
Road to Metastasis: The TWEAK Pathway as a Discriminant between Metastasizing and Non-Metastasizing Thick Melanomas

 , , ,
, , ,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
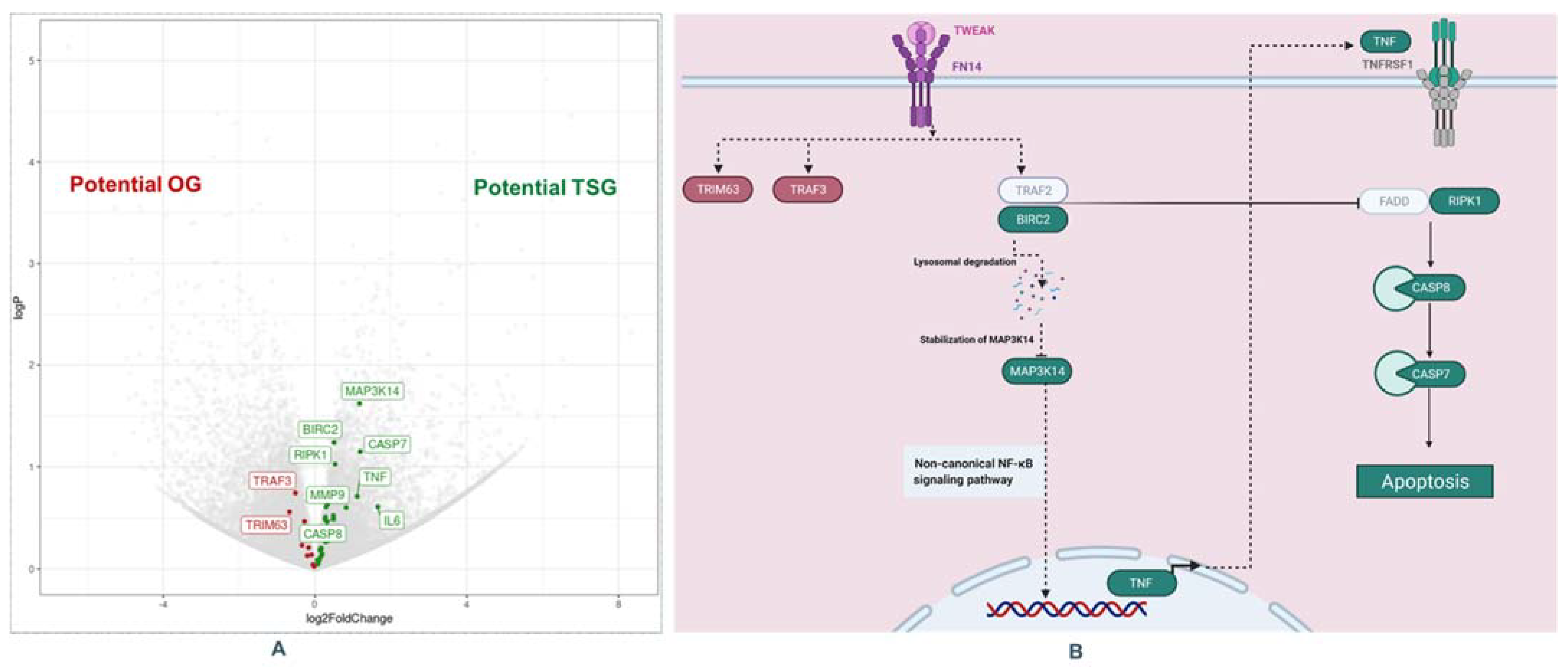
2. Results
2.1. Analysis of Clinicopathological Predictors in Thick CM
2.2. Demographic and Clinicopathological Characteristics of the Study Population for the Molecular Analysis
2.3. Molecular Analysis
2.3.1. Differentially Expressed Genes in Thick CM
2.3.2. Pathway Analysis: TNF-like Weak Inducer of Apoptosis (TWEAK) Pathway
2.3.3. Validation in the Leeds Dataset
3. Discussion
4. Materials and Methods
4.1. Patient Populations
4.2. Tumor Tissue Samples: RNA Extraction and Purification
4.3. Construction of RNA-Seq Library and Sequencing
4.4. Bioinformatics Analysis
4.5. Validation Using the Leeds Dataset
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| AKT | AKT Serine/Threonine Kinase |
| AL | acrolentiginous melanoma |
| ARID2 | AT-Rich Interaction Domain 2 |
| BIRC2 | Baculoviral IAP Repeat Containing 2 |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
| CASP7 | Caspase 7 |
| CASP8 | Caspase 8 |
| CCND1 | Cyclin D1 |
| CDK4 | Cyclin Dependent Kinase 4 |
| CDKN2a | Cyclin Dependent Kinase Inhibitor 2A |
| cIAP1 | Cellular Inhibitor of Apoptosis 1 |
| CM | Cutaneous melanoma |
| DEG | differential expressed genes |
| FADD | Fas Associated Via Death Domain |
| Fn14 | Fibroblast growth factor-inducible 14 |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| GSTP1 | Glutathione S-Transferase P |
| H & E | hematoxylin & eosin |
| HLA-DQA1 | Major Histocompatibility Complex, Class II, DQ Alpha 1 |
| IGHM | Immunoglobulin Heavy Constant Mu |
| IL6 | interleukin 6 |
| JNK | c-Jun N-terminal kinase |
| M− | non-metastatic CMs |
| M+ | metastatic CMs |
| MAP | Mitogen-Activated Protein |
| MAP3K14 | Mitogen-Activated Protein Kinase Kinase Kinase 14 |
| MDM2 | MDM2 Proto-Oncogene |
| MET | MET Proto-Oncogene, Receptor Tyrosine Kinase |
| MET, c-MET | Mesenchymal-epithelial transition tyrosine kinase receptor |
| MMP9 | expression of matrix metalloproteinase 9 |
| NIK | NFkB Inducing Kinase |
| NM | Nodular melanoma |
| NRAS | NRAS Proto-Oncogene, GTPase |
| NSCLC | Non-small-cell lung carcinoma |
| OG | oncogene |
| P53 | Tumor Protein P53 |
| PTEN | Phosphatase And Tensin Homolog |
| RAMP1 | Receptor Activity Modifying Protein 1 |
| Ras/MAPK | Ras/Mitogen-Activated Protein Kinase |
| RIPK1 | Receptor Interacting Serine/Threonine Kinase 1 |
| RNA | ribonucleic acid |
| SH3GL2 | SH3 Domain Containing GRB2 Like 2, Endophilin A1 |
| SSM | Superficial spreading melanoma |
| STK19 | Serine/Threonine Kinase 19 |
| TMOD1 | Tropomodulin 1 |
| TNF | tumor necrose factor |
| TNF-alpha | Tumor Necrosis Factor-alpha |
| TNFR | tumor necrose factor receptor |
| TNFR1 | Tumor Necrosis Factor Receptor Type 1 |
| TRAF | TNF Receptor Associated Factor |
| TRIM63 | Tripartite Motif Containing 63 |
| TSG | tumor suppressor gene |
| TWEAK | TNF-like weak inducer of apoptosis |
| TWIST2 | Twist Family BHLH Transcription Factor 2 |
References
- Arnold, M.; Holterhues, C.; Hollestein, L.M.; Coebergh, J.W.; Nijsten, T.; Pukkala, E.; Holleczek, B.; Tryggvadóttir, L.; Comber, H.; Bento, M.J.; et al. Trends in incidence and predictions of cutaneous melanoma across Europe up to 2015. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1170–1178. [Google Scholar] [CrossRef]
- Rajkumar, S.; Watson, I.R. Molecular characterisation of cutaneous melanoma: Creating a framework for targeted and immune therapies. Br. J. Cancer 2016, 115, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [Green Version]
- Spatz, A.; Shaw, H.M.; Crotty, K.A.; Thompson, J.F.; McCarthy, S.W. Analysis of histopathological factors associated with prolonged survival of 10 years or more for patients with thick melanomas (>5 mm). Histopathology 1998, 33, 406–413. [Google Scholar] [CrossRef]
- Blessing, K.; McLaren, K.M.; McLean, A.; Davidson, P. Thick malignant melanomas (greater than 3 mm Breslow) with good clinical outcome: A histological study and survival analysis. Histopathology 1991, 18, 143–148. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Baade, P.D.; Olsen, C.M. More people die from thin melanomas (1 mm) than from thick melanomas (>4 mm) in Queensland, Australia. J. Invest. Dermatol. 2015, 135, 1190–1193. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizkeleti, L.; Ecsedi, S.; Rakosy, Z.; Orosz, A.; Lazar, V.; Emri, G.; Koroknai, V.; Kiss, T.; Ádány, R.; Balázs, M. The role of CCND1 alterations during the progression of cutaneous malignant melanoma. Tumour Biol. 2012, 33, 2189–2199. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikner, A.; Ashkenazi, A. TWEAK induces apoptosis through a death-signaling complex comprising receptor-interacting protein 1 (RIP1), Fas-associated death domain (FADD), and caspase-8. J. Biol. Chem. 2011, 286, 21546–21554. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Chau, D.; Callus, B.; Wong, W.W.; Hawkins, C.J.; Schneider, P.; McKinlay, M.; Benetatos, C.A.; Condon, S.M.; Chunduru, S.K.; et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell Biol. 2008, 182, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, M.; Raju, R.; Radhakrishnan, A.; Nanjappa, V.; Muthusamy, B.; Singh, K.; Kuppusamy, D.; Lingala, B.T.; Pan, A.; Mathur, P.P.; et al. A Bioinformatics Resource for TWEAK-Fn14 Signaling Pathway. J. Signal Transduct. 2012, 2012, 376470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, R.; Laye, J.P.; Lauss, M.; Diaz, J.M.S.; O’Shea, S.J.; Pozniak, J.; Filia, A.; Harland, M.; Gascoyne, J.; Randerson-Moor, J.A.; et al. Transcriptomic Analysis Reveals Prognostic Molecular Signatures of Stage I Melanoma. Clin. Cancer Res. 2019, 25, 7424–7435. [Google Scholar] [CrossRef] [Green Version]
- Raw mRNA Expressions. The Leeds Melanoma Cohort Gene Expression Data Access Committee. Available online: https://ega-archive.org/datasets/EGAD00010001562 (accessed on 10 May 2021).
- Schmidt, A.N.; Nanney, L.B.; Boyd, A.S.; King, L.E., Jr.; Ellis, D.L. Oestrogen receptorbeta expression in melanocytic lesions. Exp. Dermatol. 2006, 15, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Glatthaar, H.; Katto, J.; Vogt, T.; Mahlknecht, U.; Glatthaar, H. Estrogen Receptor Alpha (ESR1) Single-Nucleotide Polymorphisms (SNPs) Affect Malignant Melanoma Susceptibility and Disease Course. Genet Epigenet. 2016, 8, 1–6. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Galisteo, R.; Brown, S.A.; Winkles, J.A. TWEAK activation of the non-canonical NF-kappaB signaling pathway differentially regulates melanoma and prostate cancer cell invasion. Oncotarget 2016, 7, 81474–81492. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Dai, L.; Cao, C.; Zhu, J.; Ding, C.; Xu, H.-B.; Qiu, L.; Di, W. Functional expression of TWEAK and the receptor Fn14 in human malignant ovarian tumors: Possible implication for ovarian tumor intervention. PLoS ONE 2013, 8, e57436. [Google Scholar]
- Ho, D.H.; Vu, H.; Brown, S.A.; Donohue, P.J.; Hanscom, H.N.; Winkles, J.A. Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res. 2004, 64, 8968–8972. [Google Scholar] [CrossRef] [Green Version]
- Kawakita, T.; Shiraki, K.; Yamanaka, Y.; Yamaguchi, Y.; Saitou, Y.; Enokimura, N.; Yamamoto, N.; Okano, H.; Sugimoto, K.; Murata, K.; et al. Functional expression of TWEAK in human hepatocellular carcinoma: Possible implication in cell proliferation and tumor angiogenesis. Biochem. Biophys. Res. Commun. 2004, 318, 726–733. [Google Scholar] [CrossRef]
- Kawakita, T.; Shiraki, K.; Yamanaka, Y.; Yamaguchi, Y.; Saitou, Y.; Enokimura, N.; Yamamoto, N.; Okano, H.; Sugimoto, K.; Murata, K.; et al. Functional expression of TWEAK in human colonic adenocarcinoma cells. Int. J. Oncol. 2005, 26, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.-R.; Huang, M.-T.; Chen, S.-T.; Jeng, Y.-M.; Li, Y.-J.; Liang, J.-T.; Lee, P.-H.; Chang, K.-J.; Chang, C.-C. Prognostic significance of TWEAK expression in colorectal cancer and effect of its inhibition on invasion. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), 385–394. [Google Scholar] [CrossRef]
- Yoriki, R.; Akashi, S.; Sho, M.; Nomi, T.; Yamato, I.; Hotta, K.; Takayama, T.; Matsumoto, S.; Wakatsuki, K.; Migita, K.; et al. Therapeutic potential of the TWEAK/Fn14 pathway in intractable gastrointestinal cancer. Exp. Ther. Med. 2011, 2, 103–108. [Google Scholar] [CrossRef]
- Burkly, L.C.; Michaelson, J.S.; Hahm, K.; Jakubowski, A.; Zheng, T.S. TWEAKing tissue remodeling by a multifunctional cytokine: Role of TWEAK/Fn14 pathway in health and disease. Cytokine 2007, 40, 1–16. [Google Scholar] [CrossRef]
- Fortin, S.P.; Ennis, M.J.; Savitch, B.A.; Carpentieri, D.; McDonough, W.S.; Winkles, J.A.; Loftus, J.C.; Kingsley, C.; Hostetter, G.; Tran, N.L. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol. Cancer Res. 2009, 7, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgenrath, M.; Weng, S.; Kostek, C.A.; Browning, B.; Wang, M.; Brown, S.A.; Winkles, J.A.; Michaelson, J.S.; Allaire, N.; Schneider, P.; et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. Embo J. 2006, 25, 5826–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, C.N.; Wang, Y.C.; Lund, J.K.; Chen, Y.W.; Leal, J.A.; Wiley, S.R. TWEAK induces angiogenesis and proliferation of endothelial cells. J. Biol. Chem. 1999, 274, 8455–8459. [Google Scholar] [CrossRef] [Green Version]
- Swoboda, A.; Soukup, R.; Eckel, O.; Kinslechner, K.; Wingelhofer, B.; Schörghofer, D.; Sternberg, C.; Pham, H.T.T.; Vallianou, M.; Horvath, J.; et al. STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pathway. Oncogene 2020, 40, 1091–1105. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varemo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Characteristics | Frequency (n) | Percent (%) | |
|---|---|---|---|
| Metastasis | No | 12 | 50 |
| Yes | 12 | 50 | |
| Subtype | SSM | 4 | 16.7 |
| NM | 13 | 54.2 | |
| AL | 7 | 29.2 | |
| Gender | Female | 6 | 25 |
| Male | 18 | 75 | |
| Ulceration | No | 8 | 33.3 |
| Yes | 16 | 66.7 | |
| Mitosis | No | 4 | 16.7 |
| Yes | 20 | 83.3 | |
| Age | 40–59 y | 8 | 33.3 |
| ≥60 y | 16 | 66.7 | |
| Site | Extremity | 11 | 45.8 |
| Trunk | 6 | 25 | |
| Head and neck | 7 | 29.2 | |
| Breslow thickness | 2–4 mm | 12 | 50 |
| >4 mm | 12 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güvenç, C.; Antoranz, A.; Szumera-Ciećkiewicz, A.; Teterycz, P.P.; Rutkowski, P.R.; Rawson, R.V.; Scolyer, R.A.; Thompson, J.F.; Newton-Bishop, J.; Stas, M.; et al. Road to Metastasis: The TWEAK Pathway as a Discriminant between Metastasizing and Non-Metastasizing Thick Melanomas. Int. J. Mol. Sci. 2021, 22, 10568. https://doi.org/10.3390/ijms221910568
Güvenç C, Antoranz A, Szumera-Ciećkiewicz A, Teterycz PP, Rutkowski PR, Rawson RV, Scolyer RA, Thompson JF, Newton-Bishop J, Stas M, et al. Road to Metastasis: The TWEAK Pathway as a Discriminant between Metastasizing and Non-Metastasizing Thick Melanomas. International Journal of Molecular Sciences. 2021; 22(19):10568. https://doi.org/10.3390/ijms221910568
Chicago/Turabian StyleGüvenç, Canan, Asier Antoranz, Anna Szumera-Ciećkiewicz, Pawel P. Teterycz, Piotr R. Rutkowski, Robert V. Rawson, Richard A. Scolyer, John F. Thompson, Julia Newton-Bishop, Marguerite Stas, and et al. 2021. "Road to Metastasis: The TWEAK Pathway as a Discriminant between Metastasizing and Non-Metastasizing Thick Melanomas" International Journal of Molecular Sciences 22, no. 19: 10568. https://doi.org/10.3390/ijms221910568
APA StyleGüvenç, C., Antoranz, A., Szumera-Ciećkiewicz, A., Teterycz, P. P., Rutkowski, P. R., Rawson, R. V., Scolyer, R. A., Thompson, J. F., Newton-Bishop, J., Stas, M., Boecxstaens, V., Bechter, O., Vercauteren, J., Garmyn, M., van den Oord, J., & Bosisio, F. M. (2021). Road to Metastasis: The TWEAK Pathway as a Discriminant between Metastasizing and Non-Metastasizing Thick Melanomas. International Journal of Molecular Sciences, 22(19), 10568. https://doi.org/10.3390/ijms221910568