
Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract

 and
and
Abstract
:1. Introduction
2. Results
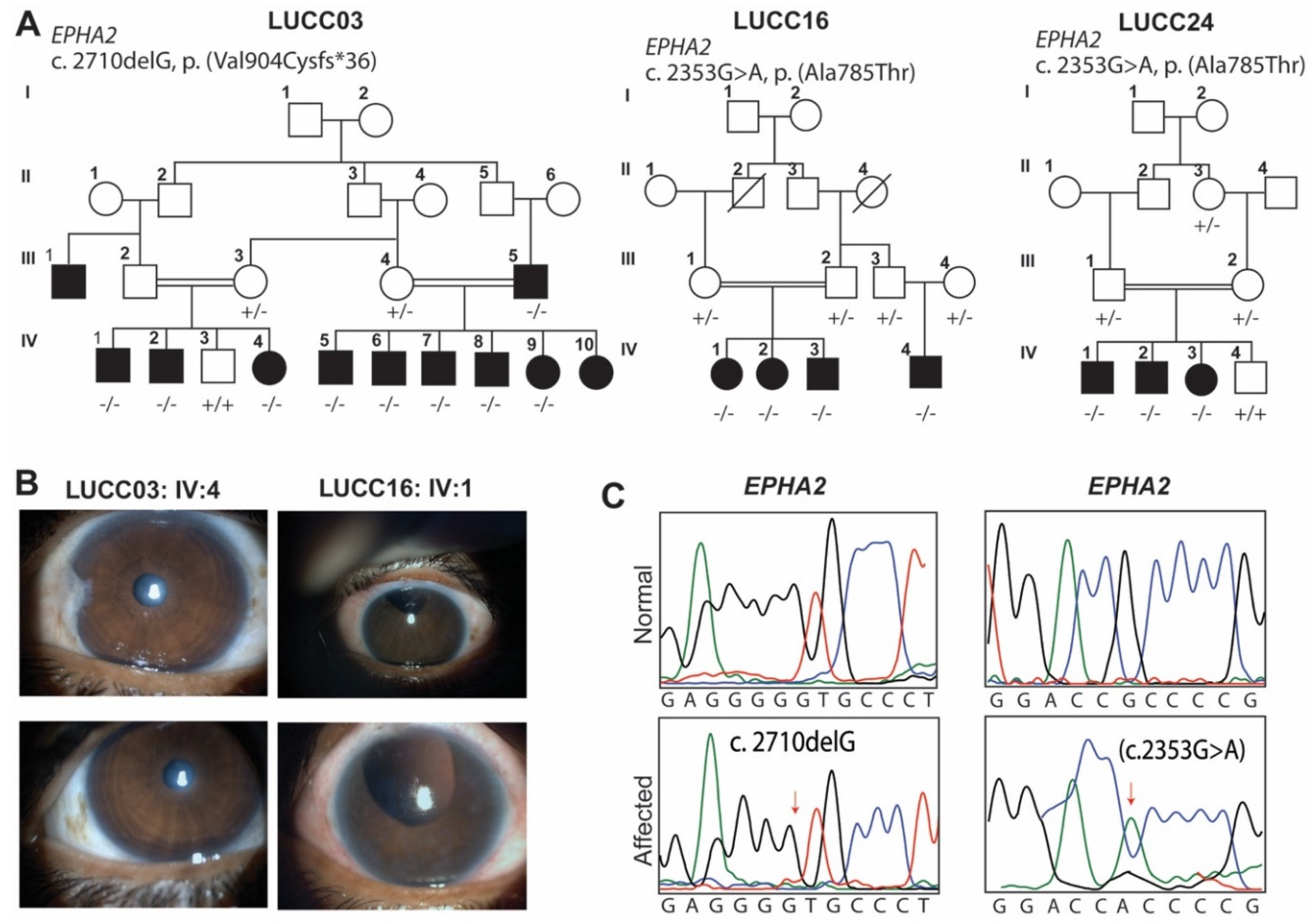
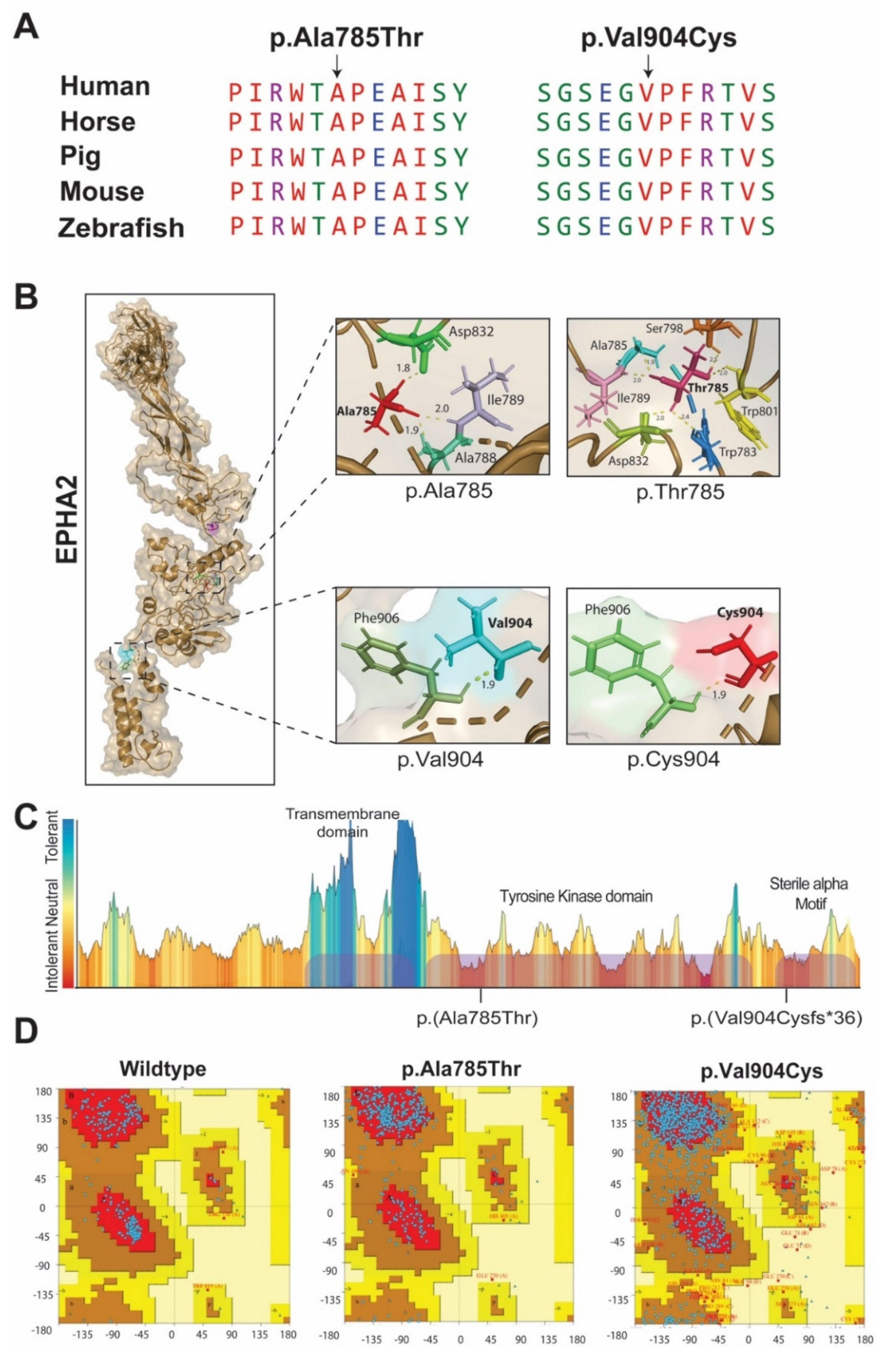
2.1. LUCC03 Family
2.2. LUCC16 and LUCC24 Families
3. Discussion
4. Material Method
4.1. Ascertainment and Clinical Evaluation
4.2. Sequencing and Bioinformatic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheeladevi, S.; Lawrenson, J.G.; Fielder, A.R.; Suttle, C.M. Global prevalence of childhood cataract: A systematic review. Eye 2016, 30, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irum, B.; Khan, S.Y.; Ali, M.; Kaul, H.; Kabir, F.; Rauf, B.; Fatima, F.; Nadeem, R.; Khan, A.O.; Obaisi, S.A.; et al. Mutation in LIM2 is responsible for autosomal recessive congenital cataracts. PLoS ONE 2016, 11, e0162620. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, V.M.; Beyer, E.C. Oxidative stress, lens gap junctions, and cataracts. Antioxid. Redox Signal. 2009, 11, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferro, E.; Capra, A.P.; Zirilli, G.; Meduri, A.; Urso, M.; Briuglia, S.; La Rosa, M.A. FTL c.-168G>C mutation in hereditary hyperferritinemia cataract syndrome: A new Italian family. Pediatric Dev. Pathol. 2018, 21, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Q.; Cabrera, P.E.; Zhong, Z.; Sun, W.; Jiao, X.; Chen, Y.; Govindarajan, G.; Naeem, M.A.; Khan, S.N.; et al. Molecular genetic analysis of pakistani families with autosomal recessive congenital cataracts by homozygosity screening. Investig. Opthalmology Vis. Sci. 2017, 58, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol. Vis. 2011, 17, 2197–2206. [Google Scholar] [PubMed]
- Ponnam, S.P.G.; Ramesha, K.; Matalia, J.; Tejwani, S.; Ramamurthy, B.; Kannabiran, C. Mutational screening of Indian families with hereditary congenital cataract. Mol. Vis. 2013, 19, 1141–1148. [Google Scholar] [PubMed]
- Li, S.; Zhang, J.; Cao, Y.; You, Y.; Zhao, X. Novel mutations identified in Chinese families with autosomal dominant congenital cataracts by targeted next-generation sequencing. BMC Med. Genet. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berry, V.; Georgiou, M.; Fujinami, K.; Quinlan, R.; Moore, A.; Michaelides, M. Inherited cataracts: Molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br. J. Ophthalmol. 2020, 104, 1331–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Hua, R.; Xiao, W.; Burdon, K.; Bhattacharya, S.S.; Craig, J.E.; Shang, D.; Zhao, X.; Mackey, D.A.; Moore, A.T.; et al. Mutations of theEPHA2receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum. Mutat. 2009, 30, E603–E611. [Google Scholar] [CrossRef] [PubMed]
- Lavker, R.M.; Kaplan, N.; Wang, J.; Peng, H. Corneal epithelial biology: Lessons stemming from old to new. Exp. Eye Res. 2020, 198, 108094. [Google Scholar] [CrossRef] [PubMed]
- Kaul, H.; Riazuddin, S.A.; Shahid, M.; Kousar, S.; Butt, N.H.; Zafar, A.U.; Khan, S.N.; Husnain, T.; Akram, J.; Hejtmancik, J.F.; et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol. Vis. 2010, 16, 511–517. [Google Scholar] [PubMed]
- Bennett, T.M.; M’Hamdi, O.; Hejtmancik, J.F.; Shiels, A. Germ-line and somatic EPHA2 coding variants in lens aging and cataract. PLoS ONE 2017, 12, e0189881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The human gene mutation database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, S.; Sheikh, S.A.; Riazuddin, S.; Waryah, A.M.; Ahmed, Z.M. INPP5K variant causes autosomal recessive congenital cataract in a Pakistani family. Clin. Genet. 2018, 93, 682–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.A.; Bowie, J.U. SAM domains: Uniform structure, diversity of function. Trends Biochem. Sci. 2003, 28, 625–628. [Google Scholar] [CrossRef]
- Lackmann, M.; Boyd, A.W. Eph, a protein family coming of age: More confusion, insight, or complexity? Sci. Signal. 2008, 1, re2. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.A.; Williams, R.M.; Hailemariam, S.; Ayala, R.; Burgers, P.; Zhang, X. Cryo-EM Structure of Nucleotide-Bound Tel1ATM Unravels the Molecular Basis of Inhibition and Structural Rationale for Disease-Associated Mutations. Structure 2019, 28, 96–104.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| cDNA Change | Protein Change | CADD | GnomAD | Mutation Taster | Polyphen2 | SIFT | ACMG Classification (Criteria Used) | Reference |
|---|---|---|---|---|---|---|---|---|
| c.2353G>A | p.(Ala785Thr) | 23.1 | 0.0003 | Disease Causing | Probably Damaging | Damaging | Uncertain Significance (BP1, PP3, PP5) | [12] |
| c.2710delG | p.(Val904Cysfs*36) | N/A | 0.000004 | Disease Causing | N/A | N/A | Pathogenic (PVS1, PM2, PP3) | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarwar, P.; Sheikh, S.A.; Waryah, Y.M.; Ujjan, I.U.; Riazuddin, S.; Waryah, A.M.; Ahmed, Z.M. Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract. Int. J. Mol. Sci. 2021, 22, 10655. https://doi.org/10.3390/ijms221910655
Jarwar P, Sheikh SA, Waryah YM, Ujjan IU, Riazuddin S, Waryah AM, Ahmed ZM. Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract. International Journal of Molecular Sciences. 2021; 22(19):10655. https://doi.org/10.3390/ijms221910655
Chicago/Turabian StyleJarwar, Priya, Shakeel Ahmed Sheikh, Yar Muhammad Waryah, Ikram Uddin Ujjan, Saima Riazuddin, Ali Muhammad Waryah, and Zubair M. Ahmed. 2021. "Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract" International Journal of Molecular Sciences 22, no. 19: 10655. https://doi.org/10.3390/ijms221910655
APA StyleJarwar, P., Sheikh, S. A., Waryah, Y. M., Ujjan, I. U., Riazuddin, S., Waryah, A. M., & Ahmed, Z. M. (2021). Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract. International Journal of Molecular Sciences, 22(19), 10655. https://doi.org/10.3390/ijms221910655