
Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells


Abstract
1. Introduction
2. Results
2.1. Pancreatic Differentiation Potential from Human Embryonic Stem Cell (hESC) Was Disrupted in Hyperglycemic Culture Conditions
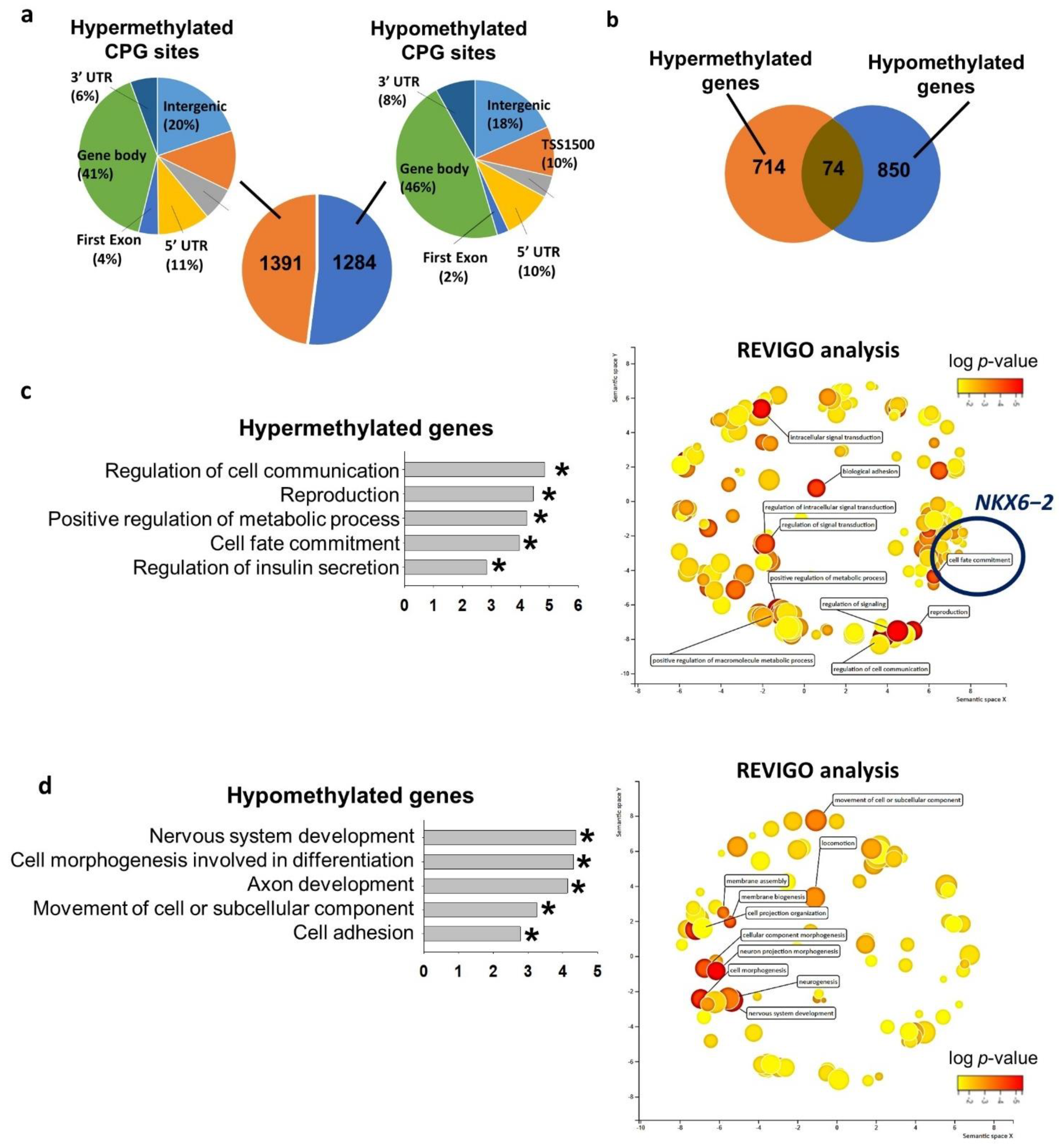
2.2. Hyperglycemia Altered Global DNA Methylome in hESC
2.3. NKX6−2 Was Hypermethylated under Hyperglycemic Condition
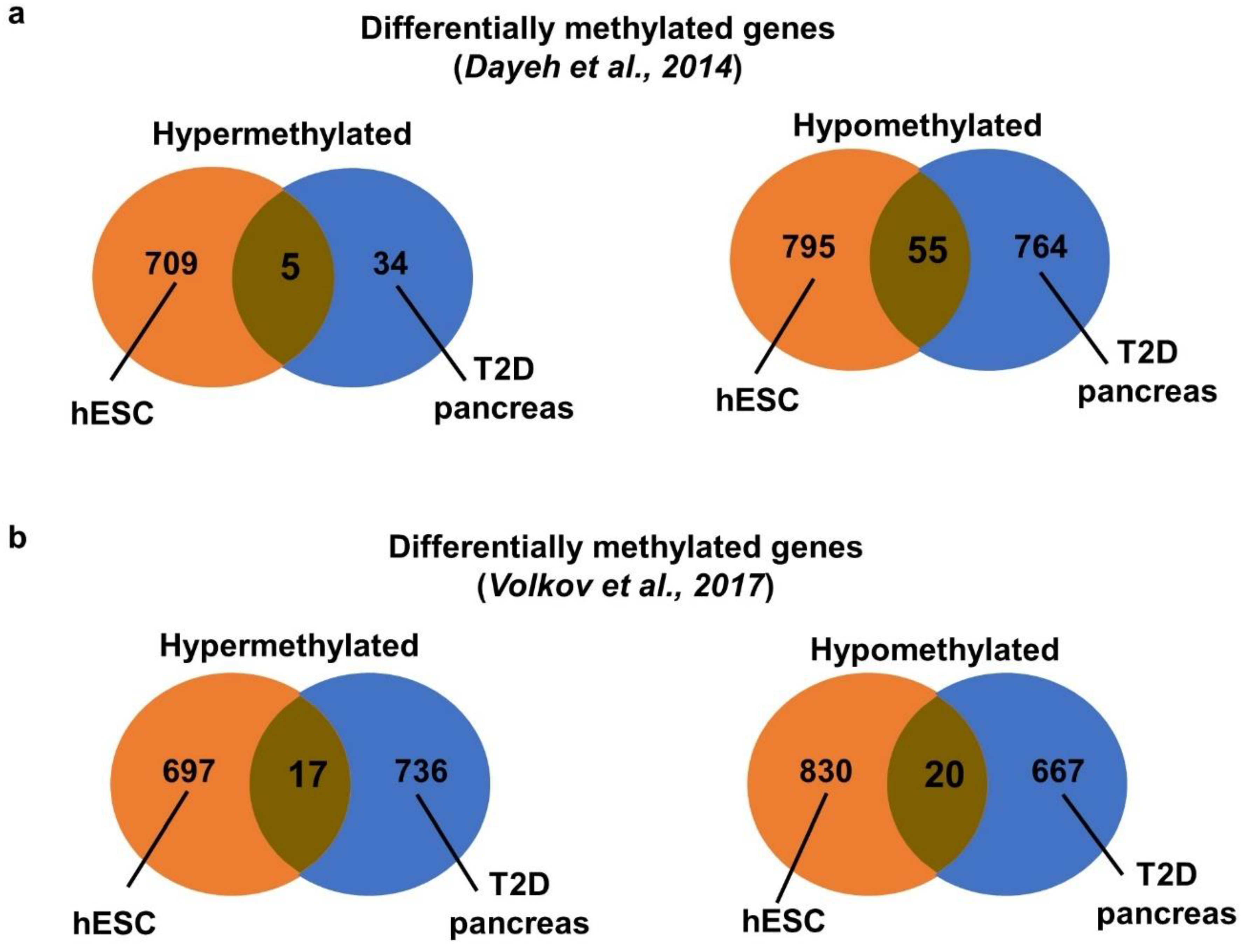
2.4. Meta-Analysis of DNA Methylomes of Hyperglycemia Treated VAL3 with Dysregulated Methylomes of Human Pancreatic Islets from T2D Patients
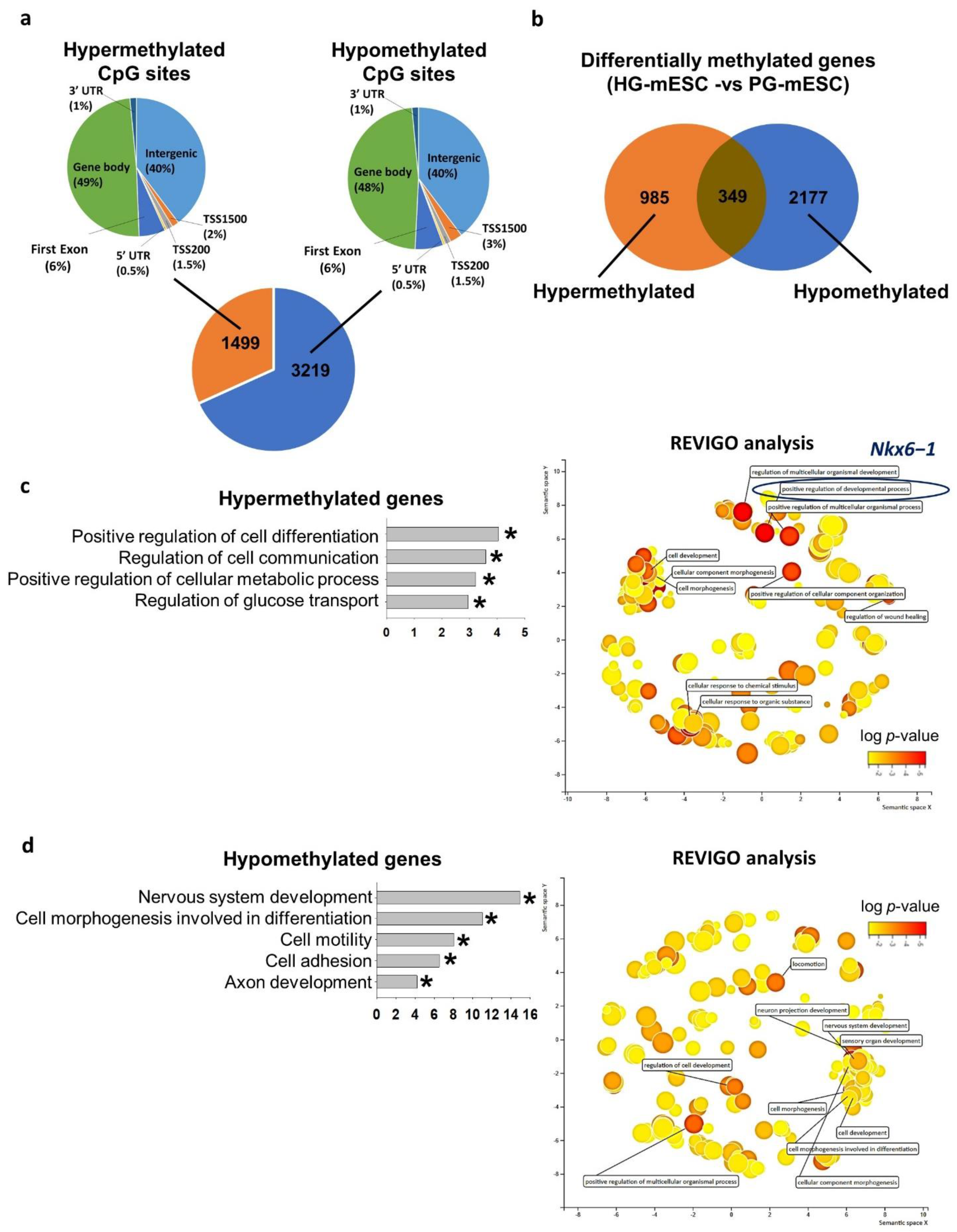
2.5. Conventional Hyperglycemic Culture Condition Dysregulated Mouse ESC DNA Methylome of Genes Related to Metabolism and Glucose Transport
3. Discussion
4. Materials and Methods
4.1. Human Embryonic Stem Cells (ESC) Culture and Hyperglycemic Conditions
4.2. Differentiation of hESC into Definitive Endoderm and Pancreatic Progenitor Cells
4.3. Bisulfite Sequencing
4.4. Derivation of Mouse Embryonic Stem Cell (mESC) Lines
4.5. Embryoid Body (EB) Formation from mESC
4.6. Genome-Wide Methylation Analysis
4.7. Quantitative Polymerase Chain Reaction (qPCR)
4.8. Chromatin Immunoprecipitation (ChIP)
4.9. Immunofluorescent Staining
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.; Winter, P.D.; Osmond, C.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Franks, P.W.; Looker, H.C.; Kobes, S.; Touger, L.; Tataranni, P.A.; Hanson, R.; Knowler, W.C. Gestational glucose tolerance and risk of type 2 diabetes in young pima indian offspring. Diabetes 2006, 55, 460–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dabelea, D. The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care 2007, 30, S169–S174. [Google Scholar] [CrossRef]
- Marco, L.J.; McCloskey, K.; Vuillermin, P.J.; Burgner, D.; Said, J.; Ponsonby, A.-L. Cardiovascular disease risk in the offspring of diabetic women: The impact of the intrauterine environment. Exp. Diabetes Res. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Dayeh, T.; Volkov, P.; Salö, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.; Wollheim, C.B.; Eliasson, L.; Rönn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef]
- Volkmar, M.; Dedeurwaerder, S.; Da Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.L.; Wang, F.-F.; Shu, J.; Tian, S.; Jiang, Y.; Zhang, D.; Wang, N.; Luo, Q.; Zhang, Y.; Jin, F.; et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 2012, 61, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.B.; Waite, S.L.; Wood, K.A.; Martin, K.L. Impact of hyperglycemia on early embryo development and embryopathy: In vitro experiments using a mouse model. Hum. Reprod. 2007, 22, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Scott-Drechsel, D.E.; Rugonyi, S.; Marks, D.L.; Thornburg, K.L.; Hinds, M.T. Hyperglycemia slows embryonic growth and suppresses cell cycle via cyclin D1 and p21. Diabetes 2013, 62, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Palanza, P.; Gioiosa, L.; Saal, F.S.V.; Parmigiani, S. Effects of developmental exposure to bisphenol A on brain and behavior in mice. Environ. Res. 2008, 108, 150–157. [Google Scholar] [CrossRef]
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.-A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010, 6, e1000811. [Google Scholar] [CrossRef]
- Chen, A.C.H.; Lee, Y.L.; Fong, S.W.; Wong, C.C.Y.; Ng, E.; Yeung, S.B.W. Hyperglycemia impedes definitive endoderm differentiation of human embryonic stem cells by modulating histone methylation patterns. Cell Tissue Res. 2017, 368, 563–578. [Google Scholar] [CrossRef]
- Valbuena, D.; Galán, A.; Sánchez, E.; Poo, M.E.; Gómez, E.; Sánchez-Luengo, S.; Melguizo, D.; García, A.; Ruiz, V.; Moreno, R.; et al. Derivation and characterization of three new Spanish human embryonic stem cell lines (VAL −3 −4 −5) on human feeder and in serum-free conditions. Reprod. Biomed. Online 2006, 13, 875–886. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Nelson, S.B.; Schaffer, A.; Sander, M. The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta-cell fate specification in Pdx1+ pancreatic progenitor cells. Development 2007, 134, 2491–2500. [Google Scholar] [CrossRef]
- Volkov, P.; Bacos, K.; Ofori, J.K.; Esguerra, J.L.S.; Eliasson, L.; Rönn, T.; Ling, C. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 2017, 66, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Braam, S.R.; Denning, C.; Matsa, E.; E Young, L.; Passier, R.; Mummery, C.L. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat. Protoc. 2008, 3, 1435–1443. [Google Scholar] [CrossRef]
- Marks, H.; Kalkan, T.; Menafra, R.; Denissov, S.; Jones, K.; Hofemeister, H.; Nichols, J.; Kranz, A.; Stewart, A.F.; Smith, A.; et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 2012, 149, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Güemes, M.; A Rahman, S.; Hussain, K. What is a normal blood glucose? Arch. Dis. Child. 2015, 101, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Pevny, L.H.; Sockanathan, S.; Placzek, M.; Lovell-Badge, R. A role for SOX1 in neural determination. Development 1998, 125, 1967–1978. [Google Scholar] [CrossRef]
- Wilson, V.; Manson, L.; Skarnes, W.C.; Beddington, R.S. The T gene is necessary for normal mesodermal morphogenetic cell movements during gastrulation. Development 1995, 121, 877–886. [Google Scholar] [CrossRef]
- Stringer, E.J.; Duluc, I.; Saandi, T.; Davidson, I.; Bialecka, M.; Sato, T.; Barker, N.; Clevers, H.; Pritchard, C.; Winton, D.J.; et al. Cdx2 determines the fate of postnatal intestinal endoderm. Development 2012, 139, 465–474. [Google Scholar] [CrossRef]
- Bermejo-Alvarez, P.; Roberts, R.; Rosenfeld, C. Effect of glucose concentration during in vitro culture of mouse embryos on development to blastocyst, success of embryo transfer, and litter sex ratio. Mol. Reprod. Dev. 2012, 79, 329–336. [Google Scholar] [CrossRef]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of in vitro fertilization with beckwith-wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Everett, L.J.; Lim, H.-W.; Patel, N.A.; Schug, J.; Kroon, E.; Kelly, O.G.; Wang, A.; D’Amour, K.A.; Robins, A.J.; et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013, 12, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, X.; Zhou, Y.; Lee, M.; Guo, L.; Han, W.; Mo, W.; Cao, W.-M.; Sun, D.; Xie, R.; et al. Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids Res. 2018, 46, 2883–2900. [Google Scholar] [CrossRef]
- Henseleit, K.D.; Nelson, S.B.; Kuhlbrodt, K.; Hennings, J.C.; Ericson, J.; Sander, M. NKX6 transcription factor activity is required for α- andβ-cell development in the pancreas. Development 2005, 132, 3139–3149. [Google Scholar] [CrossRef]
- Lu, J.; Jaafer, R.; Bonnavion, R.; Bertolino, P.; Zhang, C.-X. Transdifferentiation of pancreatic α-cells into insulin-secreting cells: From experimental models to underlying mechanisms. World J. Diabetes 2014, 5, 847–853. [Google Scholar] [CrossRef]
- Watada, H. New role for alpha cells as a source for new beta cells. J. Diabetes Investig. 2010, 2, 43–44. [Google Scholar] [CrossRef][Green Version]
- Daniil, G.; Fernandes-Rosa, F.L.; Chemin, J.; Blesneac, I.; Beltrand, J.; Polak, M.; Jeunemaitre, X.; Boulkroun, S.; Amar, L.; Strom, T.M.; et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 2016, 13, 225–236. [Google Scholar] [CrossRef]
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.-J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H mutation as a cause of aldosterone-producing adenoma. Hypertension 2020, 75, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Umpierrez, G.E.; Cantey, P.; Smiley, D.; Palacio, A.; Temponi, D.; Luster, K.; Chapman, A. Primary aldosteronism in diabetic subjects with resistant hypertension. Diabetes Care 2007, 30, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Q.; Zhou, Y.; Niu, Y.; Wang, X.; Li, X.; Zheng, H.; Wei, T.; Zhao, L.; Gao, H. S100A2 promotes glycolysis and proliferation via GLUT1 regulation in colorectal cancer. FASEB J. 2020, 34, 13333–13344. [Google Scholar] [CrossRef]
- Pan, S.-C.; Li, C.-Y.; Kuo, C.-Y.; Kuo, Y.-Z.; Fang, W.-Y.; Huang, Y.-H.; Hsieh, T.-C.; Kao, H.-Y.; Kuo, Y.; Kang, Y.-R.; et al. The p53-S100A2 positive feedback loop negatively regulates epithelialization in cutaneous wound healing. Sci. Rep. 2018, 8, 5458. [Google Scholar] [CrossRef] [PubMed]
- Klopper, J.P.; Sharma, V.; Bissonnette, R.; Haugen, B.R. Combination PPARγand RXR agonist treatment in melanoma cells: Functional importance of S100A2. PPAR Res. 2009, 2010, 1–8. [Google Scholar] [CrossRef]
- Gong, M.; Yu, Y.; Liang, L.; Vuralli, D.; Froehler, S.; Kuehnen, P.; Du Bois, P.; Zhang, J.; Cao, A.; Liu, Y.; et al. HDAC4 mutations cause diabetes and induce β-cell FoxO1 nuclear exclusion. Mol. Genet. Genom. Med. 2019, 7, e602. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Descalzo, D.L.M.; Satoorian, T.S.; Walker, L.M.; Sparks, N.R.; Pulyanina, P.Y.; Nieden, N.I.Z. Glucose-induced oxidative stress reduces proliferation in embryonic stem cells via FOXO3A/β-catenin-dependent transcription of p21cip1. Stem Cell Rep. 2016, 7, 55–68. [Google Scholar] [CrossRef]
- Yang, P.; Chen, X.; Kaushal, S.; Reece, E.A.; Yang, P. High glucose suppresses embryonic stem cell differentiation into cardiomyocytes. Stem Cell Res. Ther. 2016, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Wang, X.D.; Loeken, M.R. Mouse embryonic stem cells established in physiological-glucose media express the HighKMGlut2 glucose transporter expressed by normal embryos. Stem Cells Transl. Med. 2013, 2, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Sander, M.; Sussel, L.; Conners, J.; Scheel, D.; Kalamaras, J.; Cruz, F.D.; Schwitzgebel, V.; Hayes-Jordan, A.; German, M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000, 127, 5533–5540. [Google Scholar] [CrossRef] [PubMed]
- Kubi, J.A.; Chen, A.C.; Fong, S.W.; Lai, K.P.; Wong, C.K.; Yeung, W.S.; Lee, K.F.; Lee, Y.L. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the differentiation of embryonic stem cells towards pancreatic lineage and pancreatic beta cell function. Environ. Int. 2019, 130, 104885. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.G.; Park, J.S.; Ko, J.-H.; Kim, Y.-S. Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci. Rep. 2019, 9, 11960. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef]
- Lee, Y.L.; Peng, Q.; Fong, S.W.; Chen, A.C.H.; Lee, K.F.; Ng, E.; Nagy, A.; Yeung, W.S.B. Sirtuin 1 Facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS ONE 2012, 7, e45633. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; E Garrett-Bakelman, F.; Figueroa, M.E.; Melnick, A.; E Mason, C. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypermethylated Genes | |||
| Term | Gene Count | p-Value | |
| GO terms | GO:0010646-egulation of cell communication | 136 | 1.49 × 10−5 |
| GO:0000003-reproduction | 72 | 3.60442 × 10−5 | |
| GO:0009893-positive regulation of metabolic process | 134 | 6.12 × 10−5 | |
| GO:0045165-cell fate commitment | 21 | 1.10 × 10−4 | |
| GO:0050796-regulation of insulin secretion | 15 | 0.001501577 | |
| REVIGO | GO:0023051-regulation of signaling | 139 | 0.006234194 |
| GO:0009966-regulation of signal transduction | 126 | 0.006947825 | |
| GO:0035556-intracellular signal transduction | 123 | 0.007549951 | |
| GO:0010646-regulation of cell communication | 136 | 0.008015438 | |
| GO:0000003-reproduction | 72 | 0.011758664 | |
| GO:0010604-positive regulation of macromolecule metabolic process | 128 | 0.012049471 | |
| GO:0009893-positive regulation of metabolic process | 134 | 0.014799489 | |
| GO:0022610-biological adhesion | 84 | 0.014802392 | |
| GO:0045165-cell fate commitment | 21 | 0.019097456 | |
| GO:1902531-regulation of intracellular signal transduction | 83 | 0.019839541 | |
| Hypomethylated genes | |||
| Term | Gene Count | p-Value | |
| GO terms | GO:0007399-nervous system development | 121 | 4.14929 × 10−5 |
| GO:0000904-cell morphogenesis involved in differentiation | 53 | 4.89 × 10−5 | |
| GO:0061564-axon development | 36 | 7.21 × 10−5 | |
| GO:0006928-movement of cell or subcellular component | 98 | 5.51 × 10−4 | |
| GO:0007155-cell adhesion | 91 | 0.001670345 | |
| REVIGO | GO:0048812-neuron projection morphogenesis | 139 | 0.004559117 |
| GO:0000902-cell morphogenesis | 126 | 0.01148067 | |
| GO:0007399-nervous system development | 123 | 0.012500011 | |
| GO:0030030-cell projection organization | 136 | 0.014020097 | |
| GO:0032989-cellular component morphogenesis | 72 | 0.017652818 | |
| GO:0044091-membrane biogenesis | 128 | 0.022253644 | |
| GO:0022008-neurogenesis | 134 | 0.032864231 | |
| GO:0071709-membrane assembly | 84 | 0.033846 | |
| GO:0006928-movement of cell or subcellular component | 21 | 0.038433186 | |
| GO:0040011-locomotion | 83 | 0.046574919 | |
| Hypermethylated Genes | |||
| Term | Gene Count | p-Value | |
| GO terms | GO:0045597-positive regulation of cell differentiation | 62 | 9.01 × 10−5 |
| GO:0010646-regulation of cell communication | 140 | 2.56 × 10−4 | |
| GO:0031325-positive regulation of cellular metabolic process | 137 | 6.07 × 10−4 | |
| GO:0010827-regulation of glucose transport | 11 | 0.00112593 | |
| REVIGO | GO:0051094-positive regulation of developmental process | 82 | 0.002840517 |
| GO:2000026-regulation of multicellular organismal development | 112 | 0.002906425 | |
| GO:0061041-regulation of wound healing | 18 | 0.006799059 | |
| GO:0071310-cellular response to organic substance | 118 | 0.006814995 | |
| GO:0048468-cell development | 125 | 0.007998872 | |
| GO:0051240-positive regulation of multicellular organismal process | 94 | 0.008416542 | |
| GO:0032989-cellular component morphogenesis | 84 | 0.008509034 | |
| GO:0070887-cellular response to chemical stimulus | 139 | 0.008815601 | |
| GO:0051130-positive regulation of cellular component organization | 77 | 0.01068532 | |
| GO:0000902-cell morphogenesis | 78 | 0.011784721 | |
| Hypomethylated Genes | |||
| Term | Gene Count | p-Value | |
| GO terms | GO:0007399-nervous system development | 291 | 1.22096 × 10−15 |
| GO:0000904-cell morphogenesis involved in differentiation | 124 | 9.93 × 10−12 | |
| GO:0048870-cell motility | 171 | 8.82 × 10−9 | |
| GO:0007155-cell adhesion | 187 | 3.11 × 10−7 | |
| GO:0061564-axon development | 63 | 6.29 × 10−5 | |
| REVIGO | GO:0007399-nervous system development | 291 | 3.33609 × 10−7 |
| GO:0048468-cell development | 286 | 1.76681 × 10−6 | |
| GO:0000902-cell morphogenesis | 177 | 1.27546 × 10−5 | |
| GO:0032989-cellular component morphogenesis | 187 | 1.4186 × 10−5 | |
| GO:0000904-cell morphogenesis involved in differentiation | 124 | 1.66519 × 10−5 | |
| GO:0051240-positive regulation of multicellular organismal process | 204 | 4.87473 × 10−5 | |
| GO:0031175-neuron projection development | 131 | 0.000104272 | |
| GO:0007423-sensory organ development | 95 | 0.000163232 | |
| GO:0040011-locomotion | 192 | 0.00016505 | |
| GO:0060284-regulation of cell development | 138 | 0.00018271 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, A.C.H.; Huang, W.; Fong, S.W.; Chan, C.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells. Int. J. Mol. Sci. 2021, 22, 10729. https://doi.org/10.3390/ijms221910729
Chen ACH, Huang W, Fong SW, Chan C, Lee KC, Yeung WSB, Lee YL. Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells. International Journal of Molecular Sciences. 2021; 22(19):10729. https://doi.org/10.3390/ijms221910729
Chicago/Turabian StyleChen, Andy Chun Hang, Wen Huang, Sze Wan Fong, Chris Chan, Kai Chuen Lee, William Shu Biu Yeung, and Yin Lau Lee. 2021. "Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells" International Journal of Molecular Sciences 22, no. 19: 10729. https://doi.org/10.3390/ijms221910729
APA StyleChen, A. C. H., Huang, W., Fong, S. W., Chan, C., Lee, K. C., Yeung, W. S. B., & Lee, Y. L. (2021). Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells. International Journal of Molecular Sciences, 22(19), 10729. https://doi.org/10.3390/ijms221910729