
Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis


Abstract
1. Introduction
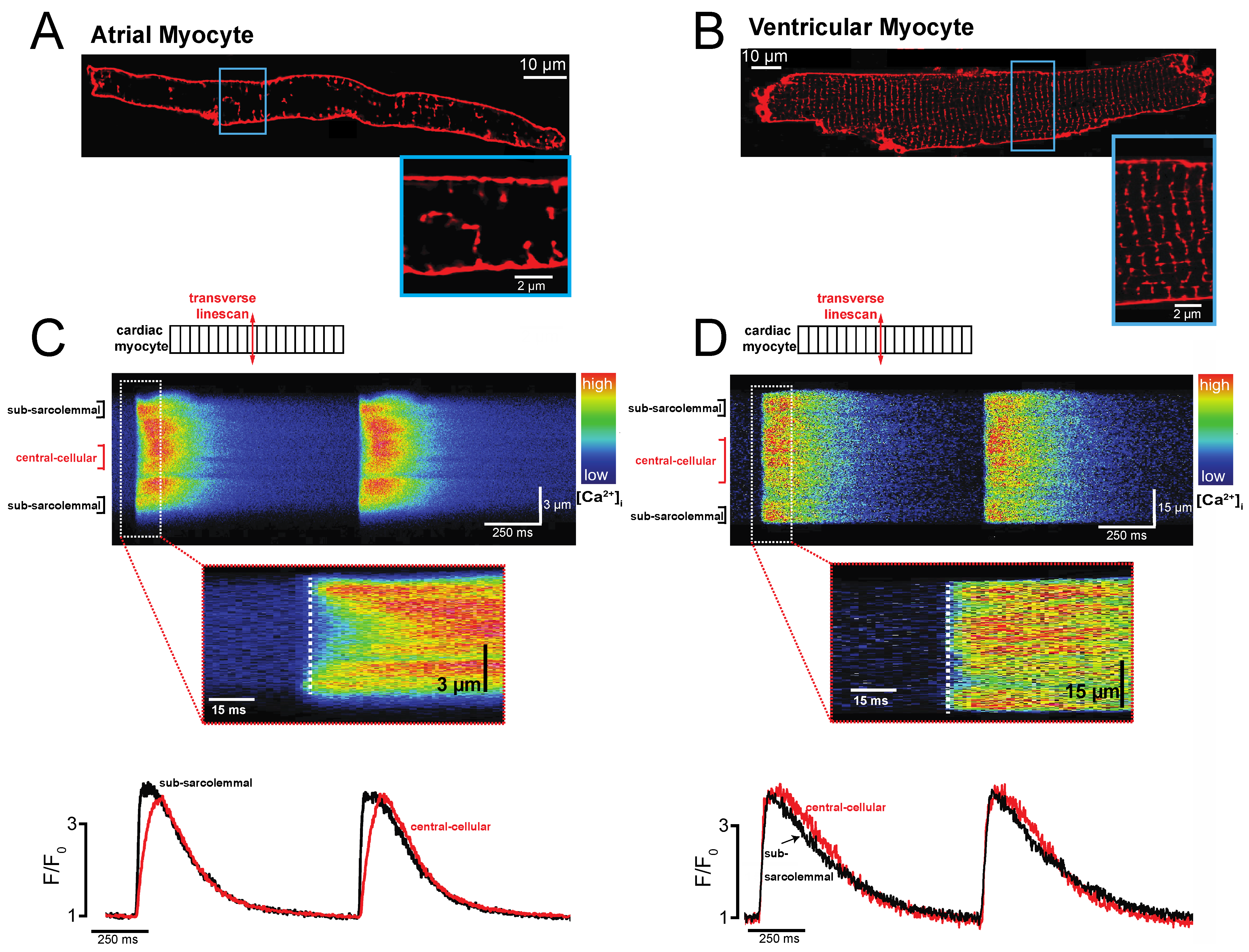
2. Atrial-Specific Intracellular Ca2+ Signaling
Mechanisms of Ca2+- and Na+-Based Arrhythmogenicity
3. Changes in Na+ and Ca2+ Homeostasis during Atrial Fibrillation: Timing Matters
3.1. Changes in Intracellular Ca2+ and Na+ Signaling at the Onset of AF: Paroxysmal Atrial Fibrillation
3.2. Adaptation of Intracellular Ca2+ and Na+ Signaling during Persistent AF
3.2.1. Reduced L-Type Ca2+ Current
3.2.2. Small Ca2+ Transient
3.2.3. Upregulated Na+/Ca2+ Exchanger
3.2.4. Remodeling of the RyR2 Macro-Complex
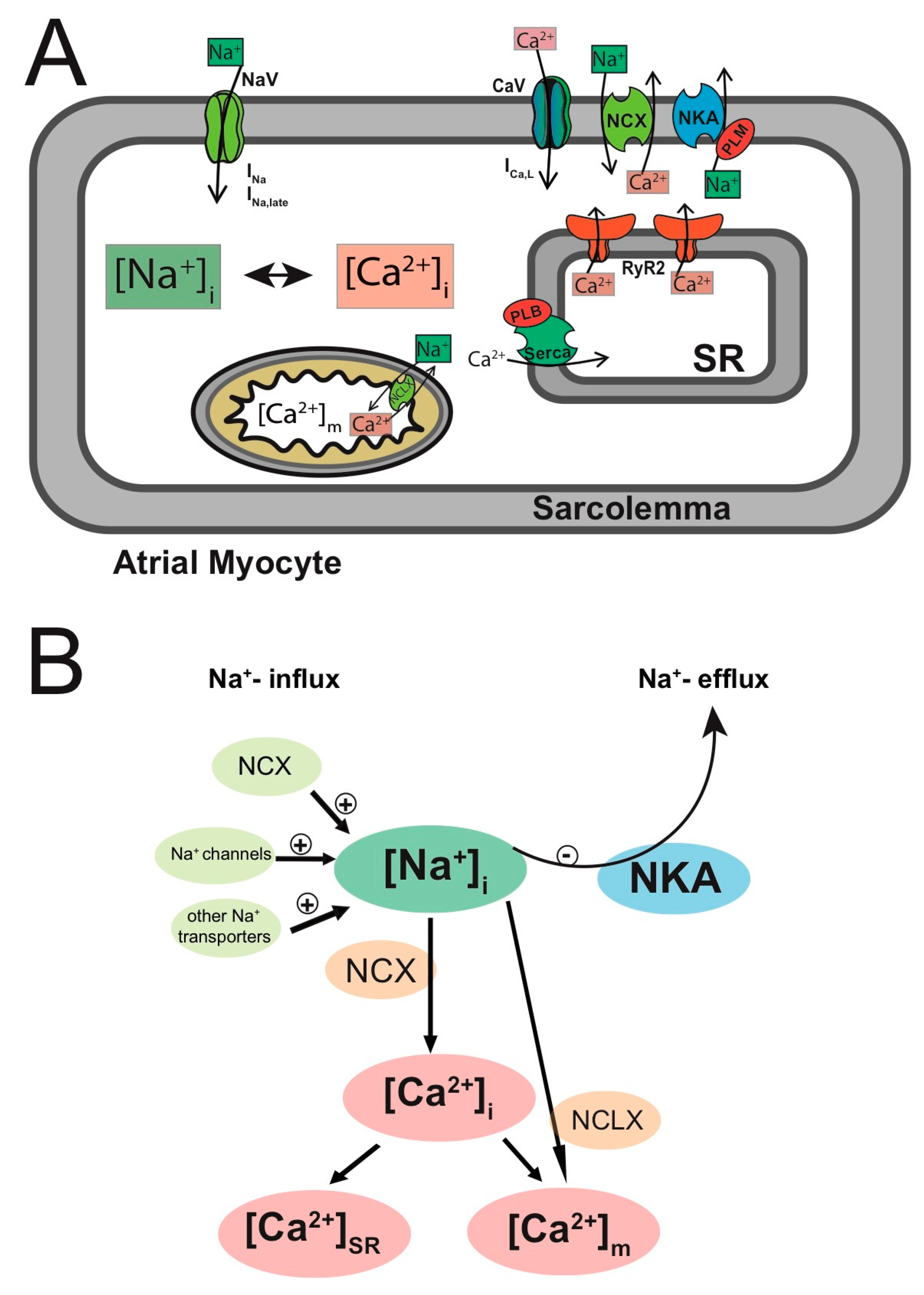
3.2.5. Remodeling of [Na+]i in Persistent AF
4. INa,L as an Antiarrhythmic Target in Atrial Arrhythmias
Results of Clinical Trials Using Ranolazine in AF Treatment
5. Outlook
Funding
Conflicts of Interest
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H., Jr.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A Global Burden of Disease 2010 Study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2014, 64, e1–e76. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Bax, J.; Blomstrom-Lundquist, C.; Calkins, H.; Camm, A.J.; Cappato, R.; Cosio, F.; Crijns, H.; Diener, H.C.; Goette, A.; et al. Early and comprehensive management of atrial fibrillation: Proceedings from the 2nd AFNET/EHRA consensus conference on atrial fibrillation entitled ‘research perspectives in atrial fibrillation’. Eurpoace 2009, 7, 860–885. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef]
- Schotten, U.; Ausma, J.; Stellbrink, C.; Sabatschus, I.; Vogel, M.; Frechen, D.; Schoendube, F.; Hanrath, P.; Allessie, M.A. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation 2001, 103, 691–698. [Google Scholar] [CrossRef]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef]
- Gaspo, R.; Bosch, R.F.; Talajic, M.; Nattel, S. Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation 1997, 96, 4027–4035. [Google Scholar] [CrossRef]
- Greiser, M. Calcium signalling silencing in atrial fibrillation. J. Physiol. 2017, 595, 4009–4017. [Google Scholar] [CrossRef]
- Greiser, M.; Kerfant, B.G.; Williams, G.S.; Voigt, N.; Harks, E.; Dibb, K.M.; Giese, A.; Meszaros, J.; Verheule, S.; Ravens, U.; et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J. Clin. Investig. 2014, 124, 4759–4772. [Google Scholar] [CrossRef]
- Christ, T.; Rozmaritsa, N.; Engel, A.; Berk, E.; Knaut, M.; Metzner, K.; Canteras, M.; Ravens, U.; Kaumann, A. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 11193–11198. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Wakili, R.; Yeh, Y.H.; Qi, X.Y.; Greiser, M.; Chartier, D.; Nishida, K.; Maguy, A.; Villeneuve, L.R.; Boknik, P.; Voigt, N.; et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ. Arrhythm. Electrophysiol. 2010, 3, 530–541. [Google Scholar] [CrossRef]
- Bers, D.M.; Barry, W.H.; Despa, S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 2003, 57, 897–912. [Google Scholar] [CrossRef]
- Despa, S.; Bers, D.M. Na/K pump current and [Na](i) in rabbit ventricular myocytes: Local [Na](i) depletion and Na buffering. Biophys. J. 2003, 84, 4157–4166. [Google Scholar] [CrossRef]
- Bers, D.M.; Despa, S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef]
- Despa, S.; Bers, D.M. Na+ transport in the normal and failing heart-remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Chen-Izu, Y.; Shaw, R.M.; Pitt, G.S.; Yarov-Yarovoy, V.; Sack, J.T.; Abriel, H.; Aldrich, R.W.; Belardinelli, L.; Cannell, M.B.; Catterall, W.A.; et al. Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. 2015, 593, 1347–1360. [Google Scholar] [CrossRef]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.D.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; et al. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kerfant, B.G.; Greiser, M. Enhanced late Na+ currents in atrial fibrillation new drug target or just an epiphenomenon? J. Am. Coll. Cardiol. 2010, 55, 2343–2345. [Google Scholar] [CrossRef]
- Greiser, M.; Schotten, U. Dynamic remodeling of intracellular Ca2+ signaling during atrial fibrillation. J. Mol. Cell. Cardiol. 2013, 58, 134–142. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Benitah, J.P.; Alvarez, J.L.; Gómez, A.M. L-type Ca2+ current in ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 26–36. [Google Scholar] [CrossRef]
- Fabiato, A.; Fabiato, F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. 1975, 249, 469–495. [Google Scholar] [CrossRef]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Greiser, M.; Lederer, W.J.; Schotten, U. Alterations of atrial Ca2+ handling as cause and consequence of atrial fibrillation. Cardiovasc. Res. 2011, 89, 722–733. [Google Scholar] [CrossRef][Green Version]
- Bootman, M.D.; Higazi, D.R.; Coombes, S.; Roderick, H.L. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J. Cell Sci. 2006, 119, 3915–3925. [Google Scholar] [CrossRef]
- Bootman, M.D.; Smyrnias, I.; Thul, R.; Coombes, S.; Roderick, H.L. Atrial cardiomyocyte calcium signalling. Biochim. Biophys. Acta 2011, 1813, 922–934. [Google Scholar] [CrossRef]
- Hüser, J.; Lipsius, S.L.; Blatter, L.A. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J. Physiol. 1996, 494, 641–651. [Google Scholar] [CrossRef]
- Kirk, M.M.; Izu, L.T.; Chen-Izu, Y.; McCulle, S.L.; Wier, W.G.; Balke, C.W.; Shorofsky, S.R. Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J. Physiol. 2003, 547, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhang, R.; Kim, B.; Ma, A.; Philipson, K.D.; Goldhaber, J.I. Heterogeneity of transverse-axial tubule system in mouse atria: Remodeling in atrial-specific Na+-Ca2+ exchanger knockout mice. J. Mol. Cell. Cardiol. 2017, 108, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Aistrup, G.L.; Supple, S.; Frank, C.; Singh, J.; Tai, S.; Zhao, A.; Chicos, L.; Marszalec, W.; Guo, A.; et al. Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm 2017, 14, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Soeller, C.; Cannell, M.B. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ. Res. 1999, 84, 266–275. [Google Scholar] [CrossRef]
- Huxley, A.F.; Taylor, R.E. Function of Krause’s membrane. Nature 1955, 176, 1068. [Google Scholar] [CrossRef]
- Brandenburg, S.; Kohl, T.; Williams, G.S.; Gusev, K.; Wagner, E.; Rog-Zielinska, E.A.; Hebisch, E.; Dura, M.; Didie, M.; Gotthardt, M.; et al. Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Investig. 2016, 126, 3999–4015. [Google Scholar] [CrossRef]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.; Steinbrecher, J.H.; Streich, J.H.; Korff, B.; Tuan, H.T.; Hagen, B.; et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef]
- Jones, P.P.; MacQuaide, N.; Louch, W.E. Dyadic Plasticity in Cardiomyocytes. Front. Physiol. 2018, 9, 1773. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef]
- Franzini-Armstrong, C.; Protasi, F.; Tijskens, P. The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 2005, 1047, 76–85. [Google Scholar] [CrossRef]
- Berlin, J.R. Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells. Am. J. Physiol. 1995, 269, H1165–H1170. [Google Scholar] [CrossRef]
- Venetucci, L.A.; Trafford, A.W.; Eisner, D.A. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: Threshold sarcoplasmic reticulum calcium content is required. Circ. Res. 2007, 100, 105–111. [Google Scholar] [CrossRef]
- Wier, W.G.; Cannell, M.B.; Berlin, J.R.; Marban, E.; Lederer, W.J. Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science 1987, 235, 325–328. [Google Scholar] [CrossRef]
- Berlin, J.R.; Cannell, M.B.; Lederer, W.J. Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ. Res. 1989, 65, 115–126. [Google Scholar] [CrossRef]
- Wier, W.G.; Kort, A.A.; Stern, M.D.; Lakatta, E.G.; Marban, E. Cellular calcium fluctuations in mammalian heart: Direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc. Natl. Acad. Sci. USA 1983, 80, 7367–7371. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Bers, B.D. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004, 14, 61–66. [Google Scholar] [CrossRef]
- Patterson, E.; Jackman, W.M.; Beckman, K.J.; Lazzara, R.; Lockwood, D.; Scherlag, B.J.; Wu, R.; Po, S. Spontaneous pulmonary vein firing in man: Relationship to tachycardia-pause early afterdepolarizations and triggered arrhythmia in canine pulmonary veins in vitro. J. Cardiovasc. Electrophysiol. 2007, 10, 1067–1075. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Philipson, K.D.; Nicoll, D.A. Sodium-calcium exchange: A molecular perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef]
- Smith, T. Digitalis: Mechanisms of action and clinical use. N. Engl. J. Med. 1988, 318, 358–365. [Google Scholar]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; De Pover, A. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L., Jr.; Roden, D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef]
- Aharonovitz, O.; Demaurex, N.; Woodside, M.; Grinstein, S. ATP dependence is not an intrinsic property of Na+/H+ exchanger NHE1: Requirement for an ancillary factor. Am. J. Physiol. 1999, 276, C1303–C1311. [Google Scholar] [CrossRef]
- Wan, E.; Abrams, J.; Weinberg, R.L.; Katchman, A.N.; Bayne, J.; Zakharov, S.I.; Yang, L.; Morrow, J.P.; Garan, H.; Marx, S.O. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J. Clin. Investig. 2016, 126, 112–122. [Google Scholar] [CrossRef]
- Nabauer, M.; Gerth, A.; Limbourg, T.; Schneider, S.; Oeff, M.; Kirchhof, P.; Goette, A.; Lewalter, T.; Ravens, U.; Meinertz, T.; et al. The Registry of the German Competence NETwork on Atrial Fibrillation: Patient characteristics and initial management. Europace 2009, 11, 423–434. [Google Scholar] [CrossRef]
- Kirchhof, P.; Breithardt, G.; Aliot, E.; Al Khatib, S.; Apostolakis, S.; Auricchio, A.; Bailleul, C.; Bax, J.; Benninger, G.; Blomstrom-Lundqvist, C.; et al. Personalized management of atrial fibrillation: Proceedings from the fourth Atrial Fibrillation competence NETwork/European Heart Rhythm Association consensus conference. Europace 2013, 15, 1540–1556. [Google Scholar] [CrossRef]
- Elvan, A.; Linnenbank, A.C.; van Bemmel, M.W.; Misier, A.R.; Delnoy, P.P.; Beukema, W.P.; de Bakker, J.M. Dominant frequency of atrial fibrillation correlates poorly with atrial fibrillation cycle length. Circ. Arrhythm. Electrophysiol. 2009, 2, 634–644. [Google Scholar] [CrossRef][Green Version]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.; Nattel, S.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef]
- Van Wagoner, D.R.; Pond, A.L.; McCarthy, P.M.; Trimmer, J.S.; Nerbonne, J.M. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 1997, 80, 772–781. [Google Scholar] [CrossRef]
- Qi, X.Y.; Vahdahi Hassani, F.; Hoffmann, D.; Xiao, J.; Xiong, F.; Villeneuve, L.R.; Ljubojevic-Holzer, S.; Kamler, M.; Abu-Taha, I.; Heijman, J.; et al. Inositol Trisphosphate Receptors and Nuclear Calcium in Atrial Fibrillation. Circ. Res. 2020, 128, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Wakili, R.; Qi, X.Y.; Chartier, D.; Boknik, P.; Kääb, S.; Ravens, U.; Coutu, P.; Dobrev, D.; Nattel, S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 2008, 1, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qi, Y.; Li, J.J.; He, W.J.; Gao, X.H.; Zhang, Y.; Sun, X.; Tong, J.; Zhang, J.; Deng, X.L.; et al. Stretch-induced sarcoplasmic reticulum calcium leak is causatively associated with atrial fibrillation in pressure-overloaded hearts. Cardiovasc. Res. 2021, 117, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Verhaert, D.V.M.; Brunner-La Rocca, H.P.; van Veldhuisen, D.J.; Vernooy, K. The bidirectional interaction between atrial fibrillation and heart failure: Consequences for the management of both diseases. Europace 2021, 23 (Suppl. 2), ii40–ii45. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Islam, M.A.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ and Na+ pump rate in rat and rabbit ventricular myocytes. J. Physiol. 2002, 539, 133–143. [Google Scholar] [CrossRef]
- Glitsch, H.G. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol. Rev. 2001, 81, 1791–1826. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.; Cohen, I.S.; Mathias, R.T. Transmural gradients in Na/K pump activity and [Na+] I in canine ventricle. Biophys. J. 2005, 89, 1700–1709. [Google Scholar] [CrossRef]
- Lebek, S.; Pichler, K.; Reuthner, K.; Trum, M.; Tafelmeier, M.; Mustroph, J.; Camboni, D.; Rupprecht, L.; Schmid, C.; Maier, L.S.; et al. Enhanced CaMKII-Dependent Late I Na Induces Atrial Proarrhythmic Activity in Patients with Sleep-Disordered Breathing. Circ. Res. 2020, 126, 603–615. [Google Scholar] [CrossRef]
- Gaspo, R.; Sun, H.; Fareh, S.; Levi, M.; Yue, L.; Allen, B.G.; Hebert, T.E.; Nattel, S. Dihydropyridine and beta adrenergic receptor binding in dogs with tachycardia-induced atrial fibrillation. Cardiovasc. Res. 1999, 42, 434–442. [Google Scholar] [CrossRef]
- Van Wagoner, D.R.; Pond, A.L.; Lamorgese, M.; Rossie, S.S.; McCarthy, P.M.; Nerbonne, J.M. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 1999, 85, 428–436. [Google Scholar] [CrossRef]
- Schotten, U.; Duytschaever, M.; Ausma, J.; Eijsbouts, S.; Neuberger, H.R.; Allessie, M. Electrical and contractile remodeling during the first days of atrial fibrillation go hand in hand. Circulation 2003, 107, 1433–1439. [Google Scholar] [CrossRef]
- Sun, H.; Chartier, D.; Leblanc, N.; Nattel, S. Intracellular calcium changes and tachycardia-induced contractile dysfunction in canine atrial myocytes. Cardiovasc. Res. 2001, 49, 751–761. [Google Scholar] [CrossRef]
- Greiser, M.; Halaszovich, C.R.; Frechen, D.; Boknik, P.; Ravens, U.; Dobrev, D.; Lückhoff, A.; Schotten, U. Pharmacological evidence for altered src kinase regulation of I (Ca, L) in patients with chronic atrial fibrillation. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007, 375, 383–392. [Google Scholar] [CrossRef]
- Greiser, M.; Neuberger, H.R.; Harks, E.; El-Armouche, A.; Boknik, P.; de Haan, S.; Verheyen, F.; Verheule, S.; Schmitz, W.; Ravens, U.; et al. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J. Mol. Cell. Cardiol. 2009, 46, 385–394. [Google Scholar] [CrossRef]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef]
- Vagos, M.R.; Arevalo, H.; Heijman, J.; Schotten, U.; Sundnes, J. A Computational Study of the Effects of Tachycardia-Induced Remodeling on Calcium Wave Propagation in Rabbit Atrial Myocytes. Front. Physiol. 2021, 12, 651428. [Google Scholar] [CrossRef]
- Sun, H.; Gaspo, R.; Leblanc, N.; Nattel, S. Cellular mechanisms of atrial contractile dysfunction caused by sustained atrial tachycardia. Circulation 1998, 98, 719–727. [Google Scholar] [CrossRef]
- Barana, A.; Matamoros, M.; Dolz-Gaiton, P.; Perez-Hernandez, M.; Amoros, I.; Nunez, M.; Sacristan, S.; Pedraz, A.; Pinto, A.; Fernandez-Aviles, F.; et al. Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ. Arrhythm. Electrophysiol. 2014, 7, 861–868. [Google Scholar] [CrossRef]
- Canon, S.; Caballero, R.; Herraiz-Martinez, A.; Perez-Hernandez, M.; Lopez, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpon, E.; Bernad, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef]
- Christ, T.; Boknik, P.; Wöhrl, S.; Wettwer, E.; Graf, E.M.; Bosch, R.F.; Knaut, E.; Schmitz, W.; Ravens, U.; Dobrev, D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 2004, 110, 2651–2657. [Google Scholar] [CrossRef]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef]
- Vest, J.A.; Wehrens, X.H.; Reiken, S.R.; Lehnart, S.E.; Dobrev, D.; Chandra, P.; Danilo, P.; Ravens, U.; Rosen, M.R.; Marks, A.R. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005, 111, 2025–2032. [Google Scholar] [CrossRef]
- Brandenburg, S.; Pawlowitz, J.; Fakuade, F.E.; Kownatzki-Danger, D.; Kohl, T.; Mitronova, G.Y.; Scardigli, M.; Neef, J.; Schmidt, C.; Wiedmann, F.; et al. Axial Tubule Junctions Activate Atrial Ca2+ Release Across Species. Front. Physiol. 2018, 9, 1227. [Google Scholar] [CrossRef]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef]
- Mesubi, O.O.; Rokita, A.G.; Abrol, N.; Wu, Y.; Chen, B.; Wang, Q.; Granger, J.M.; Tucker-Bartley, A.; Luczak, E.D.; Murphy, K.R.; et al. Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. J. Clin. Investig. 2021, 131, e95747. [Google Scholar] [CrossRef]
- Kushnir, A.; Wajsberg, B.; Marks, A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865 Pt B, 1687–1697. [Google Scholar] [CrossRef]
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef]
- Correll, R.N.; Eder, P.; Burr, A.R.; Despa, S.; Davis, J.; Bers, D.M.; Molkentin, J.D. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014, 114, 249–256. [Google Scholar] [CrossRef]
- Poulet, C.; Wettwer, E.; Grunnet, M.; Jespersen, T.; Fabritz, L.; Matschke, K.; Knaut, M.; Ravens, U. Late Sodium Current in Human Atrial Cardiomyocytes from Patients in Sinus Rhythm and Atrial Fibrillation. PLoS ONE 2015, 10, e0131432. [Google Scholar] [CrossRef]
- Bosch, R.F.; Zeng, X.; Grammer, J.B.; Popovic, K.; Mewis, C.; Kuhlkamp, V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 1999, 44, 121–131. [Google Scholar] [CrossRef]
- Gaspo, R.; Bosch, R.F.; Bou-Abboud, E.; Nattel, S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ. Res. 1997, 81, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.J.; Kane, K.A.; Rankin, A.C. Characterisation of the Na, K pump current in atrial cells from patients with and without chronic atrial fibrillation. Cardiovasc. Res. 2003, 59, 593–602. [Google Scholar] [CrossRef]
- Garber, L.; Joca, H.C.; Boyman, L.; Lederer, W.J.; Greiser, M. Camera-based measurements of intracellular [Na+]i in murine atrial myocytes. J. Vis. Exp. 2021. (In press)
- Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 1998, 98, 2545–2552. [Google Scholar] [CrossRef]
- Horvath, B.; Banyasz, T.; Jian, Z.; Hegyi, B.; Kistamas, K.; Nanasi, P.P.; Izu, L.T.; Chen-Izu, Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013, 64, 59–68. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Burashnikov, A.; Sicouri, S.; Belardinelli, L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 2011, 8, 1281–1290. [Google Scholar] [CrossRef]
- Undrovinas, A.I.; Belardinelli, L.; Undrovinas, N.A.; Sabbah, H.N. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J. Cardiovasc. Electrophysiol. 2006, 17 (Suppl. 1), S169–S177. [Google Scholar] [CrossRef]
- Song, Y.; Shryock, J.C.; Belardinelli, L. A slowly inactivating sodium current contributes to spontaneous diastolic depolarization of atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1254–H1262. [Google Scholar] [CrossRef]
- Song, Y.; Shryock, J.C.; Belardinelli, L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2031–H2039. [Google Scholar] [CrossRef]
- Lemoine, M.D.; Duverger, J.E.; Naud, P.; Chartier, D.; Qi, X.Y.; Comtois, P.; Fabritz, L.; Kirchhof, P.; Nattel, S. Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc. Res. 2011, 92, 67–74. [Google Scholar] [CrossRef]
- Olesen, M.S.; Yuan, L.; Liang, B.; Holst, A.G.; Nielsen, N.; Nielsen, J.B.; Hedley, P.L.; Christiansen, M.; Olesen, S.P.; Haunso, S.; et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 2012, 5, 450–459. [Google Scholar] [CrossRef]
- Burashnikov, A.; Di Diego, J.M.; Zygmunt, A.C.; Belardinelli, L.; Antzelevitch, C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 2007, 116, 1449–1457. [Google Scholar] [CrossRef]
- Kumar, K.; Nearing, B.D.; Carvas, M.; Nascimento, B.C.; Acar, M.; Belardinelli, L.; Verrier, R.L. Ranolazine exerts potent effects on atrial electrical properties and abbreviates atrial fibrillation duration in the intact porcine heart. J. Cardiovasc. Electrophysiol. 2009, 20, 796–802. [Google Scholar] [CrossRef]
- Burashnikov, A.; Sicouri, S.; Di Diego, J.M.; Belardinelli, L.; Antzelevitch, C. Synergistic effect of the combination of ranolazine and dronedarone to suppress atrial fibrillation. J. Am. Coll. Cardiol. 2010, 56, 1216–1224. [Google Scholar] [CrossRef]
- Belardinelli, L.; Liu, G.; Smith-Maxwell, C.; Wang, W.Q.; El-Bizri, N.; Hirakawa, R.; Karpinski, S.; Li, C.H.; Hu, L.; Li, X.J.; et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharmacol. Exp. Ther. 2013, 344, 23–32. [Google Scholar] [CrossRef]
- Sicouri, S.; Belardinelli, L.; Antzelevitch, C. Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Heart Rhythm 2013, 10, 1036–1043. [Google Scholar] [CrossRef]
- Burashnikov, A.; Di Diego, J.M.; Goodrow, R.J., Jr.; Belardinelli, L.; Antzelevitch, C. Atria are More Sensitive Than Ventricles to GS-458967-Induced Inhibition of Late Sodium Current. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 501–508. [Google Scholar] [CrossRef]
- Carneiro, J.S.; Bento, A.S.; Bacic, D.; Nearing, B.D.; Rajamani, S.; Belardinelli, L.; Verrier, R.L. The Selective Cardiac Late Sodium Current Inhibitor GS-458967 Suppresses Autonomically Triggered Atrial Fibrillation in an Intact Porcine Model. J. Cardiovasc. Electrophysiol. 2015, 26, 1364–1369. [Google Scholar] [CrossRef]
- Luo, A.; Ma, J.; Song, Y.; Qian, C.; Wu, Y.; Zhang, P.; Wang, L.; Fu, C.; Cao, Z.; Shryock, J.C. Larger late sodium current density as well as greater sensitivities to ATX II and ranolazine in rabbit left atrial than left ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H455–H461. [Google Scholar] [CrossRef]
- Nattel, S.; Harada, M. Atrial Remodeling and Atrial Fibrillation: Recent Advances and Translational Perspectives. J. Am. Coll. Cardiol. 2014, 63, 2335–2345. [Google Scholar] [CrossRef]
- Ratte, A.; Wiedmann, F.; Kraft, M.; Katus, H.A.; Schmidt, C. Antiarrhythmic Properties of Ranolazine: Inhibition of Atrial Fibrillation Associated TASK-1 Potassium Channels. Front. Pharmacol. 2019, 10, 1367. [Google Scholar] [CrossRef]
- Zimetbaum, P.J.C. Antiarrhythmic drug therapy for atrial fibrillation. Circulation 2012, 125, 381–389. [Google Scholar] [CrossRef]
- Mann, S.A.; Heide, J.; Knott, T.; Airini, R.; Epureanu, F.B.; Deftu, A.F.; Deftu, A.T.; Radu, B.M.; Amuzescu, B. Recording of multiple ion current components and action potentials in human induced pluripotent stem cell-derived cardiomyocytes via automated patch-clamp. J. Pharmacol. Toxicol. Methods 2019, 100, 106599. [Google Scholar] [CrossRef]
- Amuzescu, B.; Airini, R.; Epureanu, F.B.; Mann, S.A.; Knott, T.; Radu, B.M. Evolution of mathematical models of cardiomyocyte electrophysiology. Math. Biosci. 2021, 334, 108567. [Google Scholar] [CrossRef]
- Chaitman, B.R.J.C. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 2006, 113, 2462–2472. [Google Scholar] [CrossRef]
- Fragakis, N.; Koskinas, K.C.; Katritsis, D.G.; Pagourelias, E.D.; Zografos, T.; Geleris, P. Comparison of effectiveness of ranolazine plus amiodarone versus amiodarone alone for conversion of recent-onset atrial fibrillation. Am. J. Cardiol. 2012, 110, 673–677. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Fragakis, N.; Katritsis, D.; Skeberis, V.; Vassilikos, V. Ranolazine enhances the efficacy of amiodarone for conversion of recent-onset atrial fibrillation. Europace 2014, 16, 973–979. [Google Scholar] [CrossRef]
- Simopoulos, V.; Tagarakis, G.I.; Daskalopoulou, S.S.; Daskalopoulos, M.E.; Lenos, A.; Chryssagis, K.; Skoularingis, I.; Molyvdas, P.A.; Tsilimingas, N.B.; Aidonidis, I. Ranolazine enhances the antiarrhythmic activity of amiodarone by accelerating conversion of new-onset atrial fibrillation after cardiac surgery. Angiology 2014, 65, 294–297. [Google Scholar] [CrossRef]
- Tsanaxidis, N.; Aidonidis, I.; Hatziefthimiou, A.; Triposkiadis, F.; Skoularigis, I. Acute Ranolazine Plus Amiodarone vs Amiodarone Alone for Conversion of Recent-Onset Atrial Fibrillation: A Prospective Clinical Study. Eur. Heart J. 2015, 36, 910–911. [Google Scholar]
- Reiffel, J.A.; Camm, A.J.; Belardinelli, L.; Zeng, D.; Karwatowska-Prokopczuk, E.; Olmsted, A.; Zareba, W.; Rosero, S.; Kowey, P. The Harmony Trial: Combined Ranolazine and Dronedarone in the Management of Paroxysmal Atrial Fibrillation: Mechanistic and Therapeutic Synergism. Circ. Arrhythm. Electrophysiol. 2015, 8, 1048–1056. [Google Scholar] [CrossRef]
- Tsanaxidis, N.; Aidonidis, I.; Hatziefthimiou, A.; Daskalopoulou, S.S.; Giamouzis, G.; Triposkiadis, F.; Skoularigis, I. Ranolazine Added to Amiodarone Facilitates Earlier Conversion of Atrial Fibrillation Compared to Amiodarone-Only Therapy. Pacing Clin. Electrophysiol. 2017, 40, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, V.; Hevas, A.; Hatziefthimiou, A.; Dipla, K.; Skoularigis, I.; Tsilimingas, N.; Aidonidis, I. Amiodarone plus Ranolazine for Conversion of Post-Cardiac Surgery Atrial Fibrillation: Enhanced Effectiveness in Reduced Versus Preserved Ejection Fraction Patients. Cardiovasc. Drugs Ther. 2018, 32, 559–565. [Google Scholar] [CrossRef] [PubMed]
- De Vecchis, R.; Ariano, C.; Giasi, A.; Cioppa, C. Antiarrhythmic effects of ranolazine used both alone for prevention of atrial fibrillation and as an add-on to intravenous amiodarone for its pharmacological cardioversion: A meta-analysis. Minerva Cardioangiol. 2018, 66, 349–359. [Google Scholar] [PubMed]
- Vassallo, P.; Trohman, R.G. Prescribing amiodarone: An evidence-based review of clinical indications. JAMA 2007, 298, 1312–1322. [Google Scholar] [CrossRef]
- Murdock, D.K.; Overton, N.; Kersten, M.; Kaliebe, J.; Devecchi, F. The effect of ranolazine on maintaining sinus rhythm in patients with resistant atrial fibrillation. Indian Pacing Electrophysiol. J. 2008, 8, 175–181. [Google Scholar]
- Miles, R.H.; Passman, R.; Murdock, D.K. Comparison of effectiveness and safety of ranolazine versus amiodarone for preventing atrial fibrillation after coronary artery bypass grafting. Am. J. Cardiol. 2011, 108, 673–676. [Google Scholar] [CrossRef]
- Murdock, D.K.; Kaliebe, J.; Larrain, G. The use of ranolazine to facilitate electrical cardioversion in cardioversion-resistant patients: A case series. Pacing Clin. Electrophysiol. 2012, 35, 302–307. [Google Scholar] [CrossRef]
- Tagarakis, G.I.; Aidonidis, I.; Daskalopoulou, S.S.; Simopoulos, V.; Liouras, V.; Daskalopoulos, M.E.; Parisis, C.; Papageorgiou, K.; Skoularingis, I.; Triposkiadis, F.; et al. Effect of ranolazine in preventing postoperative atrial fibrillation in patients undergoing coronary revascularization surgery. Curr. Vasc. Pharmacol. 2013, 11, 988–991. [Google Scholar] [CrossRef]
- Scirica, B.M.; Belardinelli, L.; Chaitman, B.R.; Waks, J.W.; Volo, S.; Karwatowska-Prokopczuk, E.; Murphy, S.A.; Cheng, M.L.; Braunwald, E.; Morrow, D.A. Effect of ranolazine on atrial fibrillation in patients with non-ST elevation acute coronary syndromes: Observations from the MERLIN-TIMI 36 trial. Europace 2015, 17, 32–37. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Maier, L.S.; Mont, L.; Schwartz, P.J.; Simonis, G.; Leschke, M.; Gronda, E.; Boriani, G.; Darius, H.; Guillamon Toran, L.; et al. Ranolazine in the treatment of atrial fibrillation: Results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following an ELectricaL CardiOversion) study. Heart Rhythm 2015, 12, 872–878. [Google Scholar] [CrossRef]
- Bekeith, S.; Meghani, M.; Shariff, M.; Asti, D.; Nalluri, N.; Agarwal, V.; Shah, N.; Soomro, A.; Khan, M.; Spagnola, J.; et al. Effect of ranolazine on the incidence of atrial fibrillation following cardiac surgery. Circulation 2015, 132, A13387. [Google Scholar]
- Hammond, D.A.; Smotherman, C.; Jankowski, C.A.; Tan, S.; Osian, O.; Kraemer, D.; DeLosSantos, M. Short-course of ranolazine prevents postoperative atrial fibrillation following coronary artery bypass grafting and valve surgeries. Clin. Res. Cardiol. 2015, 104, 410–417. [Google Scholar] [CrossRef]
- Guerra, F.; Romandini, A.; Barbarossa, A.; Belardinelli, L.; Capucci, A. Ranolazine for rhythm control in atrial fibrillation: A systematic review and meta-analysis. Int. J. Cardiol. 2017, 227, 284–291. [Google Scholar] [CrossRef]
- Gong, M.; Zhang, Z.; Fragakis, N.; Korantzopoulos, P.; Letsas, K.P.; Li, G.; Yan, G.X.; Liu, T. Role of ranolazine in the prevention and treatment of atrial fibrillation: A meta-analysis of randomized clinical trials. Heart Rhythm 2017, 14, 3–11. [Google Scholar] [CrossRef]
- Patel, N.; Kluger, J. Ranolazine for Prevention of Atrial Fibrillation after Cardiac Surgery: A Systematic Review. Cureus 2018, 10, e2584. [Google Scholar] [CrossRef]
- Mariscalco, G.; Engström, K.G. Atrial fibrillation after cardiac surgery: Risk factors and their temporal relationship in prophylactic drug strategy decision. Int. J. Cardiol. 2008, 129, 354–362. [Google Scholar] [CrossRef]
- Dobrev, D.; Aguilar, M.; Heijman, J.; Guichard, J.-B.; Nattel, S. Postoperative atrial fibrillation: Mechanisms, manifestations and management. Nat. Rev. Cardiol. 2019, 16, 417–436. [Google Scholar] [CrossRef]
- Ramirez, R.J.; Takemoto, Y.; Martins, R.P.; Filgueiras-Rama, D.; Ennis, S.R.; Mironov, S.; Bhushal, S.; Deo, M.; Rajamani, S.; Berenfeld, O.; et al. Mechanisms by Which Ranolazine Terminates Paroxysmal but Not Persistent Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2019, 12, e005557. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | Population Studied (N, Age, %Male) | Study Design | AF Detection Method, Surveillance Duration | Results |
|---|---|---|---|---|
| Murdock 2008 [126] # | Recurrent AF with failure to AF ablation or anti-arrhythmic behavior 7, 67 ± 9, 57% | Oral RN (500–1000 mg/BID) after stopping all other anti-arrhythmic therapy | Not reported |
|
| Miles 2011 [127] a | Post CABG AF 182, 66.7 ± 9.3, 70% (intervention arm) 211 64.9 ± 10.9, 77% (control arm) | Intervention arm: 1500 mg RN before surgery, 1000 mg RN BID post-op for 10–14 days Control arm: 400 mg amiodarone before surgery, 200 mg amiodarone BID post-op 10–14 days | Continuous ECG monitoring throughout hospitalization |
|
| Fragakis 2012 [117] c | New onset AF (<48 h from diagnosis) 25, 62 ± 8, 60% (intervention arm) 26, 64 ± 7, 69% (control arm) | Intervention arm:1500 mg RN daily and IV amiodarone Control arm: IV amiodarone (loading dose: 5 mg/kg in 1 h followed by 50 mg/h for 24 h or until cardioversion) | Continuous ECG in CCU for 24 h followed by >1 day inpatient |
|
| Murdock 2012 [128] # | Recurrent AF with electro-cardioversion failure 25, 62 ± 11, 76% | 2000 mg RN given after failed electrocardioversion attempt, repeat electrocardioversion after 3–4 h of administration | Not reported |
|
| Tagarakis 2013 [129] c | Post-CABG AF 34, NA, NA (intervention arm) 68, NA, NA (control arm) | Intervention arm: 375 mg RN BID 3 days prior to operation until discharge Control arm: usual care | Continuous ECG monitoring for first 24 h followed by ECG monitoring every 4 h until discharge |
|
| Koskinas 2014 [118] c | New onset AF (<48 h from diagnosis) 61, 66 ± 11, 41% (intervention arm) 60, 64 ± 9, 48% (control arm) | Intervention arm: 1500 mg RN daily and IV amiodarone Control: IV amiodarone (loading dose: 5 mg/kg in 1 h followed by 50 mg/h) | Continuous ECG monitoring in the CCU for 24 h |
|
| Simopuolos 2014 [119] c | Post-CABG AF 20, 69 ± 7, 70% (intervention arm) 21, 67 ± 8, 60% (control arm) | Intervention arm: 500 mg RN (loading dose) followed by 375 mg RN BID and IV amiodarone Control arm: IV amiodarone: 300 mg in 30 min followed by 750 mg in 24 h, then 200 mg BID for one week and then 200 mg daily for 1 week | Continuous ECG monitoring for first 24 h followed by ECG every 4 h, monitoring until discharge |
|
| Scirica 2015 (MERLIN) [130] c | Patients hospitalized for NSTEMI 3162, 17% >75 yrs, 66.8% (intervention arm) 3189, 18% >75 yrs, 63.7% (control arm) | Intervention arm: IV 200 mg RN with 80 mg/h infusion for 12–96 h, then 1000 mg oral RN BID Control arm: placebo plus standard medical intervention | Continuous ECG monitoring for 7 days Median clinical follow-up at 12 months |
|
| De Ferrari 2015 (RAFFAELLO) [131] c | Persistent AF, 2 h after successful cardioversion 65, 66.9 ± 11.8, 70.8% (375 mg RN) 60, 65.5 ± 8.5, 85% (500 mg RN) 58, 63.6 ± 11.3,79.3 (750 mg RN) 55, 65.2 ± 9.5, 74.5% (control arm) | Intervention arm: either oral 375 mg BID, 500 mg BID, or 750 mg BID ranolazine Control arm: placebo | Transtelephonic electrocardiogram for 16 weeks and 12 lead ECG at 1 week, 2 months, and 4 months |
|
| Tsanaxidis 2015 [120] *,c | New onset AF 36, 67 ± 10,25% (intervention arm) 29, 62 ± 11,55% (control arm) | Intervention arm: 1000 mg RN once + IV amiodarone Control arm: IV amiodarone (loading dose: 5 mg/kg in 1 h followed by 50 mg/h) | Not reported |
|
| Bekeith 2015 [132] *,b | POAF 27, NA, NA (intervention arm) 27, NA, NA (control arm) | Intervention arm: 1000 mg RN BID for 48 h prior to surgery and 2 weeks post-op Control: placebo | ECG monitoring in patient followed by holter monitor 2 weeks post-discharge |
|
| Hammond 2015 [133] a | POAF 69, 59.7 ± 10.8, 68.1% (intervention arm) 136, 62.2 ± 11.8, 56.6% (control arm) | Intervention arm: 1000 mg RN BID starting on day of surgery for 7 days or until discharge Control arm: standard therapy | Not reported |
|
| Reiffel 2015 (HARMONY) [121] a | Paroxysmal AF with recent dual-chamber pacemaker placement 26, 70 ± 10.8, 39% (intervention arm) 52, 73.5 ± 11.5, 44.5% (control arm) | Intervention arm: 750 mg RN BID, dronedarone, or both Control: placebo | Dual-chamber pacemaker, 4-week run-in period followed by a 12-week treatment period |
|
| Tsanaxidis 2017 [122] | New onset AF (<48 h from onset) 92, 70 ± 10, 41% (intervention arm) 81, 67 ± 11, 50.6% (control arm) | Intervention arm: 1000 mg RN once and IV amiodarone Control arm: IV amiodarone (loading dose: 5 mg/kg in 1 h followed by 50 mg/h) | Not reported |
|
| Simopoulos 2018 [123] | POAF in patients with HFrEF vs. HFpEF 511, 65 ± 9, 87% (HFrEF arm) 301, 66 ± 10, 85% (HFpEF arm) | Intervention arm: 500 mg RN followed by 375 mg RN after 6 h and 375 mg RN BID thereafter and amiodarone Control arm: IV amiodarone (300 mg in first 30 min + 1125 mg over next 36 h) | Not reported |
|
| Meta-Analysis | ||||
| Guerra 2017 [134] |
| |||
| Gong 2017 [135] |
| |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplan, A.D.; Joca, H.C.; Boyman, L.; Greiser, M. Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis. Int. J. Mol. Sci. 2021, 22, 10513. https://doi.org/10.3390/ijms221910513
Kaplan AD, Joca HC, Boyman L, Greiser M. Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis. International Journal of Molecular Sciences. 2021; 22(19):10513. https://doi.org/10.3390/ijms221910513
Chicago/Turabian StyleKaplan, Aaron D., Humberto C. Joca, Liron Boyman, and Maura Greiser. 2021. "Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis" International Journal of Molecular Sciences 22, no. 19: 10513. https://doi.org/10.3390/ijms221910513
APA StyleKaplan, A. D., Joca, H. C., Boyman, L., & Greiser, M. (2021). Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis. International Journal of Molecular Sciences, 22(19), 10513. https://doi.org/10.3390/ijms221910513