
Effects of the Selective Serotonin Reuptake Inhibitor Fluoxetine on Developing Neural Circuits in a Model of the Human Fetal Cortex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
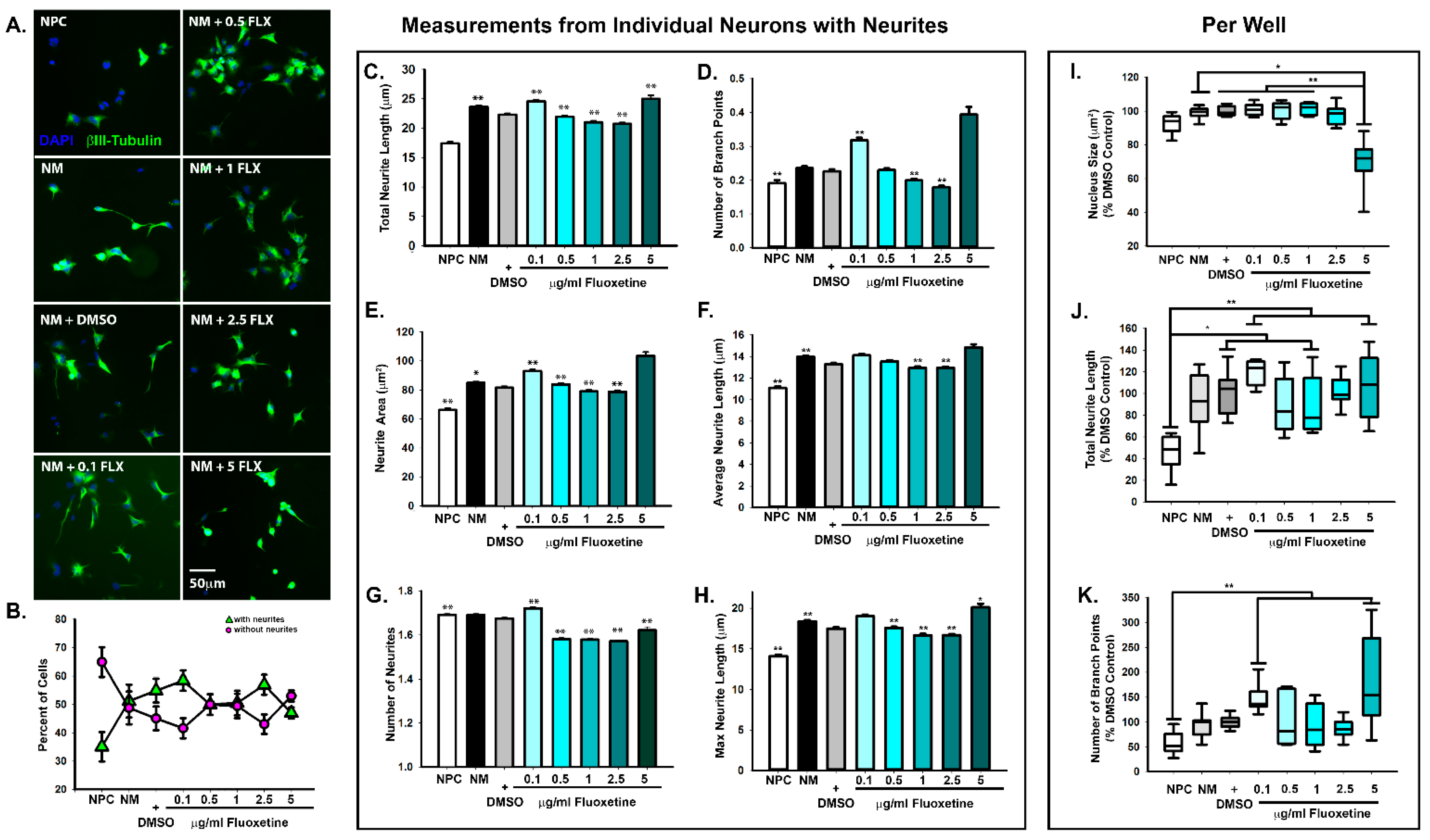
2.1. Dose-Dependent Effects of Fluoxetine on Neurite Formation during Neuronal Differentiation
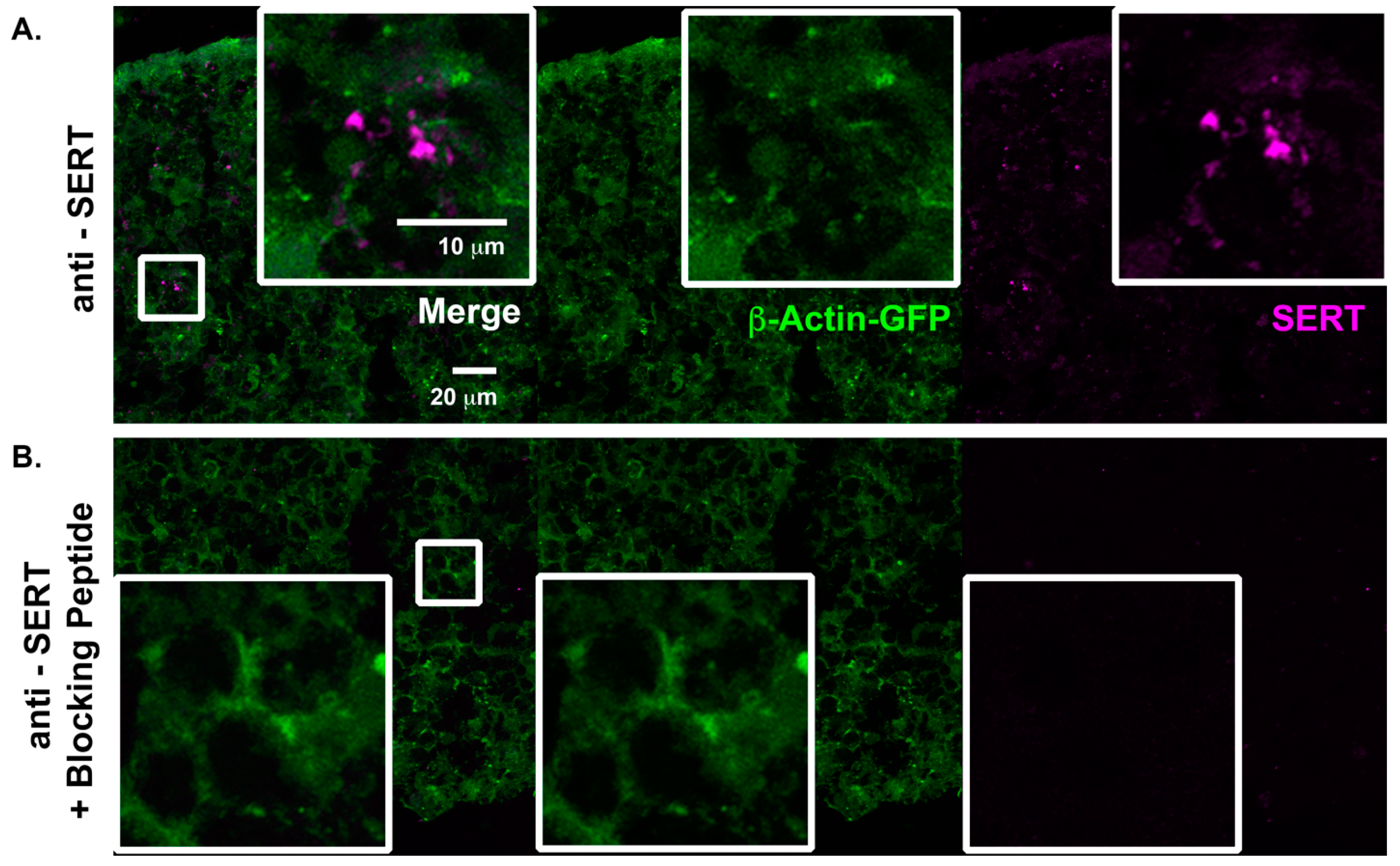
2.2. Characterization of Serotonergic Pathways in Cortical Spheroids
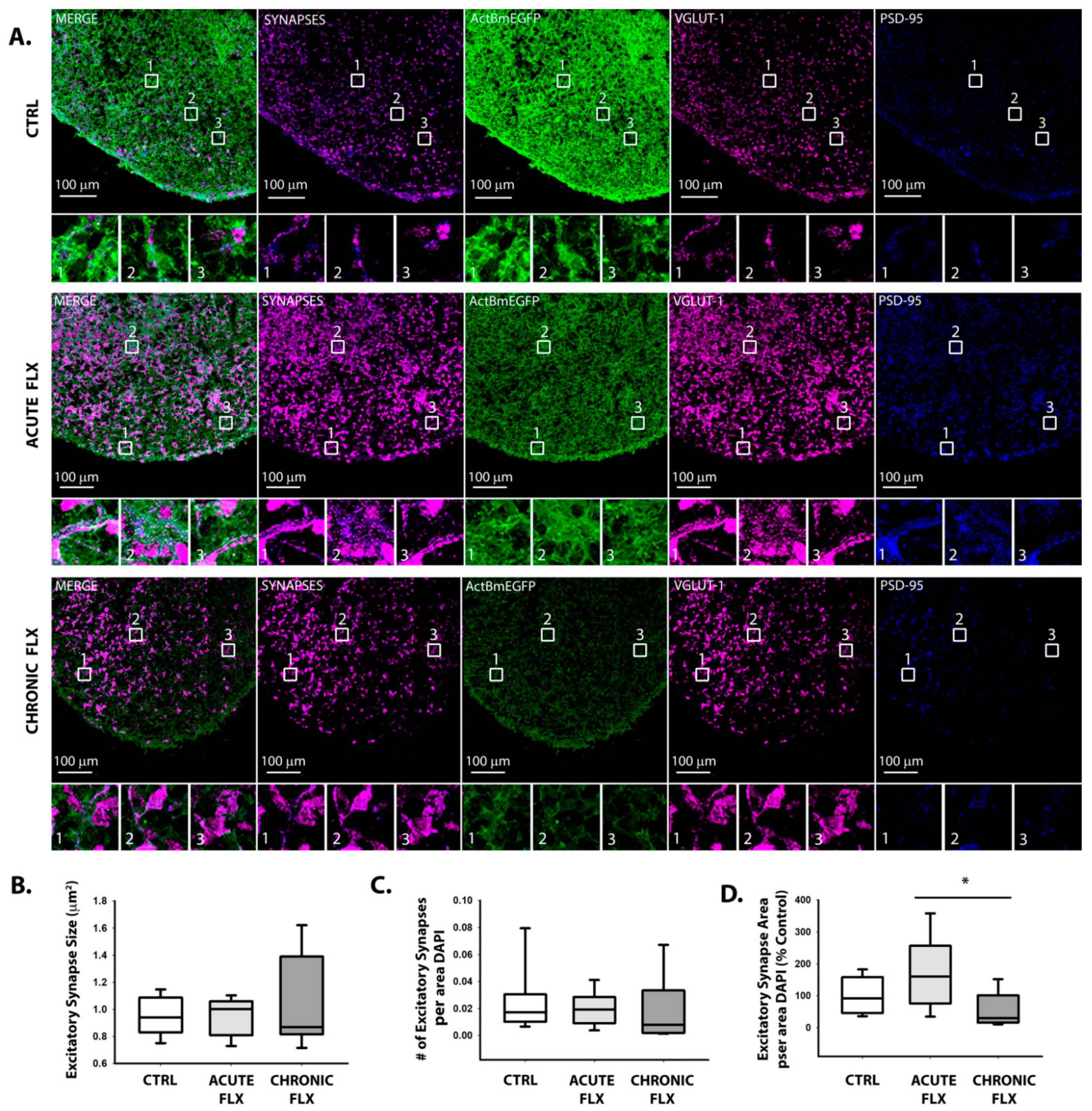
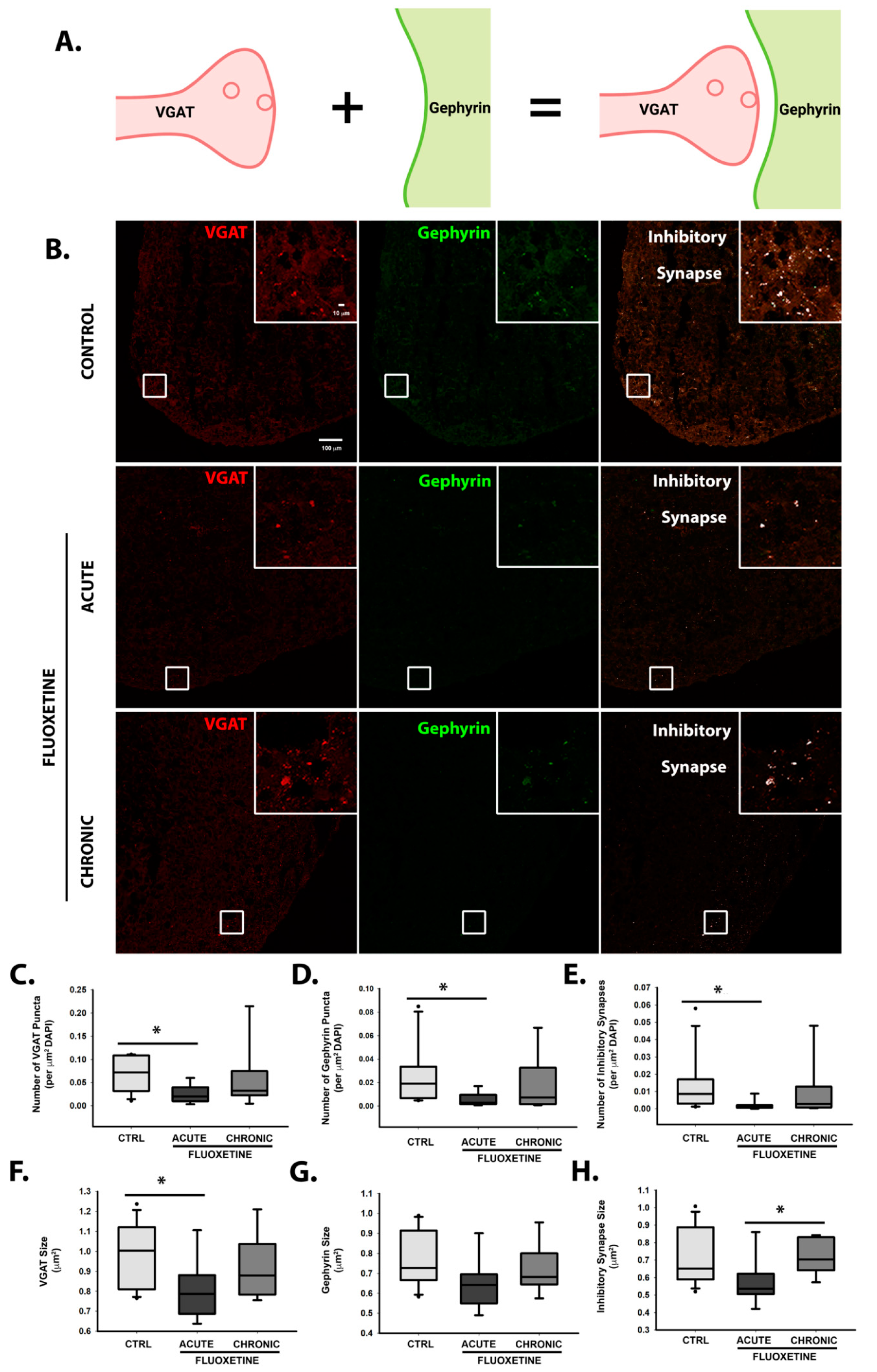
2.3. Effects of Fluoxetine on Synapse Formation
2.4. Fluoxetine Reversibly Suppresses Spontaneous Action Potential Formation
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Neural Progenitor Cell Culture
4.3. Differentiation of hiPSC-Derived NPCs into Neurons
4.4. 3-D Cortical Spheroid Culture
4.5. Drug Treatment of Cortical Spheroids
4.6. Immunohistochemistry
4.7. High Content Imaging and Analysis
4.8. Confocal Imaging
4.9. Dissociation of Human Cortical Spheroids for MEA Experiments
4.10. MEA Plate Preparation
4.11. Maestro Edge Recordings of Neural Activity
4.12. Fluoxetine MEA Experiment
4.13. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lynch, G.; Rex, C.S.; Gall, C.M. LTP consolidation: Substrates, explanatory power, and functional significance. Neuropharmacology 2007, 52, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Malenka, R.C. Understanding synapses: Past, present, and future. Neuron 2008, 60, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.S.; Newell-Litwa, K. Stem cell models of human synapse development and degeneration. Mol. Biol. Cell 2018, 29, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ziv, N.E.; Smith, S.J. Evidence for a Role of Dendritic Filopodia in Synaptogenesis and Spine Formation. Neuron 1996, 17, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Bonhoeffer, T.; Yuste, R. Spine Motility. Neuron 2002, 35, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Yuste, R.; Bonhoeffer, T. Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 2004, 5, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.; Pozzo-Miller, L. Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 2014, 601, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef]
- Forrest, M.P.; Parnell, E.; Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 2018, 19, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Emberti Gialloreti, L.; Mazzone, L.; Benvenuto, A.; Fasano, A.; Alcon, A.G.; Kraneveld, A.; Moavero, R.; Raz, R.; Riccio, M.P.; Siracusano, M.; et al. Risk and Protective Environmental Factors Associated with Autism Spectrum Disorder: Evidence-Based Principles and Recommendations. J. Clin. Med. 2019, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Maloney, S.E.; Rogers, C.E.; Constantino, J.N. Antidepressants, Pregnancy, and Autism: Setting the Record(s) Straight. Am. J. Psychiatry 2020, 177, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Adjimann, T.S.; Argañaraz, C.V.; Soiza-Reilly, M. Serotonin-related rodent models of early-life exposure relevant for neurodevelopmental vulnerability to psychiatric disorders. Transl. Psychiatry 2021, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Porcel, F.; Green, D.; Khatri, N.; Harris, S.S.; May, W.L.; Lin, R.C.S.; Paul, I.A. Neonatal exposure of rats to antidepressants affects behavioral reactions to novelty and social interactions in a manner analogous to autistic spectrum disorders. Anat. Rec. 2011, 294, 1726–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croen, L.A.; Grether, J.K.; Yoshida, C.K.; Odouli, R.; Hendrick, V. Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch. Gen. Psychiatry 2011, 68, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, R.A.; Lee, L.-C.; Crum, R.M.; Zimmerman, A.W.; Hertz-Picciotto, I. Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics 2014, 133, e1241–e1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, Y.C.; Keskin-Arslan, E.; Acar, S.; Sozmen, K. Maternal SSRI discontinuation, use, psychiatric disorder and the risk of autism in children: A meta-analysis of cohort studies. Br. J. Clin. Pharmacol. 2017, 83, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Ames, J.L.; Ladd-Acosta, C.; Fallin, M.D.; Qian, Y.; Schieve, L.A.; DiGuiseppi, C.; Lee, L.-C.; Kasten, E.P.; Zhou, G.; Pinto-Martin, J.; et al. Maternal Psychiatric Conditions, Treatment With Selective Serotonin Reuptake Inhibitors, and Neurodevelopmental Disorders. Biol. Psychiatry 2021, 90, 253–262. [Google Scholar] [CrossRef]
- Janecka, M.; Kodesh, A.; Levine, S.Z.; Lusskin, S.I.; Viktorin, A.; Rahman, R.; Buxbaum, J.D.; Schlessinger, A.; Sandin, S.; Reichenberg, A. Association of Autism Spectrum Disorder With Prenatal Exposure to Medication Affecting Neurotransmitter Systems. JAMA Psychiatry 2018, 75, 1217–1224. [Google Scholar] [CrossRef]
- Molenaar, N.M.; Bais, B.; Lambregtse-van den Berg, M.P.; Mulder, C.L.; Howell, E.A.; Fox, N.S.; Rommel, A.-S.; Bergink, V.; Kamperman, A.M. The international prevalence of antidepressant use before, during, and after pregnancy: A systematic review and meta-analysis of timing, type of prescriptions and geographical variability. J. Affect. Disord. 2020, 264, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Latendresse, G.; Elmore, C.; Deneris, A. Selective Serotonin Reuptake Inhibitors as First-Line Antidepressant Therapy for Perinatal Depression. J. Midwifery Womens. Health 2017, 62, 317–328. [Google Scholar] [CrossRef]
- Zafeiriou, D.; Ververi, A.; Vargiami, E. The Serotonergic System: Its Role in Pathogenesis and Early Developmental Treatment of Autism. Curr. Neuropharmacol. 2009, 7, 150. [Google Scholar] [CrossRef]
- Ciranna, L. Serotonin as a Modulator of Glutamate-and GABA-Mediated Neurotrans-mission: Implications in Physiological Functions and in Pathology. Curr. Neuropharmacol. 2006, 4, 101–114. [Google Scholar] [CrossRef] [Green Version]
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther 2015, 150, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, M.V.; Gulledge, A.T. Serotonin and prefrontal cortex function: Neurons, networks, and circuits. Mol. Neurobiol. 2011, 44, 449–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; Mortensen, O.V. Overview of Monoamine Transporters. Curr. Protoc. Pharmacol. 2017, 79, 12.16.1–12.16.17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Law, C.J.; Reith, M.E.A.; Wang, D.-N. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 2009, 16, 652–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, H.; Feng, N.; Wang, L.; Wang, X. Inhibitory effects of antidepressant fluoxetine on cloned Kv2.1 potassium channel expressed in HEK293 cells. Eur. J. Pharmacol. 2020, 878, 173097. [Google Scholar] [CrossRef] [PubMed]
- Warkus, E.L.L.; Marikawa, Y. Fluoxetine Inhibits Canonical Wnt Signaling to Impair Embryoid Body Morphogenesis: Potential Teratogenic Mechanisms of a Commonly Used Antidepressant. Toxicol. Sci. 2018, 165, 372–388. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Zhang, J.; Kim, H.; Tong, C.; Ying, Q.; Li, Z.; Mao, X.; Shi, G.; Yan, J.; Zhang, Z.; et al. Fluoxetine Regulates Neurogenesis In Vitro Through Modulation of GSK-3β/β-Catenin Signaling. Int. J. Neuropsychopharmacol. 2015, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Slavin, I.; Dea, S.; Arunkumar, P.; Sodhi, N.; Montefusco, S.; Siqueira-Neto, J.; Seelke, J.; Lofstrom, M.A.; Anson, B.; Zanella, F.; et al. Human iPSC-Derived 2D and 3D Platforms for Rapidly Assessing Developmental, Functional, and Terminal Toxicities in Neural Cells. Int. J. Mol. Sci 2021, 22, 1908. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.J.F.; Boulle, F.; Emerit, M.B.; Poilbout, C.; Steinbusch, H.W.M.; Van den Hove, D.L.A.; Kenis, G.; Lanfumey, L. 5-HTT independent effects of fluoxetine on neuroplasticity. Sci. Rep. 2019, 9, 6311. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-K.; Peng, C.-C.; Maner, R.S.; Zulkefli, N.D.; Huang, S.-M.; Hsieh, C.-L. Geniposide ameliorated fluoxetine-suppressed neurite outgrowth in Neuro2a neuroblastoma cells. Life Sci. 2019, 226, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Luk, C.; Richard, M.P.; Zaidi, W.; Farkas, S.; Getz, A.; Lee, A.; van Minnen, J.; Syed, N.I. Antidepressant fluoxetine suppresses neuronal growth from both vertebrate and invertebrate neurons and perturbs synapse formation between Lymnaea neurons. Eur. J. Neurosci. 2010, 31, 994–1005. [Google Scholar] [CrossRef]
- Zhong, X.; Harris, G.; Smirnova, L.; Zufferey, V.; Sá, R.d.C.; Baldino Russo, F.; Baleeiro Beltrao Braga, P.C.; Chesnut, M.; Zurich, M.-G.; Hogberg, H.T.; et al. Antidepressant Paroxetine Exerts Developmental Neurotoxicity in an iPSC-Derived 3D Human Brain Model. Front. Cell. Neurosci. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbom, L.J.; Rudisill, T.L.; Michel, N.; Litwa, K.A.; Beenhakker, M.P.; McConnell, M.J. The effect of rho kinase inhibition on morphological and electrophysiological maturity in iPSC-derived neurons. Cell Tissue Res. 2019, 375, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.; Rudisill, T.; Kirk, B.; Johnson, C.; Kemper, P.; Newell-Litwa, K. Cytoskeletal regulation of synaptogenesis in a model of human fetal brain development. J. Neurosci. Res. 2020, 98, 2148–2165. [Google Scholar] [CrossRef]
- Karson, C.N.; Newton, J.E.; Livingston, R.; Jolly, J.B.; Cooper, T.B.; Sprigg, J.; Komoroski, R.A. Human brain fluoxetine concentrations. J. Neuropsychiatry Clin. Neurosci. 1993, 5, 322–329. [Google Scholar]
- Amsterdam, J.D.; Fawcett, J.; Quitkin, F.M.; Reimherr, F.W.; Rosenbaum, J.F.; Michelson, D.; Hornig-Rohan, M.; Beasley, C.M. Fluoxetine and norfluoxetine plasma concentrations in major depression: A multicenter study. Am. J. Psychiatry 1997, 154, 963–969. [Google Scholar]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Solek, P.; Koszla, O.; Mytych, J.; Badura, J.; Chelminiak, Z.; Cuprys, M.; Fraczek, J.; Tabecka-Lonczynska, A.; Koziorowski, M. Neuronal life or death linked to depression treatment: The interplay between drugs and their stress-related outcomes relate to single or combined drug therapies. Apoptosis 2019, 24, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Jeong, Y.J.; Yu, A.-R.; Yoon, K.-S.; Choe, W.; Ha, J.; Kim, S.S.; Yeo, E.-J.; Kang, I. Fluoxetine induces apoptosis through endoplasmic reticulum stress via mitogen-activated protein kinase activation and histone hyperacetylation in SK-N-BE(2)-M17 human neuroblastoma cells. Apoptosis 2017, 22, 1079–1097. [Google Scholar] [CrossRef] [PubMed]
- Mun, A.-R.; Lee, S.-J.; Kim, G.-B.; Kang, H.-S.; Kim, J.-S.; Kim, S.-J. Fluoxetine-induced apoptosis in hepatocellular carcinoma cells. Anticancer Res. 2013, 33, 3691–3697. [Google Scholar] [PubMed]
- Chen, W.-T.; Hsu, F.-T.; Liu, Y.-C.; Chen, C.-H.; Hsu, L.-C.; Lin, S.-S. Fluoxetine induces apoptosis through extrinsic/intrinsic pathways and inhibits ERK/NF-κB-modulated anti-apoptotic and invasive potential in hepatocellular carcinoma cells in vitro. Int. J. Mol. Sci. 2019, 20, 757. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.S.; Schnellmann, R.G. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2004, 12, 129–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.A. Is there a large sample size problem? Ophthalmic Physiol. Opt. 2019, 39, 129–130. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.; Knudson, W.; Newell-Litwa, K. Hyaluronan regulates synapse formation and function in developing neural networks. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Papariello, A.; Taylor, D.; Soderstrom, K.; Litwa, K. CB(1) antagonism increases excitatory synaptogenesis in a cortical spheroid model of fetal brain development. Sci. Rep. 2021, 11, 9356. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Petit, E.I.; Dobrenis, K.; Sze, J.Y. Spatiotemporal SERT expression in cortical map development. Neurochem. Int. 2016, 98, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrand, C.; Cases, O.; Wehrlé, R.; Blakely, R.D.; Edwards, R.H.; Gaspar, P. Transient developmental expression of monoamine transporters in the rodent forebrain. J. Comp. Neurol. 1998, 401, 506–524. [Google Scholar] [CrossRef]
- Hansson, S.R.; Mezey, É.; Hoffman, B.J. Serotonin transporter messenger RNA in the developing rat brain: Early expression in serotonergic neurons and transient expression in non-serotonergic neurons. Neuroscience 1998, 83, 1185–1201. [Google Scholar] [CrossRef]
- Carson, M.J.; Thomas, E.A.; Danielson, P.E.; Sutcliffe, J.G. The 5HT5A serotonin receptor is expressed predominantly by astrocytes in which it inhibits cAMP accumulation: A mechanism for neuronal suppression of reactive astrocytes. Glia 1996, 17, 317–326. [Google Scholar] [CrossRef]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Alme, M.N.; Wibrand, K.; Dagestad, G.; Bramham, C.R. Chronic fluoxetine treatment induces brain region-specific upregulation of genes associated with BDNF-induced long-term potentiation. Neural Plast. 2007, 2007, 26496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molteni, R.; Calabrese, F.; Bedogni, F.; Tongiorgi, E.; Fumagalli, F.; Racagni, G.; Andrea Riva, M. Chronic treatment with fluoxetine up-regulates cellular BDNF mRNA expression in rat dopaminergic regions. Int. J. Neuropsychopharmacol. 2006, 9, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, C.; Castrén, E.; Kasper, S.; Lanzenberger, R. Serotonin and neuroplasticity—Links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev. 2017, 77, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansch, C.; Ziegler, G.C.; Forero, A.; Gredy, S.; Wäldchen, S.; Vitale, M.R.; Svirin, E.; Zöller, J.E.M.; Waider, J.; Günther, K.; et al. Serotonin-specific neurons differentiated from human iPSCs form distinct subtypes with synaptic protein assembly. J. Neural Transm. 2021, 128, 225–241. [Google Scholar] [CrossRef]
- Lu, J.; Zhong, X.; Liu, H.; Hao, L.; Huang, C.T.-L.; Sherafat, M.A.; Jones, J.; Ayala, M.; Li, L.; Zhang, S.-C. Generation of serotonin neurons from human pluripotent stem cells. Nat. Biotechnol. 2016, 34, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.; Haupt, A.; Tucker, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Nelson, A.; Hookway, C.; Ludmann, S.A.; Mueller, I.A.; et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol. Biol. Cell 2017, 28, 2854–2874. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tate, K.; Kirk, B.; Tseng, A.; Ulffers, A.; Litwa, K. Effects of the Selective Serotonin Reuptake Inhibitor Fluoxetine on Developing Neural Circuits in a Model of the Human Fetal Cortex. Int. J. Mol. Sci. 2021, 22, 10457. https://doi.org/10.3390/ijms221910457
Tate K, Kirk B, Tseng A, Ulffers A, Litwa K. Effects of the Selective Serotonin Reuptake Inhibitor Fluoxetine on Developing Neural Circuits in a Model of the Human Fetal Cortex. International Journal of Molecular Sciences. 2021; 22(19):10457. https://doi.org/10.3390/ijms221910457
Chicago/Turabian StyleTate, Kinsley, Brenna Kirk, Alisia Tseng, Abigail Ulffers, and Karen Litwa. 2021. "Effects of the Selective Serotonin Reuptake Inhibitor Fluoxetine on Developing Neural Circuits in a Model of the Human Fetal Cortex" International Journal of Molecular Sciences 22, no. 19: 10457. https://doi.org/10.3390/ijms221910457
APA StyleTate, K., Kirk, B., Tseng, A., Ulffers, A., & Litwa, K. (2021). Effects of the Selective Serotonin Reuptake Inhibitor Fluoxetine on Developing Neural Circuits in a Model of the Human Fetal Cortex. International Journal of Molecular Sciences, 22(19), 10457. https://doi.org/10.3390/ijms221910457