
The Multiple Faces of Integrin–ECM Interactions in Inflammatory Bowel Disease



Abstract
:1. Introduction
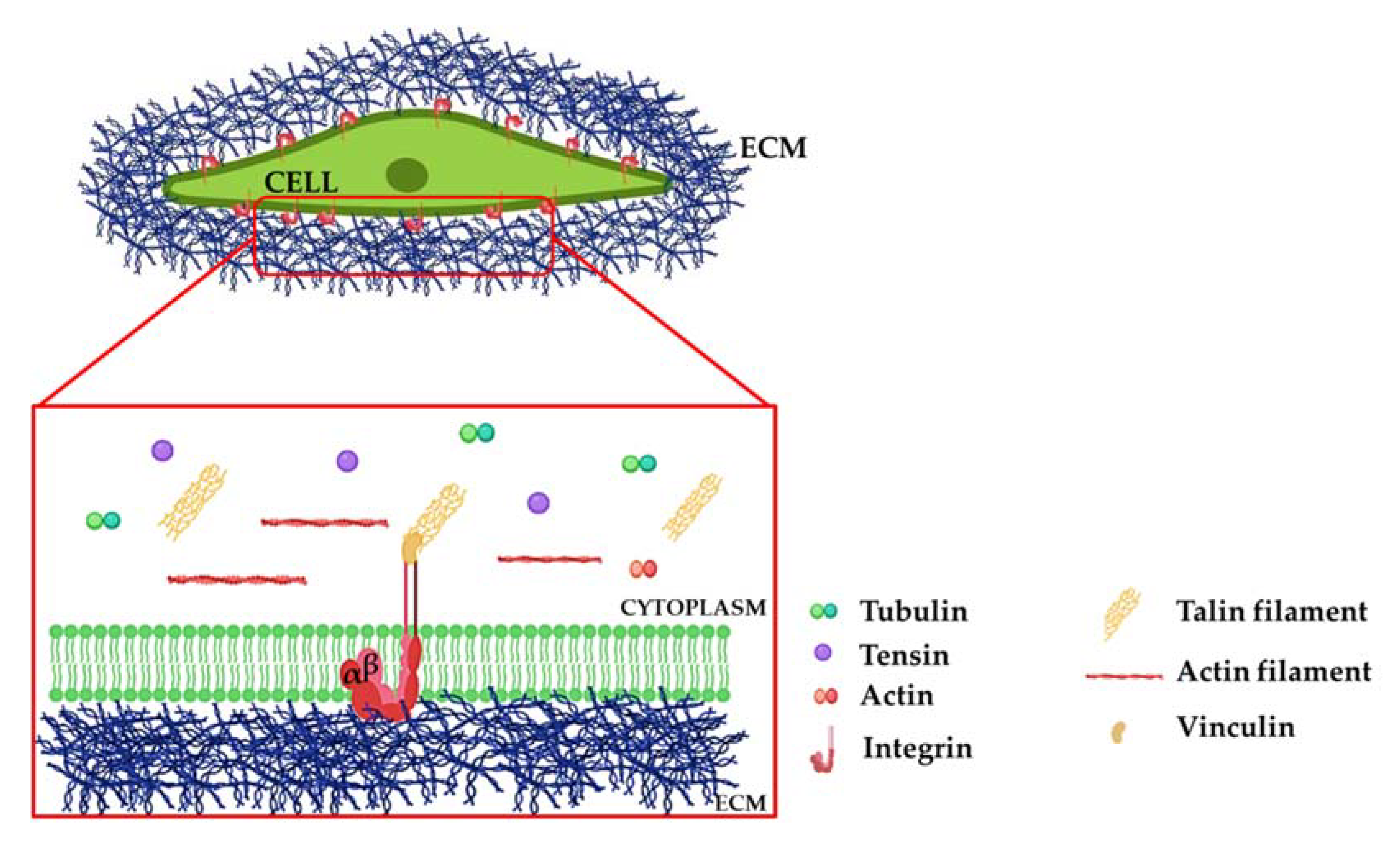
2. Integrins: A Brief Overview
Molecular Mechanisms of Integrin Signaling
3. The Role of Integrins in Inflammation-Dependent Mechanisms in IBD
4. Anti-Integrin Therapies for IBD Based on Immune-Dependent Functions
5. Integrins in IBD-Associated Fibrosis: A Key Player in Its Onset, Independent of Inflammation
5.1. Fibrosis and Tissue Stiffness
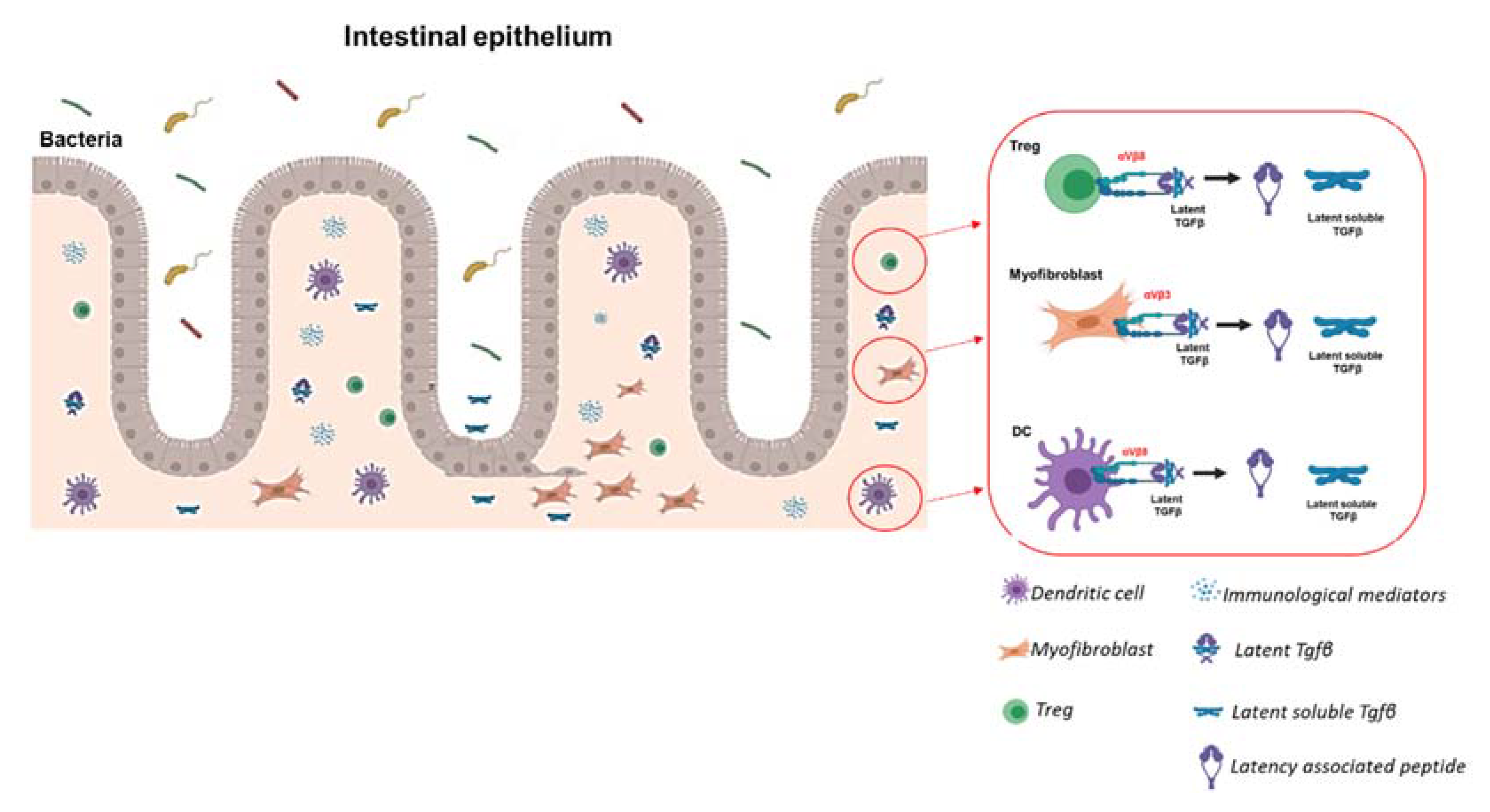
5.2. The Role of ECM in Intestinal Fibrosis: Not Only Support but Also a Reservoir of Molecular Mediators
5.3. Mechanisms of Integrin-Induced Release of Latent TGFβ
6. Functional Contribution of Integrins Expressed by Fibrosis-Associated Fibroblasts
7. Future Perspectives
8. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in IBD—A dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Etiopathogenesis of inflammatory bowel diseases. World J. Gastroenterol. 2006, 12, 4807–4812. [Google Scholar] [CrossRef]
- M’Koma, A. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest. Disord. 2018, 1, 75–105. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakatos, P.L.; Szamosi, T.; Lakatos, L. Smoking in inflammatory bowel diseases: Good, bad or ugly? World J. Gastroenterol. 2007, 13, 6134–6139. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Brenmoehl, J.; Leeb, S.; Schölmerich, J.; Rogler, G. Wound healing and fibrosis in intestinal disease. Gut 2007, 56, 130–139. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Otte, J.M.; Rosenberg, I.M.; Podolsky, D.K. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology 2003, 124, 1866–1878. [Google Scholar] [CrossRef]
- Lewis, A.; Correale, C.; Szabo, H.; Montorsi, M. Intestinal Fibrosis in Crohn’s Disease. Inflamm. Bowel Dis. 2015, 21, 1141–1150. [Google Scholar] [CrossRef]
- Cosnes, J.; Cattan, S.; Blain, A.; Beaugerie, L.; Carbonnel, F.; Parc, R.; Gendre, J.P. Long-term evolution of disease behavior of Crohn’s disease. Inflamm. Bowel Dis. 2002, 8, 244–250. [Google Scholar] [CrossRef]
- de Bruyn, J.R.; Meijer, S.L.; Wildenberg, M.E.; Bemelman, W.A.; van den Brink, G.R.; D’Haens, G.R. Development of fibrosis in acute and longstanding ulcerative colitis. J. Crohn’s Colitis 2015, 9, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfredsson, J.; Wick, M.J. Mechanism of fibrosis and stricture formation in Crohn’s disease. Scand. J. Immunol. 2020, 92, 1–23. [Google Scholar] [CrossRef]
- de Bruyn, J.R.; Becker, M.A.; Steenkamer, J.; Wildenberg, M.E.; Meijer, S.L.; Buskens, C.J.; Bemelman, W.A.; Löwenberg, M.; Ponsioen, C.Y.; van den Brink, G.R.; et al. Intestinal fibrosis is associated with lack of response to Infliximab therapy in Crohn’s disease. PLoS ONE 2018, 13, e0190999. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Fleshner, P.; Shen, B. Therapeutic armamentarium for stricturing Crohn’s disease: Medical versus endoscopic versus surgical approaches. Inflamm. Bowel Dis. 2015, 21, 2194–2213. [Google Scholar] [CrossRef] [PubMed]
- Crespi, M.; Dulbecco, P.; De Ceglie, A.; Conio, M. Strictures in Crohn’s Disease: From Pathophysiology to Treatment. Dig. Dis. Sci. 2020, 65, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.H.; Holubar, S.; Rieder, F. Fibrostenotic strictures in Crohn’s disease. Intest. Res. 2020, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, S.; Legate, K.R.; Fässler, R. Integrin-actin interactions. Cell. Mol. Life Sci. 2005, 60, 1081–1099. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFβ in fibrosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latella, G.; Di Gregorio, J.; Flati, V.; Rieder, F.; Lawrance, I.C. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 2014, 50, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Zamir, E.; Geiger, B. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 2001, 114, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, N.; Miihkinen, M.; Hamidi, H.; Alanko, J.; Mai, A.; Picas, L.; Guzmán, C.; Lévy, D.; Mattjus, P.; Goult, B.T.; et al. ProLIF—Quantitative integrin protein–protein interactions and synergistic membrane effects on proteoliposomes. J. Cell Sci. 2019, 132, jcs214270. [Google Scholar] [CrossRef] [Green Version]
- Assoian, R.K. Anchorage-dependent cell cycle progression. J. Cell Biol. 1997, 136, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Danen, E.H.J.; Yamada, K.M. Fibronectin, integrins, and growth control. J. Cell. Physiol. 2001, 189, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Assoian, R.K. Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer Metastasis Rev. 2005, 24, 383–393. [Google Scholar] [CrossRef]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; Lanerolle, P.D.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Hunter, T. Integrin signalling and tyrosine phosphorylation: Just the FAKs? Trends Cell Biol. 1998, 8, 151–157. [Google Scholar] [CrossRef]
- Tanimura, S.; Takeda, K. ERK signalling as a regulator of cell motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Merlo, A.; Herman, J.G.; Mao, L.; Lee, D.J.; Gabrielson, E.; Burger, P.C.; Baylin, S.B.; Sidransky, D. 5′ CpG Island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995, 1, 686–692. [Google Scholar] [CrossRef]
- Rampalli, A.M.; Zelenka, P.S. Insulin regulates expression of c-fos and c-jun and suppresses apoptosis of lens epithelial cells. Cell Growth Differ. 1995, 6, 945–953. [Google Scholar] [PubMed]
- Rodeck, U.; Jost, M.; DuHadaway, J.; Kari, C.; Jensen, P.J.; Risse, B.; Ewert, D.L. Regulation of Bcl-xL expression in human keratinocytes by cell-substratum adhesion and the epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5067–5072. [Google Scholar] [CrossRef] [Green Version]
- Strömblad, S.; Becker, J.C.; Yebra, M.; Brooks, P.C.; Cheresh, D.A. Suppression of p53 activity and p21(WAF1/CIP1) expression by vascular cell integrin αvβ3 during angiogenesis. J. Clin. Investig. 1996, 98, 426–433. [Google Scholar] [CrossRef]
- Howlett, A.R.; Bailey, N.; Damsky, C.; Petersen, O.W.; Bissell, M.J. Cellular growth and survival are mediated by β1 integrins in normal human breast eqithelium but not in breast carcinoma. J. Cell Sci. 1995, 108, 1945–1957. [Google Scholar] [CrossRef]
- Wewer, U.M.; Thornell, L.E.; Loechel, F.; Zhang, X.; Durkin, M.E.; Amano, S.; Burgeson, R.E.; Engvall, E.; Albrechtsen, R.; Virtanen, I.; et al. Extrasynaptic localization of laminin β2 chain in developing and adult human skeletal muscle. Am. J. Pathol. 1997, 151, 621–631. [Google Scholar]
- Farrelly, N.; Lee, Y.J.; Oliver, J.; Dive, C.; Streuli, C.H. Extracellular matrix regulates apoptosis in mammary epithelium through a control on insulin signaling. J. Cell Biol. 1999, 144, 1337–1347. [Google Scholar] [CrossRef]
- Yonekawa, K.; Harlan, J.M. Targeting leukocyte integrins in human diseases. J. Leukoc. Biol. 2005, 77, 129–140. [Google Scholar] [CrossRef]
- Minagawa, S.; Lou, J.; Seed, R.I.; Cormier, A.; Wu, S.; Cheng, Y.; Murray, L.; Tsui, P.; Connor, J.; Herbst, R.; et al. Selective targeting of TGF-β activation to treat Fibroinflammatory airway disease. Sci. Transl. Med. 2014, 6, 241ra79. [Google Scholar] [CrossRef] [Green Version]
- Erle, D.J.; Briskin, M.J.; Butcher, E.C.; Garcia-Pardo, A.; Lazarovits, A.I.; Tidswell, M. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J. Immunol. 1994, 153, 517–528. [Google Scholar] [PubMed]
- Elices, M.J.; Osborn, L.; Takada, Y.; Crouse, C.; Luhowskyj, S.; Hemler, M.E.; Lobb, R.R. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/Fibronectin binding site. Cell 1990, 60, 577–584. [Google Scholar] [CrossRef]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. α4β7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Podolsky, D.K.; Lobb, R.; King, N.; Benjamin, C.D.; Pepinsky, B.; Sehgal, P.; DeBeaumont, M. Attenuation of colitis in the cotton-top tamarin by anti-α4 integrin monoclonal antibody. J. Clin. Investig. 1993, 92, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesterberg, P.E.; Winsor-Hines, D.A.; Briskin, M.J.; Soler-Ferran, D.U.; Merrill, C.H.; Mackay, C.R.; Newman, W.A.; Ringler, D.J. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin α4β7. Gastroenterology 1996, 111, 1373–1380. [Google Scholar] [CrossRef]
- Kurmaeva, E.; Lord, J.D.; Zhang, S.; Bao, J.R.; Kevil, C.G.; Grisham, M.B.; Ostanin, D.V. T cell-associated α4 β7 but not α4 β1 integrin is required for the induction and perpetuation of chronic colitis. Mucosal Immunol. 2014, 7, 1354–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arihiro, S.; Ohtani, H.; Suzuki, M.; Murata, M.; Ejima, C.; Oki, M.; Kinouchi, Y.; Fukushima, K.; Sasaki, I.; Nakamura, S.; et al. Differential expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in ulcerative colitis and Crohn’s disease. Pathol. Int. 2002, 52, 367–374. [Google Scholar] [CrossRef]
- Lamb, C.A.; Mansfield, J.C.; Tew, G.W.; Gibbons, D.; Long, A.K.; Irving, P.; Diehl, L.; Eastham-Anderson, J.; Price, M.B.; O’Boyle, G.; et al. αEβ7 Integrin Identifies Subsets of Pro-Inflammatory Colonic CD4+ T Lymphocytes in Ulcerative Colitis. J. Crohn’s Colitis 2017, 11, 610–620. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Zundler, S.; Atreya, R.; Rath, T.; Voskens, C.; Hirschmann, S.; López-Posadas, R.; Watson, A.; Becker, C.; Schuler, G.; et al. Differential effects of α4β7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 2016, 65, 1642–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Asady, R.; Yuan, R.; Liu, K.; Wang, D.; Gress, R.E.; Lucas, P.J.; Drachenberg, C.B.; Hadley, G.A. TGF-β-dependent CD103 expression by CD8+ T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J. Exp. Med. 2005, 201, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; López-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of αeβ7 integrin suppresses accumulation of CD8 + and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briskin, M.; Winsor-Hines, D.; Shyjan, A.; Cochran, N.; Bloom, S.; Wilson, J.; McEvoy, L.M.; Butcher, E.C.; Kassam, N.; Mackay, C.R.; et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am. J. Pathol. 1997, 151, 97–110. [Google Scholar] [PubMed]
- Schön, M.P.; Arya, A.; Murphy, E.A.; Adams, C.M.; Strauch, U.G.; Agace, W.W.; Marsal, J.; Donohue, J.P.; Her, H.; Beier, D.R. Mucosal T Lymphocyte Numbers Are Selectively Reduced in Integrin Alpha E (CD103)-Deficient Mice. J. Immunol. 1999, 162, 6641–6649. [Google Scholar] [PubMed]
- Wagner, N.; Löhler, J.; Kunkel, E.J.; Ley, K.; Leung, E.; Krissansen, G.; Rajewsky, K.; Müller, W. Critical role for β7 integrins in formation of the gut-associated lymphoid tissue. Nature 1996, 382, 366–370. [Google Scholar] [CrossRef]
- Zundler, S.; Becker, E.; Spocinska, M.; Slawik, M.; Parga-Vidal, L.; Stark, R.; Wiendl, M.; Atreya, R.; Rath, T.; Leppkes, M.; et al. Hobit- and Blimp-1-driven CD4+ tissue-resident memory T cells control chronic intestinal inflammation. Nat. Immunol. 2019, 20, 288–300. [Google Scholar] [CrossRef]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat–Zucman, S.; Paul, P.; Gornet, J.M.; Douay, C.; et al. CD4+NKG2D+ T Cells in Crohn’s Disease Mediate Inflammatory and Cytotoxic Responses through MICA Interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef]
- Do, J.S.; Kim, S.; Keslar, K.; Jang, E.; Huang, E.; Fairchild, R.L.; Pizarro, T.T.; Min, B. γδ T Cells Coexpressing Gut Homing α4β7 and αE Integrins Define a Novel Subset Promoting Intestinal Inflammation. J. Immunol. 2017, 198, 908–915. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.I.; O’Connell, S.M.; Biancone, L.; Brolin, R.E.; Ebert, E.C. Spontaneous cytotoxicity of intestinal intraepithelial lymphocytes: Clues to the mechanism. Clin. Exp. Immunol. 1993, 94, 527–532. [Google Scholar] [CrossRef]
- Lúdvíksson, B.R.; Strober, W.; Nishikomori, R.; Hasan, S.K.; Ehrhardt, R.O. Administration of mAb against alpha E beta 7 prevents and ameliorates immunization-induced colitis in IL-2-/- mice. J. Immunol. 1999, 162, 4975–4982. [Google Scholar]
- Iliev, I.D.; Spadoni, I.; Mileti, E.; Matteoli, G.; Sonzogni, A.; Sampietro, G.M.; Foschi, D.; Caprioli, F.; Viale, G.; Rescigno, M.; et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut 2009, 58, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Jaensson, E.; Uronen-Hansson, H.; Pabst, O.; Eksteen, B.; Tian, J.; Coombes, J.L.; Berg, P.L.; Davidsson, T.; Powrie, F.; Johansson-Lindbom, B.; et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008, 205, 2139–2149. [Google Scholar] [CrossRef] [Green Version]
- Quan, T.E.; Cowper, S.; Wu, S.P.; Bockenstedt, L.K.; Bucala, R. Circulating fibrocytes: Collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004, 36, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Aumeunier, A.M.; Mowat, A.M.I. Intestinal CD103 + dendritic cells: Master regulators of tolerance? Trends Immunol. 2011, 32, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Nagarajan, N.; Zorlutuna, P. Effect of Substrate Stiffness on Mechanical Coupling and Force Propagation at the Infarct Boundary. Biophys. J. 2018, 115, 1966–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, E.R.; Bernardo, D.; Ng, S.C.; Rigby, R.J.; Al-Hassi, H.O.; Landy, J.; Peake, S.T.; Spranger, H.; English, N.R.; Thomas, L.V.; et al. Human gut dendritic cells drive aberrant gut-specific T-cell responses in ulcerative colitis, characterized by increased IL-4 production and loss of IL-22 and IFNγ. Inflamm. Bowel Dis. 2014, 20, 2299–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, A.; Andrew, D.P.; Jablonski-Westrich, D.; Holzmann, B.; Butcher, E.C. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 1994, 152, 3282–3293. [Google Scholar]
- Ghosh, S.; Goldin, E.; Gordon, F.H.; Malchow, H.A.; Rask-Madsen, J.; Rutgeerts, P.; Vyhnálek, P.; Zádorová, Z.; Palmer, T.; Donoghue, S. Natalizumab for Active Crohn’s Disease. N. Engl. J. Med. 2003, 348, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Feagan, B.G.; Fedorak, R.N.; Lashner, B.A.; Panaccione, R.; Present, D.H.; Spehlmann, M.E.; Rutgeerts, P.J.; Tulassay, Z.; Volfova, M.; et al. Natalizumab for the Treatment of Active Crohn’s Disease: Results of the ENCORE Trial. Gastroenterology 2007, 132, 1672–1683. [Google Scholar] [CrossRef] [Green Version]
- D’Haens, G.; Rieder, F.; Feagan, B.G.; Higgins, P.D.; Panes, J.; Maaser, C.; Rogler, G.; Löwenberg, M.; van der Voort, R.; Pinzani, M.; et al. Challenges in the Pathophysiology, Diagnosis and Management of Intestinal Fibrosis in Inflammatory Bowel Disease. Gastroenterology 2019. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 28, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Yousry, T.A.; Major, E.O.; Ryschkewitsch, C.; Fahle, G.; Fischer, S.; Hou, J.; Curfman, B.; Miszkiel, K.; Mueller-Lenke, N.; Sanchez, E.; et al. Evaluation of Patients Treated with Natalizumab for Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 2006, 354, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.S.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: Clinical features and pathogenesis. Lancet Neurol. 2010, 9, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Khoy, K.; Mariotte, D.; Defer, G.; Petit, G.; Toutirais, O.; Le Mauff, B. Natalizumab in Multiple Sclerosis Treatment: From Biological Effects to Immune Monitoring. Front. Immunol. 2020, 11, 1–7. [Google Scholar] [CrossRef]
- Card, T.; Xu, J.; Liang, H.; Bhayat, F. What Is the Risk of Progressive Multifocal Leukoencephalopathy in Patients with Ulcerative Colitis or Crohn’s Disease Treated with Vedolizumab? Inflamm. Bowel Dis. 2018, 24, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.K.; Mahadevan, U. Etrolizumab: Anti-β7—A Novel Therapy for Ulcerative Colitis. Gastroenterology 2014, 146, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Cepek, K.L.; Parker, C.M.; Madara, J.L.; Brenner, M.B. Integrin α(E)β7 mediates adhesion of T lymphocytes to epithelial cells. J. Immunol. 1993, 150, 3459–3470. [Google Scholar]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.T.; Mansfield, J.C.; Lamb, C.A.; Feagan, B.G.; Panes, J.; Salas, A.; et al. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Takazoe, M.; Watanabe, M.; Kawaguchi, T.; Matsumoto, T.; Oshitani, N.; Hiwatashi, N.; Hibi, T. S1066 Oral Alpha-4 Integrin Inhibitor (AJM300) in Patients with Active Crohn’s Disease—A Randomized, Double-Blind, Placebo-Controlled Trial. Gastroenterology 2009, 136, A-181. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Cyrille, M.; Hansen, M.B.; Feagan, B.G.; Loftus, E.V., Jr.; Rogler, G.; Vermeire, S.; Cruz, M.L.; Yang, J.; Boedigheimer, M.J.; et al. Efficacy and Safety of Abrilumab in a Randomized, Placebo-Controlled Trial for Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2019, 156, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Laukens, D. Inflammation-Independent Mechanisms of Intestinal Fibrosis: The Role of the Extracellular Matrix. In Fibrostenotic Inflammatory Bowel Disease; Springer International Publishing: Basel, Switzerland, 2018; pp. 77–95. [Google Scholar] [CrossRef]
- Wight, T.N.; Potter-Perigo, S. The extracellular matrix: An active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, 950–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, J.; Li, J.; Hagemann, T.L.; Jones, J.R.; Fraenkel, E.; Weitz, D.A.; Zhang, S.C.; Messing, A.; Feany, M.B.; et al. Tissue and cellular rigidity and mechanosensitive signaling activation in Alexander disease. Nat. Commun. 2018, 9, 1899. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Kim, K.; Johnson, L.A.; Jia, C.; Joyce, J.C.; Rangwalla, S.; Higgins, P.D.; Rubin, J.M. Noninvasive Ultrasound Elasticity Imaging (UEI) of Crohn’s Disease: Animal Model. Ultrasound Med. Biol. 2008, 34, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B. Tissue stiffness, latent TGF-β1 Activation, and mechanical signal transduction: Implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. Rep. 2009, 11, 120–126. [Google Scholar] [CrossRef]
- Johnson, L.A.; Rodansky, E.S.; Sauder, K.L.; Horowitz, J.C.; Mih, J.D.; Tschumperlin, D.J.; Higgins, P.D. Matrix Stiffness Corresponding to Strictured Bowel Induces a Fibrogenic Response in Human Colonic Fibroblasts. Inflamm. Bowel Dis. 2013, 19, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.F.; Boot-Handford, R.P.; Lamandé, S. Genetic diseases of connective tissues: Cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009, 10, 173–183. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O.; Naba, A. Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Bershadsky, A.; Kozlov, M.; Geiger, B. Adhesion-mediated mechanosensitivity: A time to experiment, and a time to theorize. Curr. Opin. Cell Biol. 2006, 18, 472–481. [Google Scholar] [CrossRef]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Hudson, N.E.; Lu, C.; Springer, T.A. Structural determinants of integrin β-subunit specificity for latent TGF-β. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
- Di Sabatino, A.; Rovedatti, L.; Rosado, M.M.; Carsetti, R.; Corazza, G.R.; MacDonald, T.T. Increased expression of mucosal addressin cell adhesion molecule 1 in the duodenum of patients with active celiac disease is associated with depletion of integrin α4β7-positive T cells in blood. Hum. Pathol. 2009, 40, 699–704. [Google Scholar] [CrossRef]
- Miyazono, K.; Heldin, C.H. Latent forms of TGF-beta: Molecular structure and mechanisms of activation. Ciba Found. Symp. 1991, 157, 81–89. [Google Scholar] [CrossRef]
- Rolli, M.; Fransvea, E.; Pilch, J.; Saven, A.; Felding-Habermann, B. Activated integrin αvβ3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9482–9487. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.G.; Cormier, A.; Ito, S.; Seed, R.I.; Bondesson, A.J.; Lou, J.; Marks, J.D.; Baron, J.L.; Cheng, Y.; Nishimura, S.L.; et al. Cryo-EM Reveals Integrin-Mediated TGF-β Activation without Release from Latent TGF-β. Cell 2020, 180, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Eslami, A.; Gallant-Behm, C.L.; Hart, D.A.; Wiebe, C.; Honardoust, D.; Gardner, H.; Häkkinen, L.; Larjava, H.S. Expression of integrin αvβ6 and TGF-β in Scarless vs Scar-forming wound healing. J. Histochem. Cytochem. 2009, 57, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Aluwihare, P.; Mu, Z.; Zhao, Z.; Yu, D.; Weinreb, P.H.; Horan, G.S.; Violette, S.M.; Munger, J.S. Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci. 2009, 122, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.B.; Cai, W.; Cao, Q.; Chen, K.; Wu, Z.; He, L.; Chen, X. 64Cu-labeled tetrameric and octameric RGD peptides for small-animal PET of tumor αvβ3 integrin expression. J. Nucl. Med. 2007, 48, 1162–1171. [Google Scholar] [CrossRef]
- Li, G.; Jin, R.; Norris, R.A.; Zhang, L.; Yu, S.; Wu, F.; Markwald, R.R.; Nanda, A.; Conway, S.J.; Smyth, S.S.; et al. Periostin mediates vascular smooth muscle cell migration through the integrins ανβ3 and ανβ5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 2010, 208, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Latella, G.; Vetuschi, A.; Sferra, R.; Speca, S.; Gaudio, E. Localization of αvβ6 integrin-TGF-β1/Smad3, mTOR and PPARγ in experimental colorectal fibrosis. Eur. J. Histochem. 2013, 57, e40. [Google Scholar] [CrossRef] [Green Version]
- Missan, D.S.; Mitchell, K.; Subbaram, S.; Di Persio, C.M. Integrin α3β1 signaling through MEK/ERK determines alternative polyadenylation of the MMP-9 mRNA transcript in immortalized mouse keratinocytes. PLoS ONE 2015, 10, e0119539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.; Franzen, A.; Karlsson, J.O.; Ericson, L.E.; Heldin, N.E.; Nilsson, M. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 2002, 115, 4227–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. αv integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Hosseini, M.; Liu, X.; Kisseleva, T.; Brenner, D.A. Human hepatic stellate cell isolation and characterization. J. Gastroenterol. 2018, 53, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Lau, W.L.; Jo, H.; Tsujino, K.; Gewin, L.; Reed, N.I.; Atakilit, A.; Nunes, A.C.; DeGrado, W.F.; Sheppard, D.; et al. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in Vivo. J. Am. Soc. Nephrol. 2017, 28, 1998–2005. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Cambier, S.; Somanath, S.; Barker, T.; Minagawa, S.; Markovics, J.; Goodsell, A.; Publicover, J.; Reichardt, L.; Jablons, D.; et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8—Mediated activation of TGF-β. J. Clin. Investig. 2011, 121, 2863–2875. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Flynn, R.S.; Grider, J.R.; Murthy, K.S.; Kellum, J.M.; Akbari, H.; Kuemmerle, J.F. Increased activation of latent TGF-β1 by αVβ3 in human Crohn’s disease and fibrosis in TNBS colitis can be prevented by cilengitide. Inflamm. Bowel Dis. 2013, 19, 2829–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahm, K.; Lukashev, M.E.; Luo, Y.; Yang, W.J.; Dolinski, B.M.; Weinreb, P.H.; Simon, K.J.; Wang, L.C.; Leone, D.R.; Lobb, R.R.; et al. αvβ6 Integrin Regulates Renal Fibrosis and Inflammation in Alport Mouse. Am. J. Pathol. 2007, 170, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Dolinski, B.M.; Kikuchi, N.; Leone, D.R.; Peters, M.G.; Weinreb, P.H.; Violette, S.M.; Bissell, D.M. Role of αvβ6 integrin in acute biliary fibrosis. Hepatology 2007, 46, 1404–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef]
- Thia, K.T.; Loftus, E.V.; Sandborn, W.J.; Yang, S.K. An update on the epidemiology of inflammatory bowel disease in Asia. Am. J. Gastroenterol. 2008, 103, 3167–3182. [Google Scholar] [CrossRef]
- Li, J.; Mao, R.; Kurada, S.; Wang, J.; Lin, S.; Chandra, J.; Rieder, F. Pathogenesis of fibrostenosing Crohn’s disease. Trans. Res. 2019, 209, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Zidar, N.; Langner, C.; Jerala, M.; Boštjančič, E.; Drobne, D.; Tomažič, A. Pathology of Fibrosis in Crohn’s Disease—Contribution to Understanding Its Pathogenesis. Front. Med. 2020, 7, 167. [Google Scholar] [CrossRef]
- Johnson, L.A.; Luke, A.; Sauder, K.; Moons, D.S.; Horowitz, J.C.; Higgins, P.D. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: Impact of a ‘top-Down’ approach to intestinal fibrosis in mice. Inflamm. Bowel Dis. 2012, 18, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Hünerwadel, A.; Fagagnini, S.; Rogler, G.; Lutz, C.; Jaeger, S.U.; Mamie, C.; Weder, B.; Ruiz, P.A.; Hausmann, M. Severity of local inflammation does not impact development of fibrosis in mouse models of intestinal fibrosis. Sci. Rep. 2018, 8, 15182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Clinical Trials | Indication in IBD |
|---|---|---|---|
| NATALIZUMAB | α4 integrin | ENCORE | Induction and maintenance in CD |
| AJM300 | α4 integrin | Phase IIa | Induction in UC |
| VEDOLIZUMAB | α4β7 integrin | Gemini1, Gemini2, Gemini3 | Induction and maintenance in UC (Gemini1) Induction and maintenance in CD (Gemini2,3) |
| ETROLIZUMAB | β7 integrin | Eucalyptus, Bergamot, Hickory | Induction in UC (Eucalyptus) Induction in CD (Bergamot) Induction in CD (Hickory) |
| ABRILUMAB | α4β7 integrin | Phase IIb | Induction in UC and CD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garlatti, V.; Lovisa, S.; Danese, S.; Vetrano, S. The Multiple Faces of Integrin–ECM Interactions in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 10439. https://doi.org/10.3390/ijms221910439
Garlatti V, Lovisa S, Danese S, Vetrano S. The Multiple Faces of Integrin–ECM Interactions in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2021; 22(19):10439. https://doi.org/10.3390/ijms221910439
Chicago/Turabian StyleGarlatti, Valentina, Sara Lovisa, Silvio Danese, and Stefania Vetrano. 2021. "The Multiple Faces of Integrin–ECM Interactions in Inflammatory Bowel Disease" International Journal of Molecular Sciences 22, no. 19: 10439. https://doi.org/10.3390/ijms221910439
APA StyleGarlatti, V., Lovisa, S., Danese, S., & Vetrano, S. (2021). The Multiple Faces of Integrin–ECM Interactions in Inflammatory Bowel Disease. International Journal of Molecular Sciences, 22(19), 10439. https://doi.org/10.3390/ijms221910439