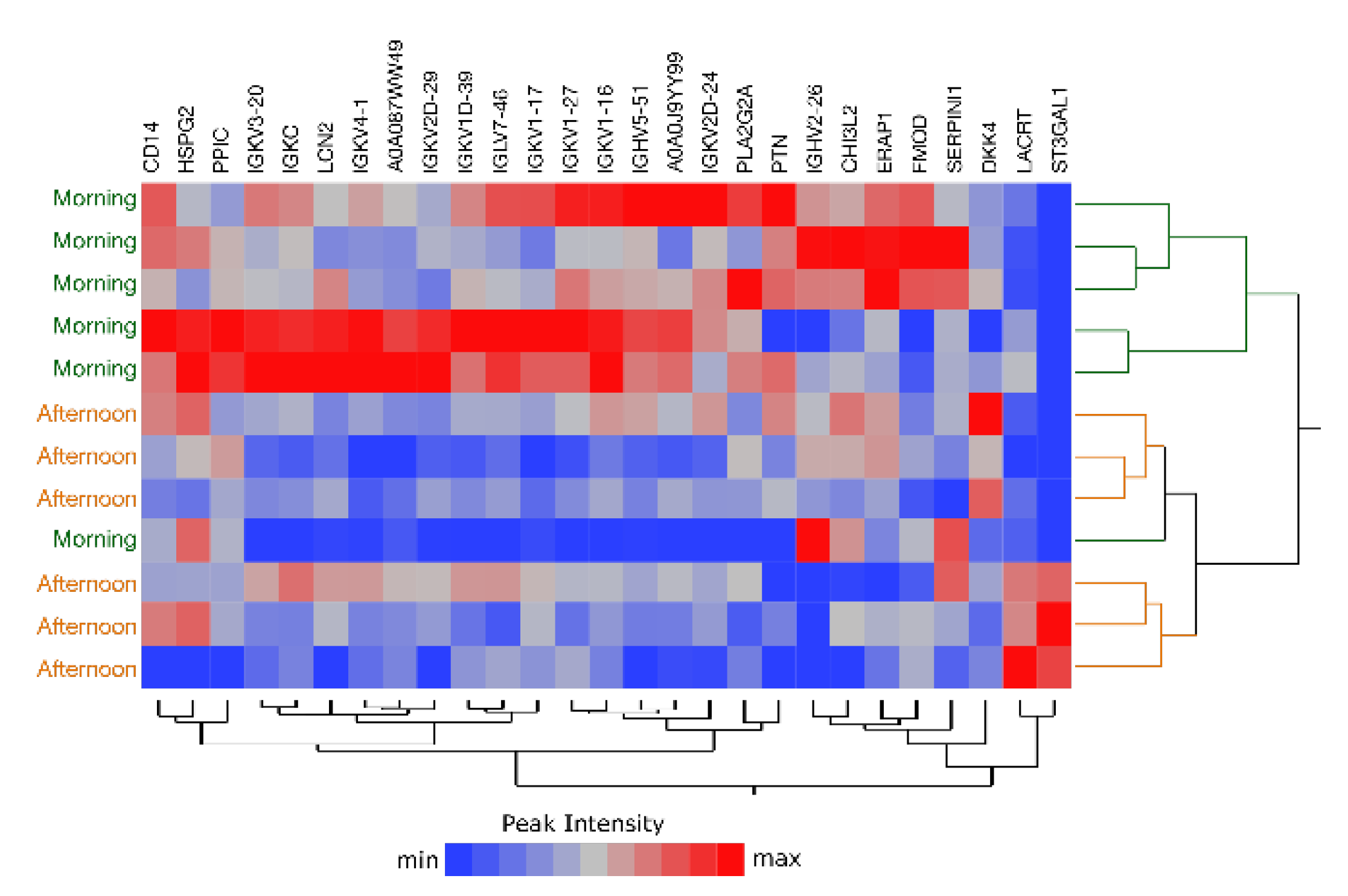
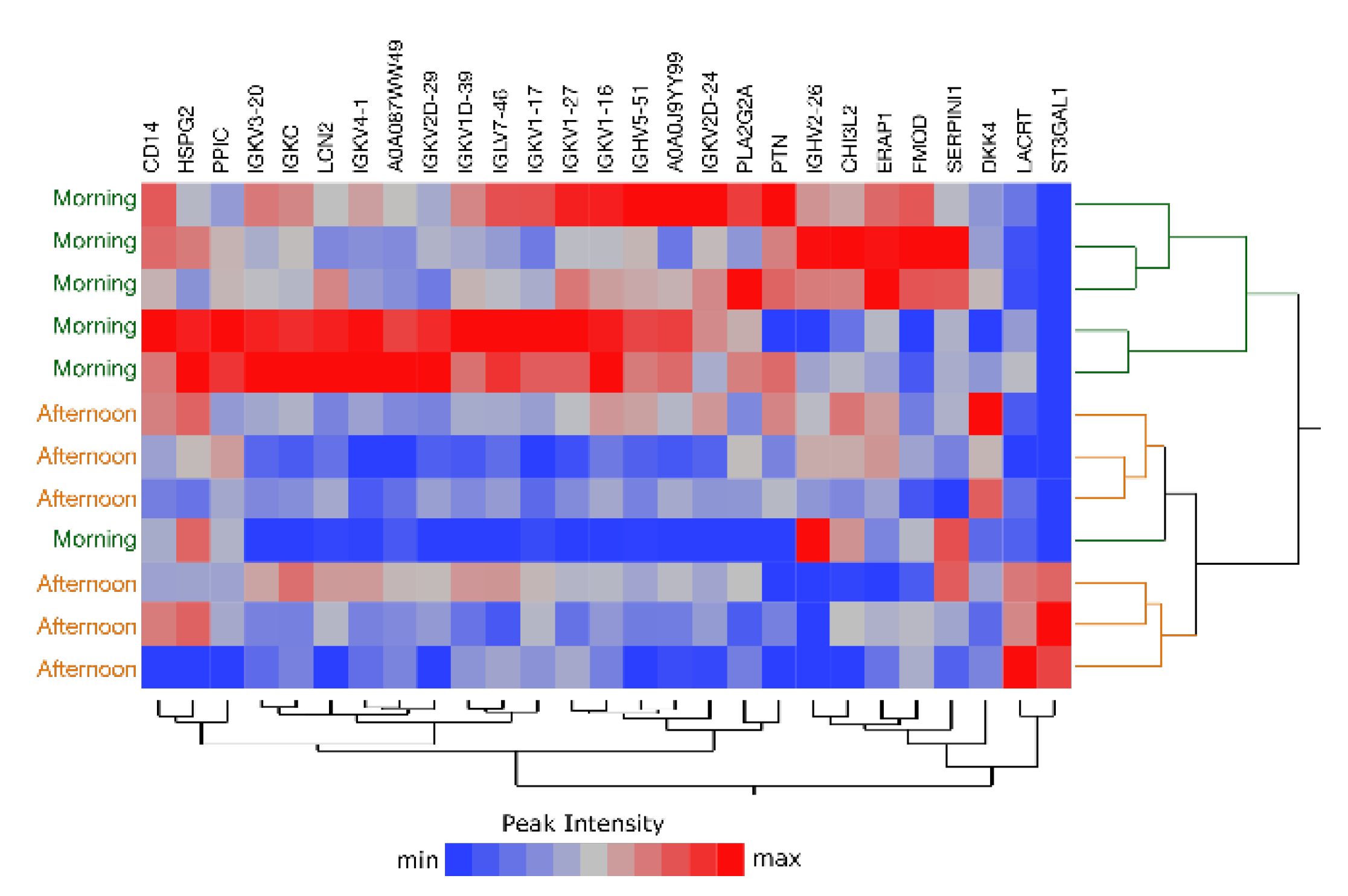
2.6. Comparison of Morning vs. Afternoon Sample Collection
3. Discussion
4. Materials and Methods
4.1. Volunteers’ Recruitment
4.2. Sample Collection
4.3. Protein Extraction
4.4. RS Analysis
4.5. FTIR Analysis
4.6. Protein Reduction, Alkylation, and Digestion
4.7. LC-MS/MS Analysis
4.8. Process and Pathway Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemp, M.A. Advances in understanding and managing dry eye disease. Am. J. Ophthalmol. 2008, 146, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Beuerman, R.W. The power of tears: How tear proteomics research could revolutionize the clinic. Expert Rev. Proteom. 2017, 14, 189–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrenson, J.G. 2-anterior eye. In Contact Lens Practice, 3rd ed.; Efron, N., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 10–27. ISBN 978-0-7020-6660-3. [Google Scholar]
- Phillips, A.; Speedwell, L. Contact Lenses, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 978-0-7020-7168-3. [Google Scholar]
- Rentka, A.; Koroskenyi, K.; Harsfalvi, J.; Szekanecz, Z.; Szucs, G.; Szodoray, P.; Kemeny-Beke, A. Evaluation of commonly used tear sampling methods and their relevance in subsequent biochemical analysis. Ann. Clin. Biochem. 2017, 54, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.; Acera, A.; Merayo-LLoves, J.; Durán, J.A.; González, N.; Rodriguez, S.; Bistolas, N.; Schumacher, S.; Bier, F.F.; Peter, H.; et al. Tear proteome analysis in ocular surface diseases using label-free LC-MS/MS and multiplexed-microarray biomarker validation. Sci. Rep. 2017, 7, 17478. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Beuerman, R.W.; Chan, C.M.; Zhao, S.Z.; Li, X.R.; Yang, H.; Tong, L.; Liu, S.; Stern, M.E.; Tan, D. Identification of tear fluid biomarkers in dry eye syndrome using ITRAQ quantitative proteomics. J. Proteom. Res. 2009, 8, 4889–4905. [Google Scholar] [CrossRef]
- Tong, L.; Zhou, L.; Beuerman, R.W.; Zhao, S.Z.; Li, X.R. Association of tear proteins with meibomian gland disease and dry eye symptoms. Br. J. Ophthalmol. 2011, 95, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Thangavelu, M.; Zhang, L.; Green, K.B.; Nichols, K.K. ITRAQ quantitative proteomics in the analysis of tears in dry eye patients. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5052–5059. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Smith, J.W.; Huang, C.-M. Mass Spectrometry-Based Label-Free Quantitative Proteomics. Available online: https://www.hindawi.com/journals/bmri/2010/840518/ (accessed on 24 November 2020).
- Li, N.; Wang, N.; Zheng, J.; Liu, X.M.; Lever, O.W.; Erickson, P.M.; Li, L. Characterization of human tear proteome using multiple proteomic analysis techniques. J. Proteom. Res. 2005, 4, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.A.; de Godoy, L.M.; Mann, M. Identification of 491 proteins in the tear fluid proteome reveals a large number of proteases and protease inhibitors. Genome Biol. 2006, 7, R72. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhao, S.Z.; Koh, S.K.; Chen, L.; Vaz, C.; Tanavde, V.; Li, X.R.; Beuerman, R.W. In-depth analysis of the human tear proteome. J. Proteom. 2012, 75, 3877–3885. [Google Scholar] [CrossRef]
- Aass, C.; Norheim, I.; Eriksen, E.F.; Thorsby, P.M.; Pepaj, M. Single unit filter-aided method for fast proteomic analysis of tear fluid. Anal. Biochem. 2015, 480, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Dor, M.; Eperon, S.; Lalive, P.H.; Guex-Crosier, Y.; Hamedani, M.; Salvisberg, C.; Turck, N. Investigation of the global protein content from healthy human tears. Exp. Eye Res. 2019, 179, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Vlahou, A. Implementation of clinical proteomics: A step closer to personalized medicine? Proteom. Clin. Appl. 2019, 13, 1800088. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzini, E.; Santambrogio, C.; De Palma, A.; Mauri, P.; Tavazzi, S.; Grandori, R. Mass spectrometry-based tear proteomics for noninvasive biomarker discovery. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef]
- Butler, H.J.; Cameron, J.M.; Jenkins, C.A.; Hithell, G.; Hume, S.; Hunt, N.T.; Baker, M.J. Shining a light on clinical spectroscopy: Translation of diagnostic IR, 2D-IR and raman spectroscopy towards the clinic. Clin. Spectrosc. 2019, 1, 100003. [Google Scholar] [CrossRef]
- Gorr, H.M.; Zueger, J.M.; Barnard, J.A. Characteristic size for onset of coffee-ring effect in evaporating lysozyme-water solution droplets. J. Phys. Chem. B 2012, 116, 12213–12220. [Google Scholar] [CrossRef] [PubMed]
- Am, M.; Ra, F.; El-Naggar, A.H.; Tm, A.; Akhtar, S. Structure and microanalysis of tear film ferning of camel tears, human tears, and refresh plus. Mol. Vis. 2018, 24, 305–314. [Google Scholar]
- Filik, J.; Stone, N. Investigation into the protein composition of human tear fluid using centrifugal filters and drop coating deposition raman spectroscopy. J. Raman Spectrosc. 2009, 40, 218–224. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Czamara, K.; Majzner, K.; Pacia, M.Z.; Kochan, K.; Kaczor, A.; Baranska, M. Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 2015, 46, 4–20. [Google Scholar] [CrossRef]
- Ami, D.; Mereghetti, P.; Foli, A.; Tasaki, M.; Milani, P.; Nuvolone, M.; Palladini, G.; Merlini, G.; Lavatelli, F.; Natalello, A. ATR-FTIR spectroscopy supported by multivariate analysis for the characterization of adipose tissue aspirates from patients affected by systemic amyloidosis. Anal. Chem. 2019, 91, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.M.; Motta, S.; Silvestre, D.D.; Falchi, F.; Dehò, G.; Mauri, P.; Sperandeo, P.; Polissi, A. Dissecting Escherichia coli outer membrane biogenesis using differential proteomics. PLoS ONE 2014, 9, e100941. [Google Scholar] [CrossRef] [Green Version]
- Ananthi, S.; Prajna, N.V.; Lalitha, P.; Valarnila, M.; Dharmalingam, K. Pathogen induced changes in the protein profile of human tears from fusarium keratitis patients. PLoS ONE 2013, 8, e53018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pult, H.; Tosatti, S.G.P.; Spencer, N.D.; Asfour, J.-M.; Ebenhoch, M.; Murphy, P.J. Spontaneous blinking from a tribological viewpoint. Ocul. Surf. 2015, 13, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Pult, H.; Purslow, C.; Murphy, P.J. Mucins and ocular signs in symptomatic and asymptomatic contact lens wear. Optom. Vis. Sci. Off. Publ. Am. Acad. Optom. 2008, 85, E930–E938. [Google Scholar] [CrossRef]
- Sonawane, S.; Khanolkar, V.; Namavari, A.; Chaudhary, S.; Gandhi, S.; Tibrewal, S.; Jassim, S.H.; Shaheen, B.; Hallak, J.; Horner, J.H.; et al. Ocular surface extracellular DNA and nuclease activity imbalance: A new paradigm for inflammation in dry eye disease. Invest. Ophthalmol. Vis. Sci. 2012, 53, 8253–8263. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Fan, X.-G.; Tang, D. Release and activity of histone in diseases. Cell Death Dis. 2014, 5, e1370. [Google Scholar] [CrossRef] [Green Version]
- Ponzini, E.; Scotti, L.; Grandori, R.; Tavazzi, S.; Zambon, A. Lactoferrin concentration in human tears and ocular diseases: A meta-analysis. Invest. Ophthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef]
- Vagge, A.; Senni, C.; Bernabei, F.; Pellegrini, M.; Scorcia, V.; Traverso, C.E.; Giannaccare, G. Therapeutic effects of lactoferrin in ocular diseases: From dry eye disease to infections. Int. J. Mol. Sci. 2020, 21, 6668. [Google Scholar] [CrossRef]
- Careba, I.; Gradinaru, D.; Chiva, A.; Totir, M.; Ciuluvica, R.; Gradinaru, S.; Ungureanu, E. Correlations between eyelid tumors and tear lipocalin, lysozyme and lactoferrin concentrations in postmenopausal women. J. Med. Life 2015, 8, 94–98. [Google Scholar] [PubMed]
- Offiah, I.; Calder, V.L. Immune mechanisms in allergic eye diseases: What is new? Curr. Opin. Allergy Clin. Immunol. 2009, 9, 477–481. [Google Scholar] [CrossRef]
- Masoudi, S.; Zhong, L.; Raftery, M.J.; Stapleton, F.J.; Willcox, M.D. Method development for quantification of five tear proteins using Selected Reaction Monitoring (SRM) mass spectrometry. Invest. Ophthalmol. Vis. Sci. 2014, 55, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Choy, C.K.M.; Cho, P.; Chung, W.-Y.; Benzie, I.F.F. Water-soluble antioxidants in human tears: Effect of the collection method. Invest. Ophthalmol. Vis. Sci. 2001, 42, 3130–3134. [Google Scholar] [PubMed]
- Van Haeringen, N.J.; Glasius, E. Lysosomal hydrolases in tears and the lacrimal gland: Effect of acetylsalicylic acid on the release from the lacrimal gland. Invest. Ophthalmol. Vis. Sci. 1980, 19, 826–829. [Google Scholar]
- Yang, X.; Pan, X.; Zhao, X.; Luo, J.; Xu, M.; Bai, D.; Hu, Y.; Liu, X.; Yu, Q.; Gao, D. Autophagy and Age-Related Eye Diseases. Available online: https://www.hindawi.com/journals/bmri/2019/5763658/ (accessed on 19 January 2021).
- Wert, K.J.; Lin, J.H.; Tsang, S.H. General pathophysiology in retinal degeneration. Cell Based Ther. Retin. Degener. Dis. 2014, 53, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Mukai, R.; Okunuki, Y.; Husain, D.; Kim, C.B.; Lambris, J.D.; Connor, K.M. The complement system is critical in maintaining retinal integrity during aging. Front. Aging Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Mauri, P.; Dehò, G. A Proteomic approach to the analysis of RNA degradosome composition in Escherichia coli. Methods Enzymol. 2008, 447, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG mapper for inferring cellular functions from protein sequences. Protein Sci. Publ. Protein Soc. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Bari, E.; Di Silvestre, D.; Mastracci, L.; Grillo, F.; Grisoli, P.; Marrubini, G.; Nardini, M.; Mastrogiacomo, M.; Sorlini, M.; Rossi, R.; et al. GMP-compliant sponge-like dressing containing MSC lyo-secretome: Proteomic network of healing in a murine wound model. Eur. J. Pharm. Biopharm. Off. J. Arbeitsgemeinschaft Pharm. Verfahrenstechnik EV 2020, 155, 37–48. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponzini, E.; Ami, D.; Duse, A.; Santambrogio, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Pezzoli, F.; Natalello, A.; Tavazzi, S.; et al. Single-Tear Proteomics: A Feasible Approach to Precision Medicine. Int. J. Mol. Sci. 2021, 22, 10750. https://doi.org/10.3390/ijms221910750
Ponzini E, Ami D, Duse A, Santambrogio C, De Palma A, Di Silvestre D, Mauri P, Pezzoli F, Natalello A, Tavazzi S, et al. Single-Tear Proteomics: A Feasible Approach to Precision Medicine. International Journal of Molecular Sciences. 2021; 22(19):10750. https://doi.org/10.3390/ijms221910750
Chicago/Turabian StylePonzini, Erika, Diletta Ami, Alessandro Duse, Carlo Santambrogio, Antonella De Palma, Dario Di Silvestre, Pierluigi Mauri, Fabio Pezzoli, Antonino Natalello, Silvia Tavazzi, and et al. 2021. "Single-Tear Proteomics: A Feasible Approach to Precision Medicine" International Journal of Molecular Sciences 22, no. 19: 10750. https://doi.org/10.3390/ijms221910750
APA StylePonzini, E., Ami, D., Duse, A., Santambrogio, C., De Palma, A., Di Silvestre, D., Mauri, P., Pezzoli, F., Natalello, A., Tavazzi, S., & Grandori, R. (2021). Single-Tear Proteomics: A Feasible Approach to Precision Medicine. International Journal of Molecular Sciences, 22(19), 10750. https://doi.org/10.3390/ijms221910750