
Bona Fide Tumor Suppressor Genes Hypermethylated in Melanoma: A Narrative Review


Abstract
1. Introduction
2. Results
2.1. WFDC1
2.2. SOCS1
2.3. TSPY—CYBA—MTA2—MX1—RPL37A—HSPB1
2.4. SYK
2.5. H-cadherin and E-cadherin
2.6. TRIM16
2.7. RUNX3
2.8. APC
2.9. MAPK13
2.10. RARβ
2.11. AGTR1
2.12. SERPINB5
2.13. 14-3-3σ
2.14. TCF21
2.15. SPINT2
3. Discussion
4. Conclusions
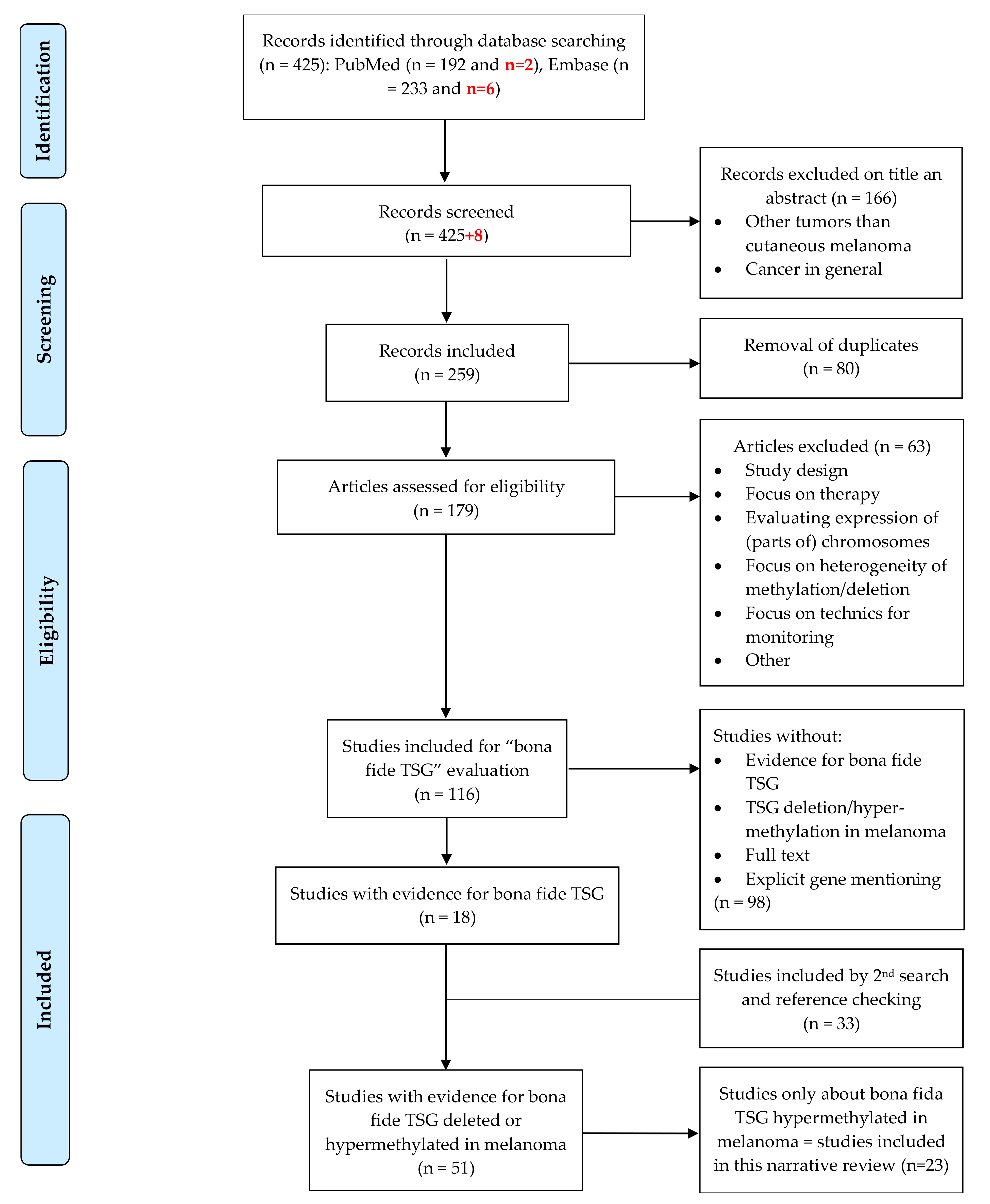
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A(G)TR1 | Angiotensin II receptor type 1 |
| AKAP12 | A-kinase anchoring protein 12 |
| APAF-1 | Apoptotic peptidase activating factor 1 |
| APC | Adenomatous polyposis coli |
| ATRA | All trans retinoic acid |
| BM | Basal membrane |
| CDH1 | Cadherin 1 |
| CDH13 | Cadherin 13 |
| CDKN2A | Cyclin dependent kinase inhibitor 2A |
| CMM | Cutaneous malignant melanoma |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CYBA | Cytochrome b-245 alpha chain |
| DAC or 5-Aza | 5-aza-deoxycytidine |
| DKK1 | Dickkopf WNT signaling pathway inhibitor 1 |
| DNA | Desoxyribonucleic acid |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| FERMT3 | Fermitin family member 3 |
| HGF | Hepatocyte growth factor |
| HSPB1 | Heat shock protein family B (small) member 1 |
| IFNβ1 | Interferon beta 1 |
| KIND3 | Kindlin-3 |
| KO | Knock-out |
| MAPK | Mitogen-activated protein kinase |
| MEN1 | Menin 1 |
| MET | Mesenchymal-epithelial transition factor |
| miRNA | MicroRNA |
| MMP-2 | Matrix metalloproteinase-2 |
| MTA2 | Metastasis associated 1 family member 2 |
| MTAP | Methylthioadenosine phosphorylase |
| MX1 | MX dynamin like GTPase 1 |
| PTEN | Phosphatase and tensin homolog |
| RARβ | Retinoic acid receptor beta |
| RASSF1A | Ras association domain family member 1 isoform A |
| RASSF6 | Ras association domain family member 6 |
| RASSF8 | Ras association domain family member 8 |
| RPL37A | Ribosomal protein L37a |
| RUNX3 | Runt related transcription factor 3 |
| SERPINB5 | Serpin family B member 5 |
| SFN | Stratifin |
| Sh | Small hairpin |
| siRNA | Small interfering ribonucleic acid |
| SOCS1 | Suppressor of cytokine signaling 1 |
| SPINT2 | Serine peptidase inhibitor, Kunitz type 2 |
| SSeCKS | Src-suppressed C-kinase substrate |
| SYK | Spleen associated tyrosine kinase |
| TCF21 | Transcription factor 21 |
| TRIM16 | Tripartite motif containing 16 |
| TSG(s) | Tumor suppressor gene(s) |
| TSPY | Testis specific protein, Y-linked 1 |
| WFDC1 | WAP four-disulfide core domain 1 |
Appendix A
References
- Guo, Y.; Long, J.; Lei, S. Promoter methylation as biomarkers for diagnosis of melanoma: A systematic review and meta-analysis. J. Cell. Physiol. 2018, 234, 7356–7367. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.H.; Scoggins, C.R. Melanoma. J. Surg. Oncol. 2019, 120, 873–881. [Google Scholar] [CrossRef]
- Gaggioli, C.; Sahai, E. Melanoma invasion-Current knowledge and future directions. Pigment Cell Res. 2007, 20, 161–172. [Google Scholar] [CrossRef]
- Sun, W.; Yang, J. Functional Mechanisms for Human Tumor Suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef]
- Swale, V.J.; Quinn, A.G. Tumour suppressor genes. Clin. Exp. Dermatol. 2000, 25, 231–235. [Google Scholar] [CrossRef]
- Wang, J.; Hua, W.; Huang, S.K.; Fan, K.; Takeshima, L.; Mao, Y.; Hoon, D.S. RASSF8 regulates progression of cutaneous melanoma through nuclear factor-kappab. Oncotarget 2015, 6, 30165–30177. [Google Scholar] [CrossRef]
- Marzese, D.M.; Scolyer, R.A.; Roqué, M.; Vargas-Roig, L.M.; Huynh, J.L.; Wilmott, J.S.; Murali, R.; Buckland, M.E.; Barkhoudarian, G.; Thompson, J.F.; et al. DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro-Oncology 2014, 16, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Katoh, D.; Nishizuka, M.; Osada, S.; Imagawa, M. FAD104, a Regulator of Adipogenesis and Osteogenesis, Interacts with the C-Terminal Region of STAT3 and Represses Malignant Transformation of Melanoma Cells. Biol. Pharm. Bull. 2016, 39, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Lian, Z.; D’Andrea, K.; Letrero, R.; Sheng, W.; Liu, S.; Diehl, J.; Pytel, D.; Barbash, O.; Schuchter, L.; et al. The FBXO4 Tumor Suppressor Functions as a Barrier to BrafV600E-Dependent Metastatic Melanoma. Mol. Cell. Biol. 2013, 33, 4422–4433. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.R.; Gupta, S.; Park, K.-H.; Chung, K.Y.; Lauss, M.; Flaherty, K.T.; Jönsson, G.; Rha, S.Y.; Tsao, H. Promoter Methylation of PTEN Is a Significant Prognostic Factor in Melanoma Survival. J. Investig. Dermatol. 2016, 136, 1002–1011. [Google Scholar] [CrossRef]
- Aguissa-Touré, A.-H.; Li, G. Genetic alterations of PTEN in human melanoma. Cell. Mol. Life Sci. 2011, 69, 1475–1491. [Google Scholar] [CrossRef] [PubMed]
- Terzian, T.; Torchia, E.C.; Dai, D.; Robinson, S.E.; Murao, K.; Stiegmann, R.A.; Gonzalez, V.; Boyle, G.; Powell, M.B.; Pollock, P.; et al. p53 prevents progression of nevi to melanoma predominantly through cell cycle regulation. Pigment Cell Melanoma Res. 2010, 23, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Jorapur, A.; Shain, A.H.; Lang, U.E.; Torres, R.; Zhang, Y.; McNeal, A.S.; Botton, T.; Lin, J.; Donne, M.; et al. Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation. Cancer Cell 2018, 34, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Dacosta, S.A.; Schumaker, L.M.; Ellis, M.J. Mannose 6-phosphate/insulin-like growth factor 2 receptor, a bona fide tumor suppressor gene or just a promising candidate? J. Mammary Gland Biol. Neoplasia 2000, 5, 85–94. [Google Scholar] [CrossRef]
- Kadariya, Y.; Cheung, M.; Xu, J.; Pei, J.; Sementino, E.; Menges, C.W.; Cai, K.Q.; Rauscher, F.J.; Klein-Szanto, A.J.; Testa, J.R. Bap1 Is a Bona Fide Tumor Suppressor: Genetic Evidence from Mouse Models Carrying Heterozygous Germline Bap1 Mutations. Cancer Res. 2016, 76, 2836–2844. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, H.; Lin, S.Y.; Goss, J.A.; Du, C.; Li, K. Mcph1/Brit1 deficiency promotes genomic instability and tumor formation in a mouse model. Oncogene 2015, 34, 4368–4378. [Google Scholar] [CrossRef]
- Yuan, B.-Z.; Zhou, X.; Durkin, M.E.; Zimonjic, D.B.; Gumundsdottir, K.; Eyfjord, J.E.; Thorgeirsson, S.S.; Popescu, N.C. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene 2003, 22, 445–450. [Google Scholar] [CrossRef]
- Vorvis, C.; Hatziapostolou, M.; Joshi, S.; Koutsioumpa, M.; Williams, J.; Donahue, T.R.; Poultsides, G.A.; Eibl, G.; Iliopoulos, D. Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am. J. Physiol. Liver Physiol. 2016, 310, G1124–G1137. [Google Scholar] [CrossRef]
- Regneri, J.; Klotz, B.; Wilde, B.; Kottler, V.A.; Hausmann, M.; Kneitz, S.; Regensburger, M.; Maurus, K.; Götz, R.; Lu, Y.; et al. Analysis of the putative tumor suppressor gene cdkn2ab in pigment cells and melanoma of Xiphophorus and medaka. Pigment Cell Melanoma Res. 2018, 32, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Wu, C.-F.; Rajasekaran, N.; Shin, Y.K. Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview. Cell. Physiol. Biochem. 2018, 51, 2647–2693. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef]
- Liang, G.; Weisenberger, D.J. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 2017, 12, 416–432. [Google Scholar] [CrossRef]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Liu, S.; Howell, P.; Ren, S.; Fodstad, O.; Zhang, G.; Samant, R.; Shevde, L.; Xi, Y.; Pannell, L.K.; Riker, A.I. Expression and functional analysis of the WAP four disulfide core domain 1 gene in human melanoma. Clin. Exp. Metastasis 2009, 26, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Yang, J.; Chen, X.; Li, J.; Li, X.; Wang, L.; Tan, Y.; Xiong, W.; Zhou, M.; McCarthy, J.B.; et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J. Cell. Physiol. 2011, 226, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Parrillas, V.; Muñoz, L.M.; Holgado, B.L.; Kumar, A.; Cascio, G.; Lucas, P.; Rodríguez-Frade, J.M.; Malumbres, M.; Carrera, A.C.; Van Wely, K.H.; et al. Suppressor of cytokine signaling 1 blocks mitosis in human melanoma cells. Cell. Mol. Life Sci. 2013, 70, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Tagami-Nagata, N.; Serada, S.; Fujimoto, M.; Tanemura, A.; Nakatsuka, R.; Ohkawara, T.; Murota, H.; Kishimoto, T.; Katayama, I.; Naka, T. Suppressor of cytokine signaling-1 (SOCS-1) induces significant preclinical anti-tumor effect in malignant melanoma cells. Exp. Dermatol. 2015, 24, 864–871. [Google Scholar] [CrossRef]
- Gallagher, W.M.; Bergin, O.E.; Rafferty, M.; Kelly, Z.D.; Nolan, I.-M.; Fox, E.J.; Culhane, A.C.; McArdle, L.; Fraga, M.; Hughes, L.; et al. Multiple markers for melanoma progression regulated by DNA methylation: Insights from transcriptomic studies. Carcinogenesis 2005, 26, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Bailet, O.; Fenouille, N.; Abbe, P.; Robert, G.; Rocchi, S.; Gonthier, N.; Denoyelle, C.; Ticchioni, M.; Ortonne, J.-P.; Ballotti, R.; et al. Spleen Tyrosine Kinase Functions as a Tumor Suppressor in Melanoma Cells by Inducing Senescence-like Growth Arrest. Cancer Res. 2009, 69, 2748–2756. [Google Scholar] [CrossRef]
- Kuphal, S.; Martyn, A.C.; Pedley, J.; Crowther, L.M.; Bonazzi, V.F.; Parsons, P.G.; Bosserhoff, A.K.; Hayward, N.K.; Boyle, G.M. H-Cadherin expression reduces invasion of malignant melanoma. Pigment Cell Melanoma Res. 2009, 22, 296–306. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Biondo, C.; Beninati, C.; Teti, D.; Venza, I. DNA methylation-induced E-cadherin silencing is correlated with the clinicopathological features of melanoma. Oncol. Rep. 2016, 35, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Sutton, S.K.; Cheung, B.B.; Massudi, H.; Tan, O.; Koach, J.; Mayoh, C.; Carter, D.R.; Marshall, G.M. Heterozygous loss of keratinocyte TRIM16 expression increases melanocytic cell lesions and lymph node metastasis. J. Cancer Res. Clin. Oncol. 2019, 145, 2241–2250. [Google Scholar] [CrossRef]
- Sutton, S.K.; Koach, J.; Tan, O.; Liu, B.; Carter, D.R.; Wilmott, J.; Yosufi, B.; Haydu, L.E.; Mann, G.; Thompson, J.; et al. TRIM16 inhibits proliferation and migration through regulation of interferon beta 1 in melanoma cells. Oncotarget 2014, 5, 10127–10139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Wang, L.; Zeng, X.; Fujita, T.; Liu, W. Runx3 inhibits melanoma cell migration through regulation of cell shape change. Cell Biol. Int. 2017, 41, 1048–1055. [Google Scholar] [CrossRef]
- Kang, S.; Wang, Z.; Li, B.; Gao, X.; He, W.; Cao, S.; Cai, Y.; Chen, H. Anti-tumor effects of resveratrol on malignant melanoma is associated with promoter demethylation of RUNX3 gene. Die Pharm. 2019, 74, 163–167. [Google Scholar]
- Worm, J.; Christensen, C.; Grønbæk, K.; Tulchinsky, E.; Guldberg, P. Genetic and epigenetic alterations of the APC gene in malignant melanoma. Oncogene 2004, 23, 5215–5226. [Google Scholar] [CrossRef]
- Gao, L.; Smit, M.A.; Oord, J.J.V.D.; Goeman, J.J.; Verdegaal, E.M.E.; Van Der Burg, S.H.; Stas, M.; Beck, S.; Gruis, N.A.; Tensen, C.; et al. Genome-wide promoter methylation analysis identifies epigenetic silencing ofMAPK13in primary cutaneous melanoma. Pigment Cell Melanoma Res. 2013, 26, 542–554. [Google Scholar] [CrossRef]
- Dahl, C.; Christensen, C.; Jönsson, G.; Lorentzen, A.; Skjødt, M.L.; Borg, Å.; Pawelec, G.; Guldberg, P. Mutual Exclusivity Analysis of Genetic and Epigenetic Drivers in Melanoma Identifies a Link Between p14ARF and RARβ Signaling. Mol. Cancer Res. 2013, 11, 1166–1178. [Google Scholar] [CrossRef]
- Renziehausen, A.; Wang, H.; Rao, B.; Weir, L.; Nigro, C.L.; Lattanzio, L.; Merlano, M.; Rioja, A.V.; Fernandez-Carranco, M.D.C.; Hajji, N.; et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2018, 38, 2320–2336. [Google Scholar] [CrossRef]
- Denk, A.E.; Bettstetter, M.; Wild, P.J.; Hoek, K.; Bataille, F.; Dietmaier, W.; Bosserhoff, A. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Res. 2007, 20, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Ibrahim, S.M.; Vera, J.; Kunz, M. 14-3-3sigma gene silencing during melanoma progression and its role in cell cycle control and cellular senescence. Mol. Cancer 2009, 8, 53. [Google Scholar] [CrossRef]
- Arab, K.; Smith, L.T.; Gast, A.; Weichenhan, D.; Huang, J.P.-H.; Claus, R.; Hielscher, T.; Espinosa, A.V.; Ringel, M.D.; Morrison, C.D.; et al. Epigenetic deregulation of TCF21 inhibits metastasis suppressor KISS1 in metastatic melanoma. Carcinogenesis 2011, 32, 1467–1473. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, H.E.; Min, M.; Mahalingam, M.; Alani, R.; Ryu, B. Epigenetic silencing of putative tumor suppressor SPINT2/HAI-2 in melanoma. Pigment Cell Melanoma Res. 2012, 25, 864. Available online: http://www.embase.com/search/results?subaction=viewrecord&from=export&id=L71000997 (accessed on 1 May 2020).
- Mezzanotte, J.J.; Hill, V.; Schmidt, M.L.; Shinawi, T.; Tommasi, S.; Krex, D.; Schackert, G.; Pfeifer, G.P.; Latif, F.; Clark, G.J. RASSF6 exhibits promoter hypermethylation in metastatic melanoma and inhibits invasion in melanoma cells. Epigenetics 2014, 9, 1496–1503. [Google Scholar] [CrossRef]
- Hoeller, C.; Thallinger, C.; Pratscher, B.; Bister, M.D.; Schicher, N.; Loewe, R.; Heere-Ress, E.; Roka, F.; Sexl, V.; Pehamberger, H. The Non-Receptor-Associated Tyrosine Kinase Syk is a Regulator of Metastatic Behavior in Human Melanoma Cells. J. Investig. Dermatol. 2005, 124, 1293–1299. [Google Scholar] [CrossRef]
- Kitago, M.; Martinez, S.R.; Nakamura, T.; Sim, M.S.; Hoon, D.S. Regulation of RUNX3 tumor suppressor gene expression in cutaneous melanoma. Clin. Cancer Res. 2009, 15, 2988–2994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, G.; Cheng, Y.; Martinka, M.; Li, G. Prognostic significance of RUNX3 expression in human melanoma. Cancer 2011, 117, 2719–2727. [Google Scholar] [CrossRef]
- Abildgaard, C.; Dahl, C.; Abdul-Al, A.; Christensen, A.; Guldberg, P. Inhibition of retinoic acid receptor β signaling confers glycolytic dependence and sensitization to dichloroacetate in melanoma cells. Oncotarget 2017, 8, 84210–84223. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Yu, N.-Z.; Long, F.; Feng, C.; Wang, X.-J. Effects of CDKN2A (p16INK4A/p14ARF) Over-Expression on Proliferation and Migration of Human Melanoma A375 Cells. Cell. Physiol. Biochem. 2016, 40, 1367–1376. [Google Scholar] [CrossRef]
- Nash, K.T.; Phadke, P.A.; Navenot, J.-M.; Hurst, D.; Accavitti-Loper, M.A.; Sztul, E.; Vaidya, K.S.; Frost, A.R.; Kappes, J.C.; Peiper, S.C.; et al. Requirement of KISS1 Secretion for Multiple Organ Metastasis Suppression and Maintenance of Tumor Dormancy. J. Natl. Cancer Inst. 2007, 99, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Mattei, F.; Schiavoni, G.; Sestili, P.; Spadaro, F.; Fragale, A.; Sistigu, A.; Lucarini, V.; Spada, M.; Sanchez, M.; Scala, S.; et al. IRF-8 Controls Melanoma Progression by Regulating the Cross Talk between Cancer and Immune Cells within the Tumor Microenvironment. Neoplasia 2012, 14, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hinoda, Y.; Sato, S.; Itoh, F.; Adachi, M.; Hareyama, M.; Imai, K. Reduced Invasive and Metastatic Potentials ofKAI1-transfected Melanoma Cells. Jpn. J. Cancer Res. 1998, 89, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Shevde, L.A.; Samant, R.S.; Goldberg, S.F.; Sikanetab, T.; Alessandrinib, A.; Donahue, H.J.; Mauger, D.T.; Welch, D. Suppression of Human Melanoma Metastasis by the Metastasis Suppressor Gene, BRMS1. Exp. Cell Res. 2002, 273, 229–239. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| TSG | Protein | Location | Hallmark of Cancer | First Author | Year |
|---|---|---|---|---|---|
| WFDC1 | WAP four-disulfide core domain 1 | 16q24.1 | Tumor growth ↑ | Liu S. [26] | 2009 |
| RASSF1A | Ras association domain family member 1, isoform A | 3p21.31 | Apoptosis ↓ Proliferation ↑ Tumor growth ↑ | Yi M. [27] | 2011 |
| RASSF8 | Ras association domain family member 8 | 12p12.1 | Proliferation ↑ Migration ↑ Invasion ↑ Apoptosis ↓ Cell/tumor growth↑ | Wang J. [27] | 2015 |
| RASSF6 | Ras association domain family member 6 | 4q13.3 | Growth ↑ Invasion ↑ Extravasation ↑ | Mezzanotte J. [27] | 2014 |
| SOCS1 | Suppressor of cytokine signaling 1 | 16p13.13 | Cell growth ↑ | Parrillas V. [28] | 2012 |
| Cell growth ↑ Apoptosis ↓ | Tagami- Nagata N. [29] | 2015 | |||
| TSPY | Testis-specific protein, Y-linked 1 | Yp11.2 | Tumor growth ↑ Migration ↑ | Gallagher W. [30] | 2005 |
| CYBA | Cytochrome b-245 alpha chain | 16q24.2 | |||
| MTA2 | Metastasis associated 1 family member 2 | 11q12.3 | |||
| MX1 | MX dynamin like GTPase 1 | 21q22.3 | |||
| RPL37A | Ribosomal protein L37a | 2q35 | |||
| HSPB1 | Heat shock protein family B (small) member 1 | 7q11.23 | |||
| SYK | Spleen associated tyrosine kinase | 9q22.2 | Tumor growth ↑ Invasion ↑ Metastasis ↑ | Hoeller C. [30] | 2005 |
| Tumor growth ↑ Migration ↑ Invasion ↑ Proliferation ↑ Senescence ↓ | Bailet O. [31] | 2009 | |||
| CDH13 | Cadherin 13, T-cadherin, H-cadherin | 16q23.3 | Migration ↑ Invasion ↑ Attachment independent growth ↑ | Kuphal S. [32] | 2009 |
| CDH1 | Cadherin 1, E-cadherin | 16q22.1 | Invasion ↑ | Venza M. [33] | 2016 |
| TRIM16 | Tripartite motif containing 16 | 17p12 | Proliferation ↑ Migration ↑ | Sutton S. K. [34] | 2014 |
| Radial migration↑ Proliferation ↑ Tumor growth ↑ Metastasis ↑ | Sutton S. K [35] | 2019 | |||
| RUNX3 | Runt-related transcription factor 3 | 1p36.11 | Cell migration ↑ Invasion ↑ | Zhang X. [36] | 2017 |
| Proliferation ↑ Tumor growth ↑ | Kang S. [37] | 2019 | |||
| APC | APC | 5q22.2 | Proliferation ↑ Invasion ↓ (!) | Worm J. [38] | 2004 |
| MAPK13 | Mitogen-activated protein kinase 13 | 6p21.31 | Proliferation ↑ | Gao L. [39] | 2013 |
| RAR | Retinoic acid receptor β | 3p24.2 | Senescence ↓ Proliferation ↑ | Dahl C. [40] | 2013 |
| AGTR1 | Angiotensin II receptor type 1 | 3q24 | Proliferation ↑ | Renzie-hausen A. [41] | 2019 |
| SERPINB5 | Maspin | 18q21.3-q23 | Invasion↑ Cell adhesion (BM-ECM) ↓ | Denk A. E. [42] | 2007 |
| 14-3-3 | 14-3-3σ, Stratifin (SFN) | 1p36.11 | Proliferation↑ (loss of cell cycle control) Senescence↓ Migration↑ | Schultz J. [43] | 2009 |
| TCF21 | Transcription factor 21 | 6q23.2 | Mesenchymal-epithelial transition ↓ | Arab K. [44] | 2011 |
| SPINT2 | Serine peptidase inhibitor Kunitz type 2 | 19q13.2 | Migration ↑ Proliferation ↑ Invasive growth↑ | Hwang S. [45] | 2015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güvenç, C.; Neckebroeck, F.; Antoranz, A.; Garmyn, M.; van den Oord, J.; Bosisio, F.M. Bona Fide Tumor Suppressor Genes Hypermethylated in Melanoma: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 10674. https://doi.org/10.3390/ijms221910674
Güvenç C, Neckebroeck F, Antoranz A, Garmyn M, van den Oord J, Bosisio FM. Bona Fide Tumor Suppressor Genes Hypermethylated in Melanoma: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(19):10674. https://doi.org/10.3390/ijms221910674
Chicago/Turabian StyleGüvenç, Canan, Fien Neckebroeck, Asier Antoranz, Marjan Garmyn, Joost van den Oord, and Francesca Maria Bosisio. 2021. "Bona Fide Tumor Suppressor Genes Hypermethylated in Melanoma: A Narrative Review" International Journal of Molecular Sciences 22, no. 19: 10674. https://doi.org/10.3390/ijms221910674
APA StyleGüvenç, C., Neckebroeck, F., Antoranz, A., Garmyn, M., van den Oord, J., & Bosisio, F. M. (2021). Bona Fide Tumor Suppressor Genes Hypermethylated in Melanoma: A Narrative Review. International Journal of Molecular Sciences, 22(19), 10674. https://doi.org/10.3390/ijms221910674