
Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
Clinical Observations and Synaptic Pathologies of Neuropsychiatric Disorders
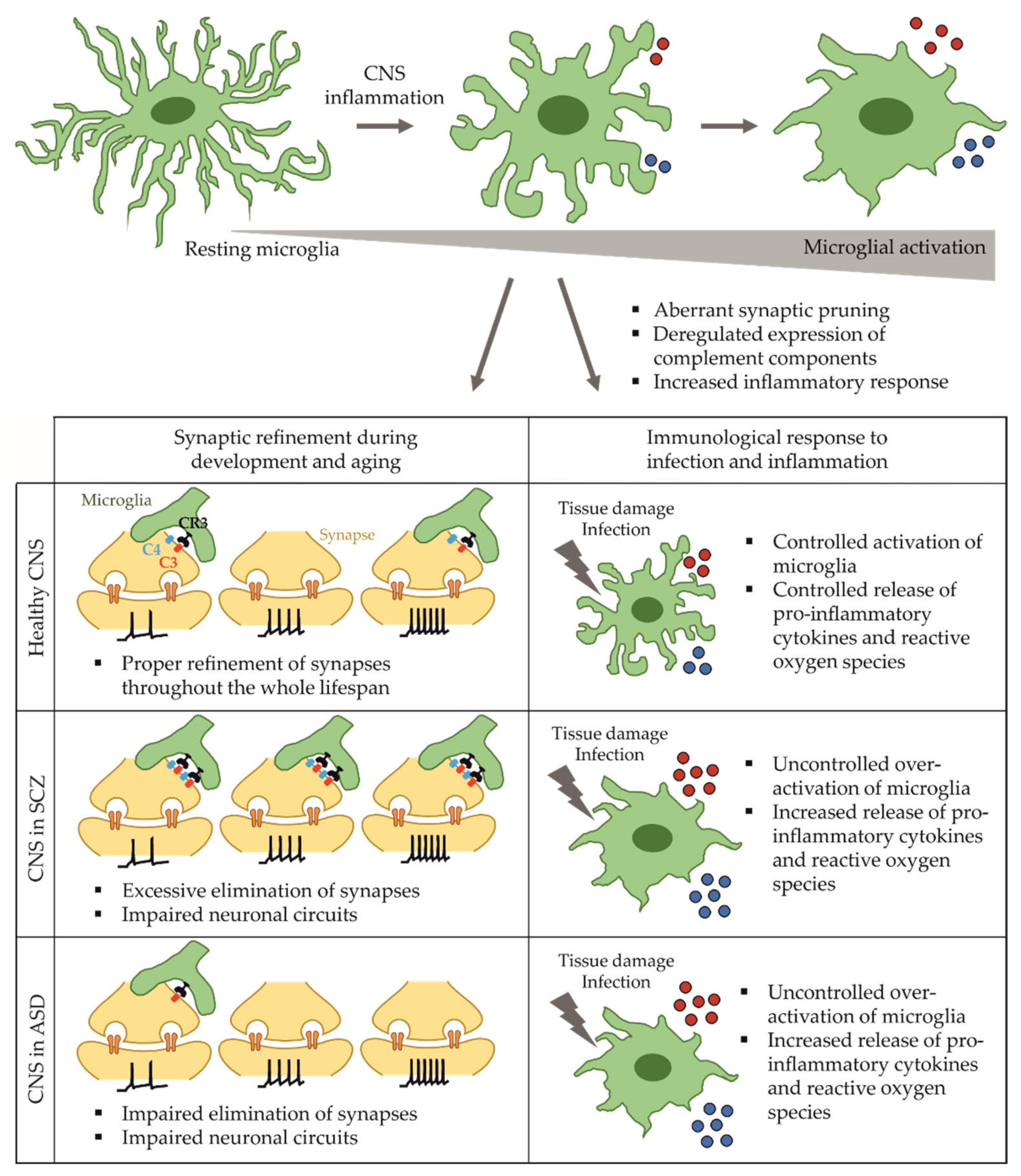
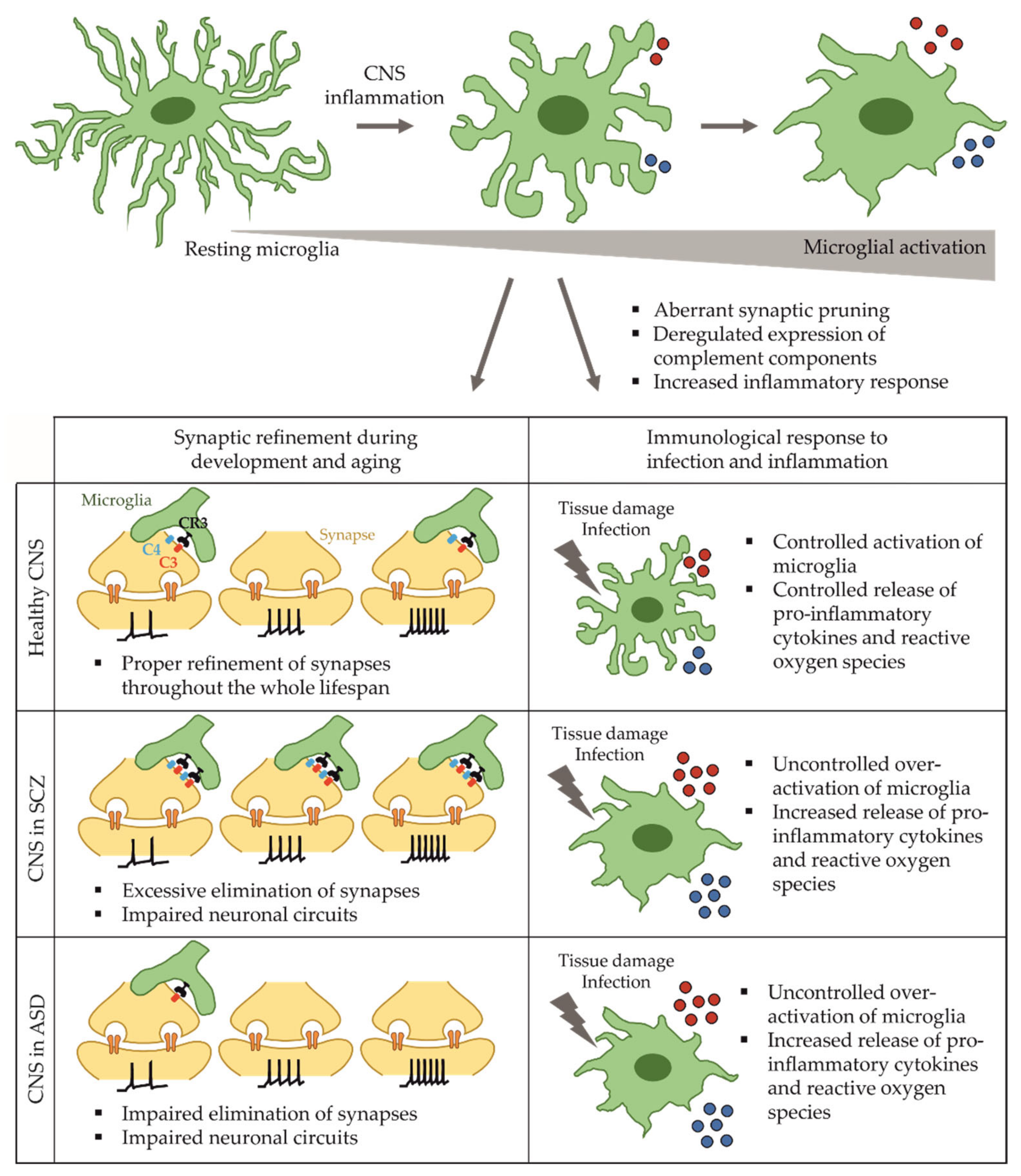
2. Microglia in Health and Disease
2.1. Functions of Microglia in the Developing and Adult Brain
2.2. Microglia in Schizophrenia
2.3. Microglia-Based Models for Schizophrenia
2.4. Microglia Dysfunction in Autism Spectrum Disorder
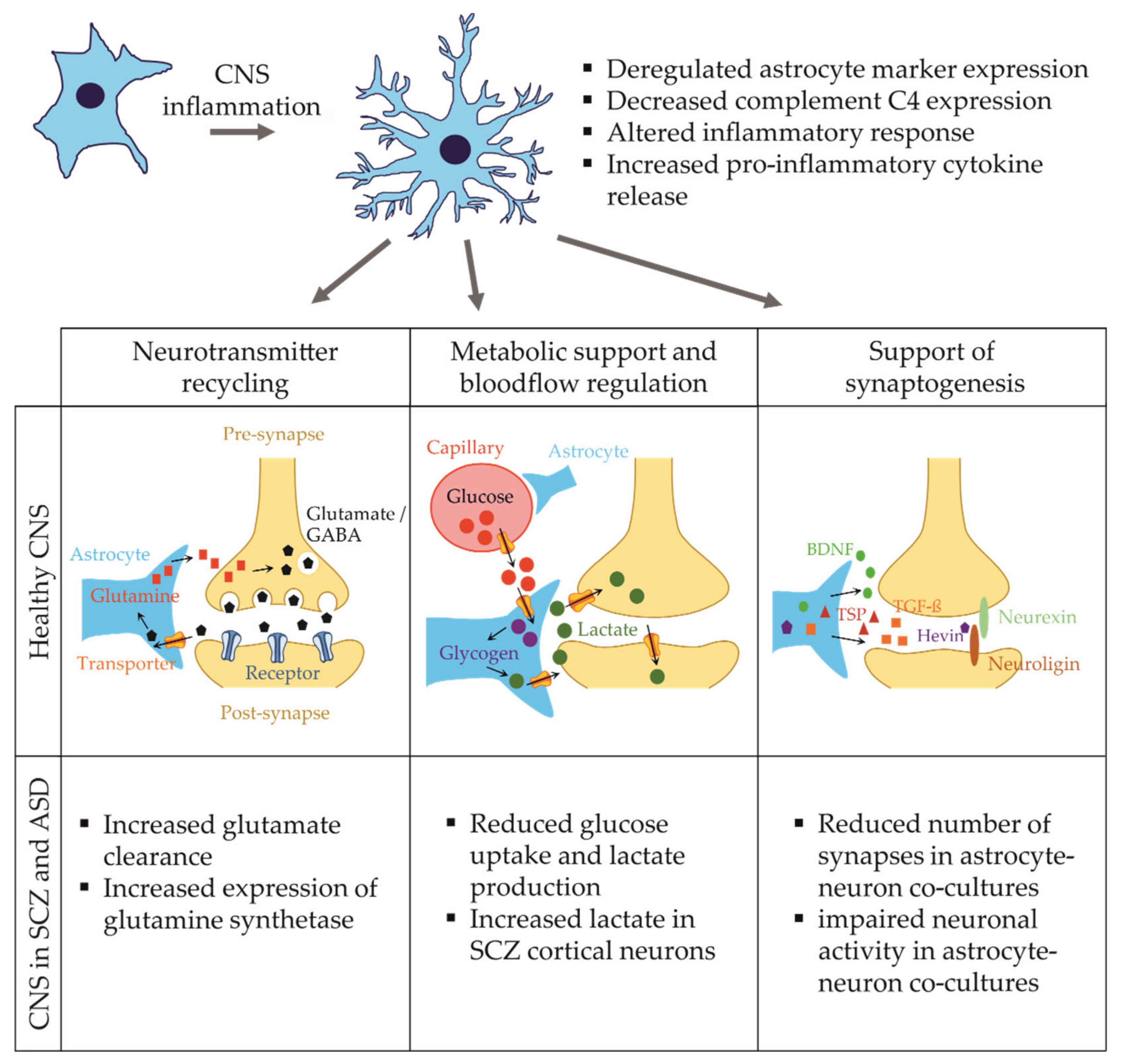
3. Astrocytes in Health and Disease
3.1. Functions of Astrocytes in the Developing and Adult Brain
3.2. Astrocytes in Schizophrenia
3.3. Astrocyte-Based Models for Schizophrenia
3.4. Astrocyte Dysfunction in Autism Spectrum Disorder
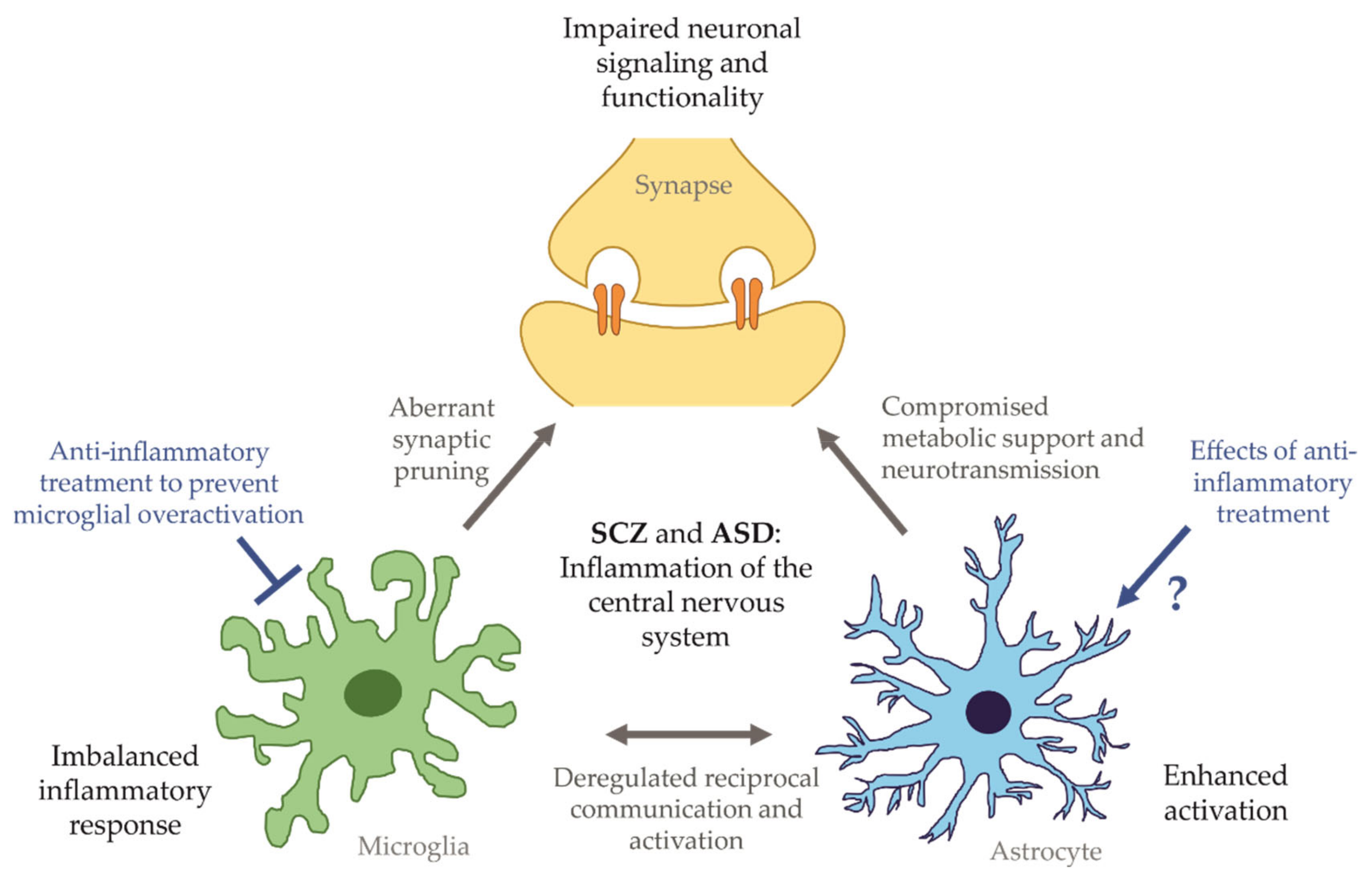
4. Lessons from iPSC Models for Research and Drug Development
4.1. Multi-Culture Models to Study Reciprocal Cross-Talk in Diseases of the Central Nervous System
4.2. Opportunities and Limitations of iPSC-Based Models for Drug Development
4.3. Brain Organoids to Study 3D Neuron-Glia Interactions In Vitro
4.4. Brain Organoids for Neuropsychiatric Research and Drug Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Kessler, R.C.; Birnbaum, H.; Demler, O.; Falloon, I.R.; Gagnon, E.; Guyer, M.; Howes, M.J.; Kendler, K.S.; Shi, L.; Walters, E.; et al. The prevalence and correlates of nonaffective psychosis in the national comorbidity survey replication (ncs-r). Biol. Psychiatry 2005, 58, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, united states, 2014. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Picchioni, M.M.; Murray, R.M. Schizophrenia. BMJ 2007, 335, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R.; Pallagrosi, M. Autism spectrum disorders and schizophrenia spectrum disorders: Excitation/inhibition imbalance and developmental trajectories. Front. Psychiatry 2017, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Minshew, N.J.; Allen, D.N.; Seaton, B.E. High-functioning autism and schizophrenia: A comparison of an early and late onset neurodevelopmental disorder. Arch. Clin. Neuropsychol. 2002, 17, 461–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordjman, S.; Celume, M.P.; Denis, L.; Motillon, T.; Keromnes, G. Reframing schizophrenia and autism as bodily self-consciousness disorders leading to a deficit of theory of mind and empathy with social communication impairments. Neurosci. Biobehav. Rev. 2019, 103, 401–413. [Google Scholar] [CrossRef]
- Carbon, M.; Correll, C.U. Thinking and acting beyond the positive: The role of the cognitive and negative symptoms in schizophrenia. CNS Spectr. 2014, 19 (Suppl. S1), 38–52. [Google Scholar] [CrossRef]
- Li, R.; Ma, X.; Wang, G.; Yang, J.; Wang, C. Why sex differences in schizophrenia? J. Transl. Neurosci. 2016, 1, 37–42. [Google Scholar]
- Johnson, C.P.; Myers, S.M. Identification and evaluation of children with autism spectrum disorders. Pediatrics 2007, 120, 1183–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child. Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glausier, J.R.; Lewis, D.A. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef] [Green Version]
- Dienel, S.J.; Lewis, D.A. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Grunwald, L.M.; Stock, R.; Haag, K.; Buckenmaier, S.; Eberle, M.C.; Wildgruber, D.; Storchak, H.; Kriebel, M.; Weissgraeber, S.; Mathew, L.; et al. Comparative characterization of human induced pluripotent stem cells (hipsc) derived from patients with schizophrenia and autism. Transl. Psychiatry 2019, 9, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, X.; Wu, H.; Xun, G.; Ou, J.; Zhang, Q.; Li, H.; Bai, T.; Hu, Z.; Zou, X.; et al. Genotype and phenotype correlations for shank3 de novo mutations in neurodevelopmental disorders. Am. J. Med. Genet. Part A 2018, 176, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.; Beltrao-Braga, P.; Trujillo, C.A.; Mendes, A.P.D.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef]
- Laskaris, L.E.; Di Biase, M.A.; Everall, I.; Chana, G.; Christopoulos, A.; Skafidas, E.; Cropley, V.L.; Pantelis, C. Microglial activation and progressive brain changes in schizophrenia. Br. J. Pharmacol. 2016, 173, 666–680. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Onwordi, E.C.; Halff, E.F.; Whitehurst, T.; Mansur, A.; Cotel, M.C.; Wells, L.; Creeney, H.; Bonsall, D.; Rogdaki, M.; Shatalina, E.; et al. Synaptic density marker sv2a is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat. Commun. 2020, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.; Bonoldi, I.; Kalk, N.; Turkheimer, F.; et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: An [(11)c]pbr28 pet brain imaging study. Am. J. Psychiatry 2016, 173, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Stremmel, C.; Schuchert, R.; Wagner, F.; Thaler, R.; Weinberger, T.; Pick, R.; Mass, E.; Ishikawa-Ankerhold, H.C.; Margraf, A.; Hutter, S.; et al. Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat. Commun. 2018, 9, 75. [Google Scholar] [CrossRef]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Bruttger, J.; Karram, K.; Wortge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-cell rna sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates cns synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.A.; Kun, J.; Torok, G.; Bulyaki, E.; Borhegyi, Z.; Gulyassy, P.; Kis, V.; Szocsics, P.; Micsonai, A.; Matko, J.; et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. USA 2018, 115, 6303–6308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormel, P.R.; Bottcher, C.; Gigase, F.A.J.; Missall, R.D.; van Zuiden, W.; Fernandez Zapata, M.C.; Ilhan, D.; de Goeij, M.; Udine, E.; Sommer, I.E.C.; et al. A characterization of the molecular phenotype and inflammatory response of schizophrenia patient-derived microglia-like cells. Brain Behav. Immun. 2020, 90, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.K.; Miller, B.J. Meta-analysis of cerebrospinal fluid cytokine and tryptophan catabolite alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder, and depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Bechter, K.; Reiber, H.; Herzog, S.; Fuchs, D.; Tumani, H.; Maxeiner, H.G. Cerebrospinal fluid analysis in affective and schizophrenic spectrum disorders: Identification of subgroups with immune responses and blood-csf barrier dysfunction. J. Psychiatry Res. 2010, 44, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pahlajani, S.; De Sanctis, V.; Stern, J.N.H.; Najjar, A.; Chong, D. Neurovascular unit dysfunction and blood-brain barrier hyperpermeability contribute to schizophrenia neurobiology: A theoretical integration of clinical and experimental evidence. Front. Psychiatry 2017, 8, 83. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondelli, V.; Di Forti, M.; Morgan, B.P.; Murray, R.M.; Pariante, C.M.; Dazzan, P. Baseline high levels of complement component 4 predict worse clinical outcome at 1-year follow-up in first-episode psychosis. Brain Behav. Immun. 2020, 88, 913–915. [Google Scholar] [CrossRef]
- Kim, H.S.; Suh, Y.H. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009, 196, 168–179. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, Z.; Li, Z.; Yang, Y.; Zheng, Z.; Li, Y.; Xie, Y.; Li, Z. Minocycline as adjunct therapy for a male patient with deficit schizophrenia. Neuropsychiatr. Dis Treat. 2018, 14, 2697–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levkovitz, Y.; Mendlovich, S.; Riwkes, S.; Braw, Y.; Levkovitch-Verbin, H.; Gal, G.; Fennig, S.; Treves, I.; Kron, S. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J. Clin. Psychiatry 2010, 71, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Sommer, I.E.; van Westrhenen, R.; Begemann, M.J.; de Witte, L.D.; Leucht, S.; Kahn, R.S. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: An update. Schizophr. Bull. 2014, 40, 181–191. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique tgf-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janabi, N.; Peudenier, S.; Heron, B.; Ng, K.H.; Tardieu, M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the sv40 large t antigen. Neurosci. Lett. 1995, 195, 105–108. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Coburn, M.; Tu, C.H.; Hasselmann, J.; Davtyan, H.; Blurton-Jones, M. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegener. 2018, 13, 67. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [Green Version]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Hung, Y.S.; Fuh, J.L.; Chen, N.J.; Chu, Y.S.; Chen, S.C.; Fann, M.J.; Wong, Y.H. Efficient conversion of human induced pluripotent stem cells into microglia by defined transcription factors. Stem Cell Rep. 2021, 16, 1363–1380. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. Ipsc-derived human microglia-like cells to study neurological diseases. Neuron 2017, 94, 278–293.e279. [Google Scholar] [CrossRef] [Green Version]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46. [Google Scholar] [CrossRef]
- Wurm, J.; Konttinen, H.; Andressen, C.; Malm, T.; Spittau, B. Microglia development and maturation and its implications for induction of microglia-like cells from human ipscs. Int. J. Mol. Sci. 2021, 22, 3088. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Sheridan, S.D.; Gracias, J.; Xuan, D.; Fu, T.; Perlis, R.H. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 2017, 22, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Abramson, I.; Semendeferi, K.; Courchesne, E.; Everall, I.P. Abnormal microglial-neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 2012, 1456, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Ansel, A.; Rosenzweig, J.P.; Zisman, P.D.; Melamed, M.; Gesundheit, B. Variation in gene expression in autism spectrum disorders: An extensive review of transcriptomic studies. Front. Neurosci. 2016, 10, 601. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.P.; Burger, R.A.; Odell, D.; Torres, A.R.; Warren, W.L. Decreased plasma concentrations of the c4b complement protein in autism. Arch. Pediatr. Adolesc. Med. 1994, 148, 180–183. [Google Scholar] [CrossRef]
- Odell, D.; Maciulis, A.; Cutler, A.; Warren, L.; McMahon, W.M.; Coon, H.; Stubbs, G.; Henley, K.; Torres, A. Confirmation of the association of the c4b null allelle in autism. Hum. Immunol. 2005, 66, 140–145. [Google Scholar] [CrossRef]
- Eftekharian, M.M.; Ghafouri-Fard, S.; Noroozi, R.; Omrani, M.D.; Arsang-Jang, S.; Ganji, M.; Gharzi, V.; Noroozi, H.; Komaki, A.; Mazdeh, M.; et al. Cytokine profile in autistic patients. Cytokine 2018, 108, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, F.; Quintana, F.J. The role of astrocytes in cns inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The transcription factor nfia controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molofsky, A.V.; Deneen, B. Astrocyte development: A guide for the perplexed. Glia 2015, 63, 1320–1329. [Google Scholar] [CrossRef]
- Nixdorf-Bergweiler, B.E.; Albrecht, D.; Heinemann, U. Developmental changes in the number, size, and orientation of gfap-positive cells in the ca1 region of rat hippocampus. Glia 1994, 12, 180–195. [Google Scholar] [CrossRef]
- Allen, N.J. Role of glia in developmental synapse formation. Curr. Opin. Neurobiol. 2013, 23, 1027–1033. [Google Scholar] [CrossRef]
- Fan, S.; Gangwar, S.P.; Machius, M.; Rudenko, G. Interplay between hevin, sparc, and mdgas: Modulators of neurexin-neuroligin transsynaptic bridges. Structure 2021, 29, 664–678.e666. [Google Scholar] [CrossRef] [PubMed]
- Sultan, S.; Li, L.; Moss, J.; Petrelli, F.; Casse, F.; Gebara, E.; Lopatar, J.; Pfrieger, F.W.; Bezzi, P.; Bischofberger, J.; et al. Synaptic integration of adult-born hippocampal neurons is locally controlled by astrocytes. Neuron 2015, 88, 957–972. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, S.J.; MacVicar, B.A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Min, R.; van der Knaap, M.S. Genetic defects disrupting glial ion and water homeostasis in the brain. Brain Pathol. 2018, 28, 372–387. [Google Scholar] [CrossRef] [Green Version]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through megf10 and mertk pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Haydon, P.G.; Nedergaard, M. How do astrocytes participate in neural plasticity? Cold Spring Harb. Perspect. Biol. 2014, 7, a020438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; Steve White, H. Glutamate and gaba synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef]
- Spangaro, M.; Bosia, M.; Zanoletti, A.; Bechi, M.; Cocchi, F.; Pirovano, A.; Lorenzi, C.; Bramanti, P.; Benedetti, F.; Smeraldi, E.; et al. Cognitive dysfunction and glutamate reuptake: Effect of eaat2 polymorphism in schizophrenia. Neurosci. Lett. 2012, 522, 151–155. [Google Scholar] [CrossRef]
- Roussos, P.; Giakoumaki, S.G.; Adamaki, E.; Georgakopoulos, A.; Robakis, N.K.; Bitsios, P. The association of schizophrenia risk d-amino acid oxidase polymorphisms with sensorimotor gating, working memory and personality in healthy males. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2011, 36, 1677–1688. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Kim, S.K.; Kim, J.W.; Kang, W.S.; Chung, J.H. Association of thrombospondin 1 gene with schizophrenia in korean population. Mol. Biol. Rep. 2012, 39, 6875–6880. [Google Scholar] [CrossRef]
- Liu, J.; Shi, Y.; Tang, J.; Guo, T.; Li, X.; Yang, Y.; Chen, Q.; Zhao, X.; He, G.; Feng, G.; et al. Snps and haplotypes in the s100b gene reveal association with schizophrenia. Biochem. Biophys. Res. Commun. 2005, 328, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Akamatsu, W.; Okada, Y.; Ohnishi, T.; Balan, S.; Hisano, Y.; Iwayama, Y.; Toyota, T.; Matsumoto, T.; Itasaka, N.; et al. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl. Psychiatry 2016, 6, e934. [Google Scholar] [CrossRef] [Green Version]
- Steffek, A.E.; McCullumsmith, R.E.; Haroutunian, V.; Meador-Woodruff, J.H. Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr. Res. 2008, 103, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Barley, K.; Dracheva, S.; Byne, W. Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr. Res. 2009, 112, 54–64. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in schizophrenia: Pathogenetic aspects and therapeutic considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Pakkenberg, B. Pronounced reduction of total neuron number in mediodorsal thalamic nucleus and nucleus accumbens in schizophrenics. Arch. Gen. Psychiatry 1990, 47, 1023–1028. [Google Scholar] [CrossRef]
- Williams, M.R.; Marsh, R.; Macdonald, C.D.; Jain, J.; Pearce, R.K.; Hirsch, S.R.; Ansorge, O.; Gentleman, S.M.; Maier, M. Neuropathological changes in the nucleus basalis in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 485–495. [Google Scholar] [CrossRef]
- Williams, M.R.; Hampton, T.; Pearce, R.K.; Hirsch, S.R.; Ansorge, O.; Thom, M.; Maier, M. Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Galvin, K.; O’Sullivan, B.; MacDonald, C.D.; Ching, E.W.; Turkheimer, F.; Howes, O.D.; Pearce, R.K.; Hirsch, S.R.; Maier, M. Neuropathological changes in the substantia nigra in schizophrenia but not depression. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Bruton, C.J.; Crow, T.J.; Frith, C.D.; Johnstone, E.C.; Owens, D.G.; Roberts, G.W. Schizophrenia and the brain: A prospective clinico-neuropathological study. Psychol. Med. 1990, 20, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Ao, Y.; Sofroniew, M.V. Heterogeneity of reactive astrocytes. Neurosci. Lett. 2014, 565, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.R. Neuropathology of schizophrenia. Arch. Gen. Psychiatry 1982, 39, 1131–1139. [Google Scholar] [CrossRef]
- Kolomeets, N.S. [Astroglia of the hippocampus in schizophrenia]. Zhurnal Nevrologii I Psikhiatrii Imeni S.S. Korsakova 2008, 108, 70–76. [Google Scholar]
- Krencik, R.; Weick, J.P.; Liu, Y.; Zhang, Z.-J.; Zhang, S.-C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Russo, F.B.; Freitas, B.C.; Pignatari, G.C.; Fernandes, I.R.; Sebat, J.; Muotri, A.R.; Beltrão-Braga, P.C.B. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol. Psychiatry 2018, 83, 569–578. [Google Scholar] [CrossRef]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, M.; Kondo, T.; Inoue, H. Modeling neurological disorders with human pluripotent stem cell-derived astrocytes. Int. J. Mol. Sci. 2019, 20, 3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef]
- McGivern, J.V.; Patitucci, T.N.; Nord, J.A.; Barabas, M.A.; Stucky, C.L.; Ebert, A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 2013, 61, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of tdp-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human ipsc glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 2017, 21, 195–208.e196. [Google Scholar] [CrossRef]
- Liu, Z.; Osipovitch, M.; Benraiss, A.; Huynh, N.P.T.; Foti, R.; Bates, J.; Chandler-Militello, D.; Findling, R.L.; Tesar, P.J.; Nedergaard, M.; et al. Dysregulated glial differentiation in schizophrenia may be relieved by suppression of smad4- and rest-dependent signaling. Cell Rep. 2019, 27, 3832–3843.e3836. [Google Scholar] [CrossRef] [Green Version]
- Akkouh, I.A.; Hribkova, H.; Grabiec, M.; Budinska, E.; Szabo, A.; Kasparek, T.; Andreassen, O.A.; Sun, Y.M.; Djurovic, S. Derivation and molecular characterization of a morphological subpopulation of human ipsc astrocytes reveal a potential role in schizophrenia and clozapine response. Schizophr. Bull. 2021, sbab092. [Google Scholar] [CrossRef]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through disc1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Jouroukhin, Y.; Kageyama, Y.; Misheneva, V.; Shevelkin, A.; Andrabi, S.; Prandovszky, E.; Yolken, R.H.; Dawson, V.L.; Dawson, T.M.; Aja, S.; et al. Disc1 regulates lactate metabolism in astrocytes: Implications for psychiatric disorders. Transl. Psychiatry 2018, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Zhu, S.; Shevelkin, A.; Ross, C.A.; Pletnikov, M. Disc1, astrocytes and neuronal maturation: A possible mechanistic link with implications for mental disorders. J. Neurochem. 2016, 138, 518–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O’Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, ipscs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgkinson, C.A.; Goldman, D.; Jaeger, J.; Persaud, S.; Kane, J.M.; Lipsky, R.H.; Malhotra, A.K. Disrupted in schizophrenia 1 (disc1): Association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am. J. Hum. Genet. 2004, 75, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Nwulia, E.; Chang, J.; Balkissoon, R.; Ishizuka, K.; Chen, H.; Zandi, P.; McInnis, M.G.; Sawa, A. Differential expression of disrupted-in-schizophrenia (disc1) in bipolar disorder. Biol. Psychiatry 2006, 60, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Tondo, L.; Vázquez, G.H.; Baldessarini, R.J. Depression and mania in bipolar disorder. Curr. Neuropharmacol. 2017, 15, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Fries, G.R.; Walss-Bass, C.; Bauer, M.E.; Teixeira, A.L. Revisiting inflammation in bipolar disorder. Pharmacol. Biochem. Behav. 2019, 177, 12–19. [Google Scholar] [CrossRef]
- Toker, L.; Mancarci, B.O.; Tripathy, S.; Pavlidis, P. Transcriptomic evidence for alterations in astrocytes and parvalbumin interneurons in subjects with bipolar disorder and schizophrenia. Biol. Psychiatry 2018, 84, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Vadodaria, K.C.; Mendes, A.P.D.; Mei, A.; Racha, V.; Erikson, G.; Shokhirev, M.N.; Oefner, R.; Heard, K.J.; McCarthy, M.J.; Eyler, L.; et al. Altered neuronal support and inflammatory response in bipolar disorder patient-derived astrocytes. Stem Cell Rep. 2021, 16, 825–835. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in autism spectrum disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Ahlsen, G.; Rosengren, L.; Belfrage, M.; Palm, A.; Haglid, K.; Hamberger, A.; Gillberg, C. Glial fibrillary acidic protein in the cerebrospinal fluid of children with autism and other neuropsychiatric disorders. Biol. Psychiatry 1993, 33, 734–743. [Google Scholar] [CrossRef]
- Liao, X.; Liu, Y.; Fu, X.; Li, Y. Postmortem studies of neuroinflammation in autism spectrum disorder: A systematic review. Mol. Neurobiol. 2020, 57, 3424–3438. [Google Scholar] [CrossRef]
- Mansur, F.; Teles e Silva, A.L.; Gomes, A.K.S.; Magdalon, J.; de Souza, J.S.; Griesi-Oliveira, K.; Passos-Bueno, M.R.; Sertié, A.L. Complement c4 is reduced in ipsc-derived astrocytes of autism spectrum disorder subjects. Int. J. Mol. Sci. 2021, 22, 7579. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.C.; Zhong, X.; Mohamed, A.; Li, R.; Liu, Y.; Dong, Q.; Ananiev, G.E.; Mok, J.C.C.; Lin, B.R.; Lu, J.; et al. Mutant astrocytes differentiated from rett syndrome patients-specific ipscs have adverse effects on wild-type neurons. Hum. Mol. Genet. 2014, 23, 2968–2980. [Google Scholar] [CrossRef]
- Dong, Q.; Kim, J.; Nguyen, L.; Bu, Q.; Chang, Q. An astrocytic influence on impaired tonic inhibition in hippocampal ca1 pyramidal neurons in a mouse model of rett syndrome. J. Neurosci. 2020, 40, 6250–6261. [Google Scholar] [CrossRef]
- Okabe, Y.; Takahashi, T.; Mitsumasu, C.; Kosai, K.-i.; Tanaka, E.; Matsuishi, T. Alterations of gene expression and glutamate clearance in astrocytes derived from an mecp2-null mouse model of rett syndrome. PLoS ONE 2012, 7, e35354. [Google Scholar]
- Ehinger, Y.; Matagne, V.; Cunin, V.; Borloz, E.; Seve, M.; Bourgoin-Voillard, S.; Borges-Correia, A.; Villard, L.; Roux, J.-C. Analysis of astroglial secretomic profile in the mecp2-deficient male mouse model of rett syndrome. Int. J. Mol. Sci. 2021, 22, 4316. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; Maussion, G.; Chaineau, M.; Sigutova, V.; Rouleau, G.; Durcan, T.M.; Stifani, S. Characterization of human ipsc-derived astrocytes with potential for disease modeling and drug discovery. Neurosci. Lett. 2020, 731, 135028. [Google Scholar] [CrossRef]
- Hyvärinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-stimulation with il-1β and tnf-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci. Rep. 2019, 9, 16944. [Google Scholar] [CrossRef]
- Drago, F.; Lombardi, M.; Prada, I.; Gabrielli, M.; Joshi, P.; Cojoc, D.; Franck, J.; Fournier, I.; Vizioli, J.; Verderio, C. Atp modifies the proteome of extracellular vesicles released by microglia and influences their action on astrocytes. Front. Pharmacol. 2017, 8, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model c3 production in alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- Li, J.; Ryan, S.K.; Deboer, E.; Cook, K.; Fitzgerald, S.; Lachman, H.M.; Wallace, D.C.; Goldberg, E.M.; Anderson, S.A. Mitochondrial deficits in human ipsc-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry 2019, 9, 302. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.X.; Di Giorgio, F.P.; Yao, J.; Marchetto, M.C.; Brennand, K.; Wright, R.; Mei, A.; McHenry, L.; Lisuk, D.; Grasmick, J.M.; et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2014, 2, 295–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.E.; Schrode, N.; Flaherty, E.; Brennand, K.J. New considerations for hipsc-based models of neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 49–66. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from escs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human es cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-organization of polarized cerebellar tissue in 3d culture of human pluripotent stem cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3d culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling zikv exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Pasca, S.P. Human astrocyte maturation captured in 3d cerebral cortical spheroids derived from pluripotent stem cells. Neuron 2017, 95, 779–790.e776. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Boreland, A.J.; Li, X.; Erickson, C.; Jin, M.; Atkins, C.; Pang, Z.P.; Daniels, B.P.; Jiang, P. Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modeling brain development and pathology. Stem Cell Rep. 2021, 16, 1923–1937. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Stachowiak, E.K.; Benson, C.A.; Narla, S.T.; Dimitri, A.; Chuye, L.E.B.; Dhiman, S.; Harikrishnan, K.; Elahi, S.; Freedman, D.; Brennand, K.J.; et al. Cerebral organoids reveal early cortical maldevelopment in schizophrenia-computational anatomy and genomics, role of fgfr1. Transl Psychiatry 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Kathuria, A.; Lopez-Lengowski, K.; Jagtap, S.S.; McPhie, D.; Perlis, R.H.; Cohen, B.M.; Karmacharya, R. Transcriptomic landscape and functional characterization of induced pluripotent stem cell-derived cerebral organoids in schizophrenia. JAMA Psychiatry 2020, 77, 745–754. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. Foxg1-dependent dysregulation of gaba/glutamate neuron differentiation in autism spectrum disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. Crispr/cas9-mediated heterozygous knockout of the autism gene chd8 and characterization of its transcriptional networks in cerebral organoids derived from ips cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaduri, A.; Andrews, M.G.; Mancia Leon, W.; Jung, D.; Shin, D.; Allen, D.; Jung, D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef]
- Gordon, A.; Yoon, S.J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Nzou, G.; Wicks, R.T.; Wicks, E.E.; Seale, S.A.; Sane, C.H.; Chen, A.; Murphy, S.V.; Jackson, J.D.; Atala, A.J. Human cortex spheroid with a functional blood brain barrier for high-throughput neurotoxicity screening and disease modeling. Sci. Rep. 2018, 8, 7413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, H.; Grabos, M.; Becker, K.J.; Kagermeier, T.E.; Wu, J.; Otto, M.; Peischard, S.; Zeuschner, D.; TsyTsyura, Y.; Disse, P.; et al. A fully automated high-throughput workflow for 3d-based chemical screening in human midbrain organoids. Elife 2020, 9, e52904. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heider, J.; Vogel, S.; Volkmer, H.; Breitmeyer, R. Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development. Int. J. Mol. Sci. 2021, 22, 10254. https://doi.org/10.3390/ijms221910254
Heider J, Vogel S, Volkmer H, Breitmeyer R. Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development. International Journal of Molecular Sciences. 2021; 22(19):10254. https://doi.org/10.3390/ijms221910254
Chicago/Turabian StyleHeider, Johanna, Sabrina Vogel, Hansjürgen Volkmer, and Ricarda Breitmeyer. 2021. "Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development" International Journal of Molecular Sciences 22, no. 19: 10254. https://doi.org/10.3390/ijms221910254
APA StyleHeider, J., Vogel, S., Volkmer, H., & Breitmeyer, R. (2021). Human iPSC-Derived Glia as a Tool for Neuropsychiatric Research and Drug Development. International Journal of Molecular Sciences, 22(19), 10254. https://doi.org/10.3390/ijms221910254