
Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties

Abstract
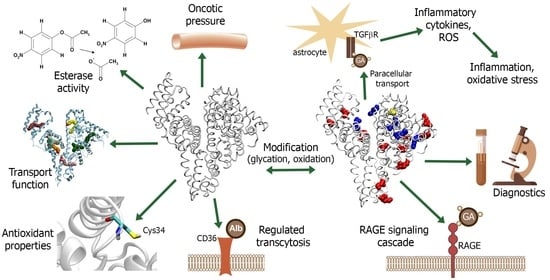
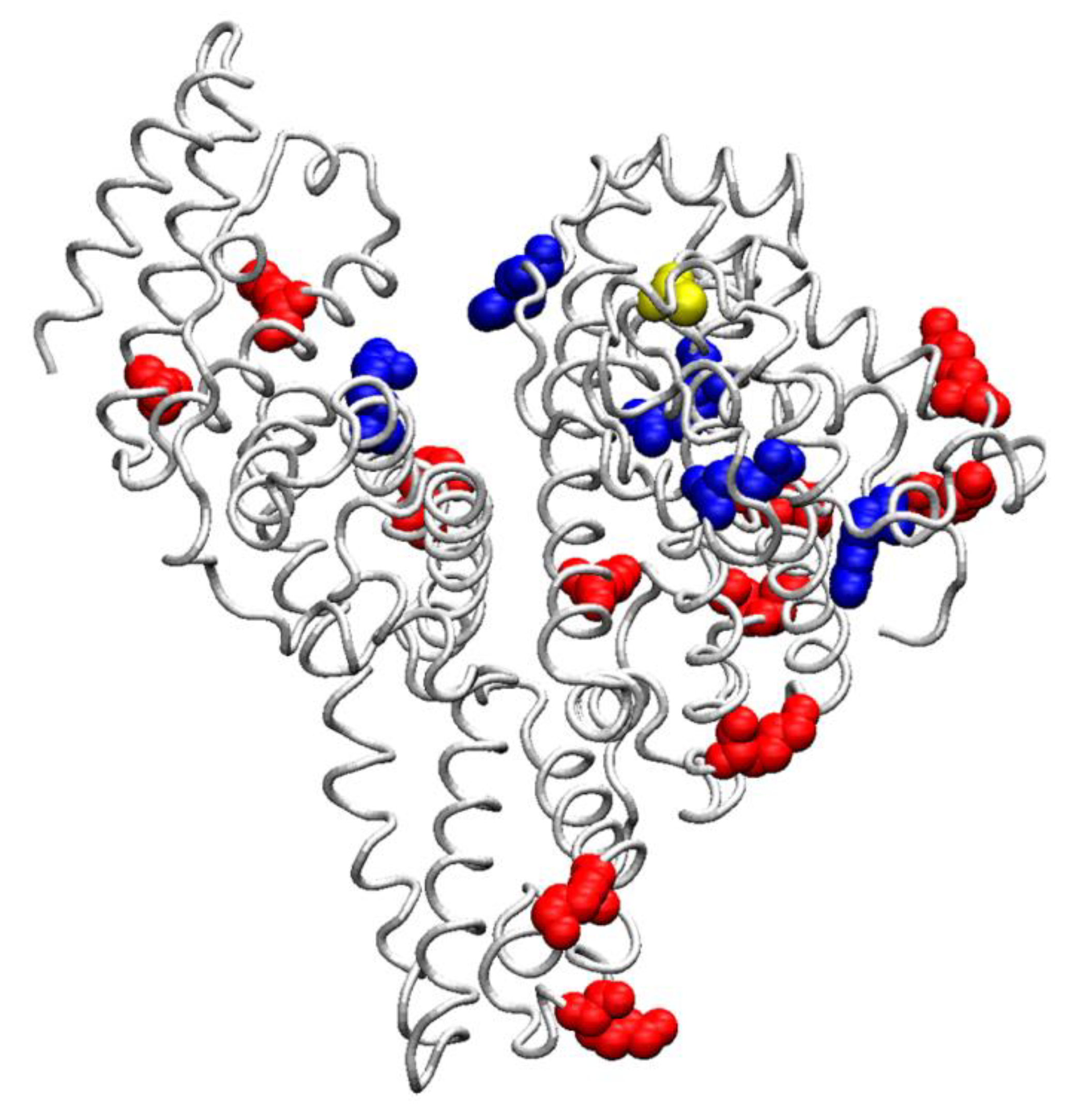
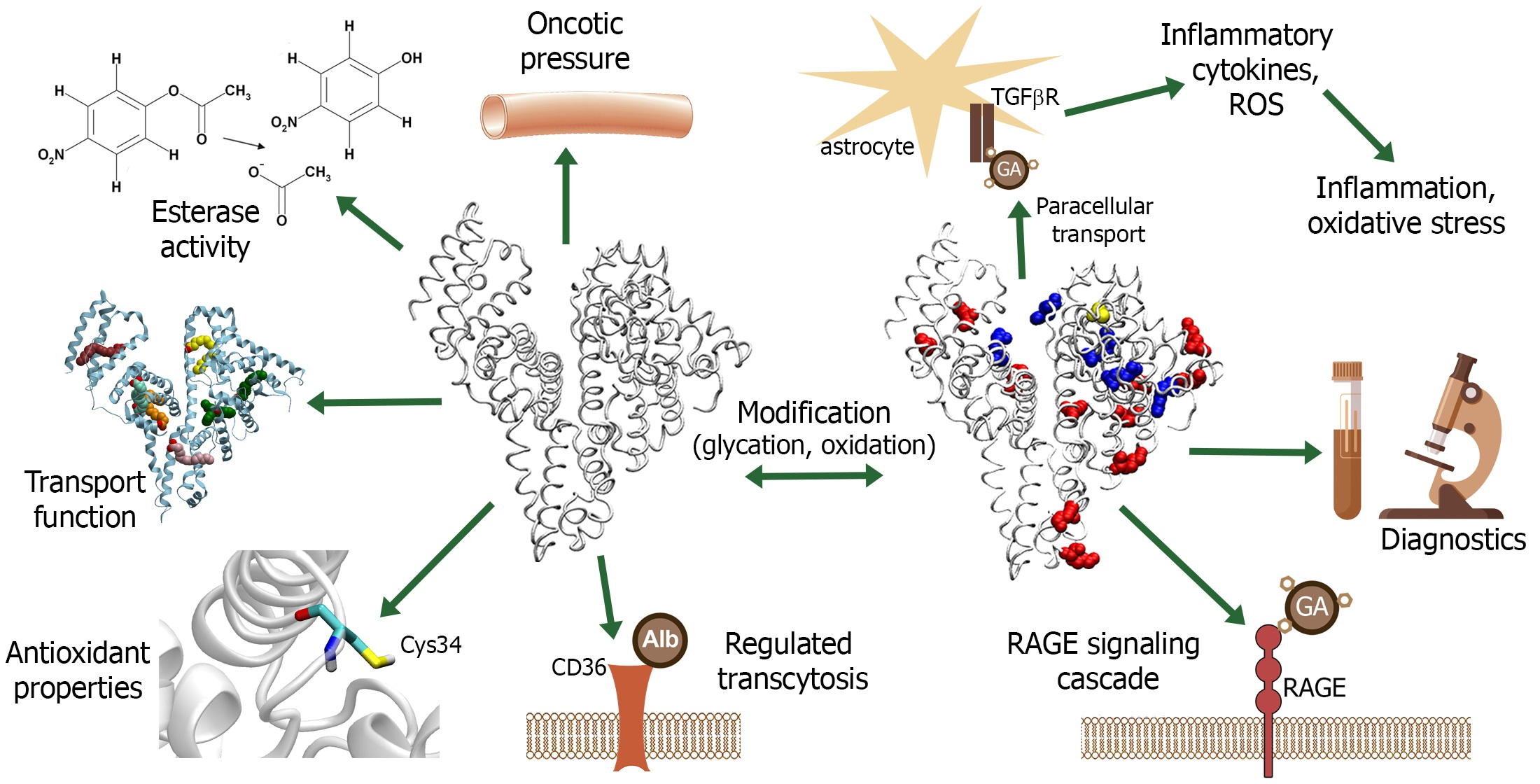{kind=link}
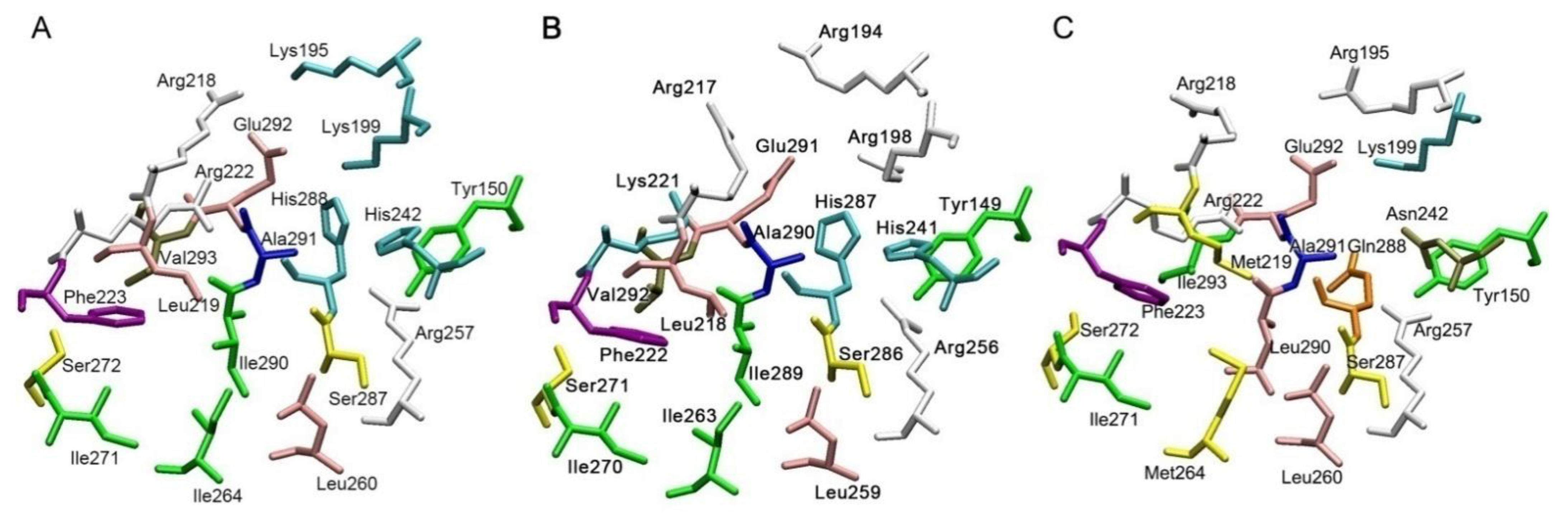
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Historical Aspects, Origin and Destination, and Evolutionary and Genetic Features of Albumin
2. Transporting Function and Structural Characteristics of Albumins of Different Species
2.1. Binding and Transporting Properties of Albumin
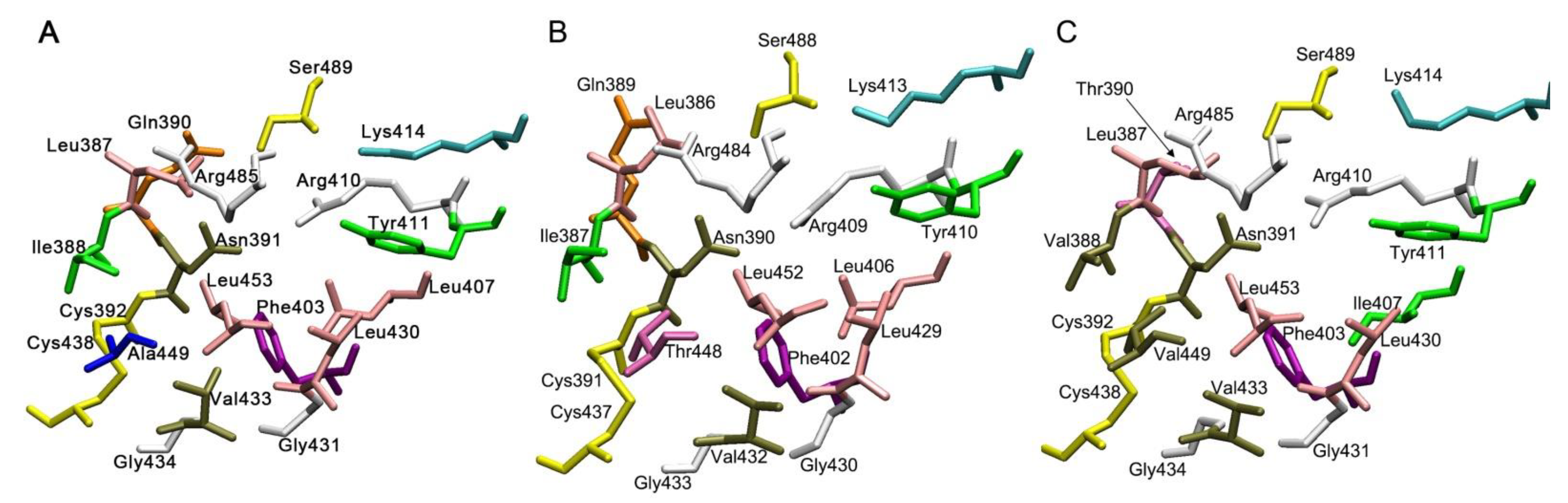
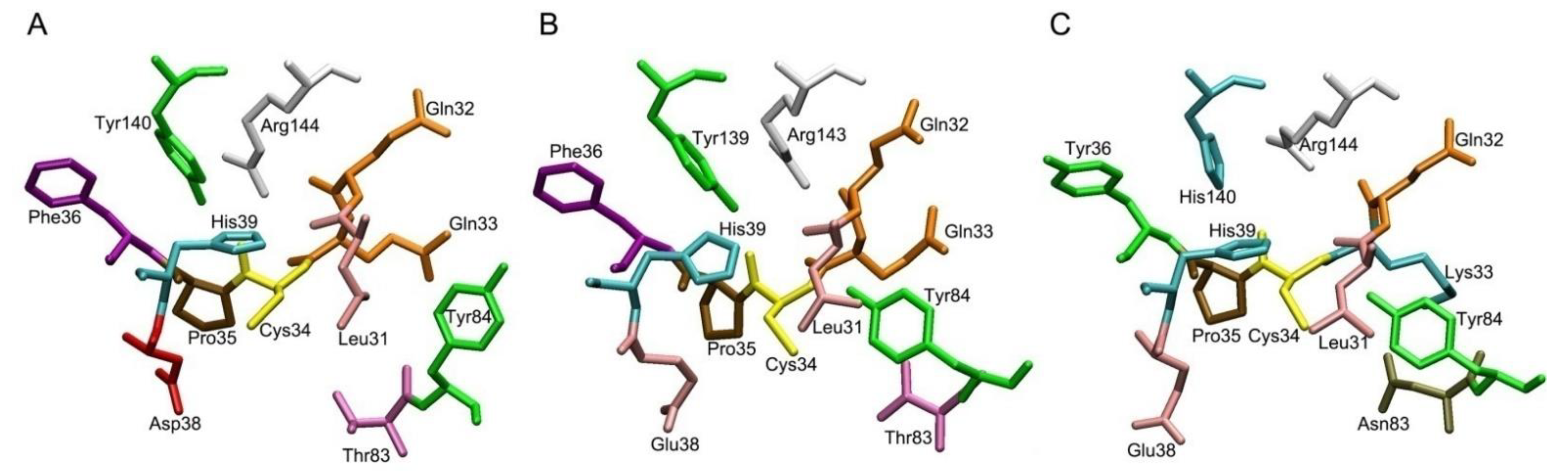
2.2. Comparative Characteristics of Human, Bovine and Rat Albumin
2.3. Albumins of Other Species
3. Enzymatic Activity of Albumin
4. Redox Modulation and Redox Activity of Albumin
4.1. Antioxidant Properties of Albumin
4.2. Practical Aspects of Redox Status of Albumin
5. GA: Biomarker and Pathogenetic Factors of DM
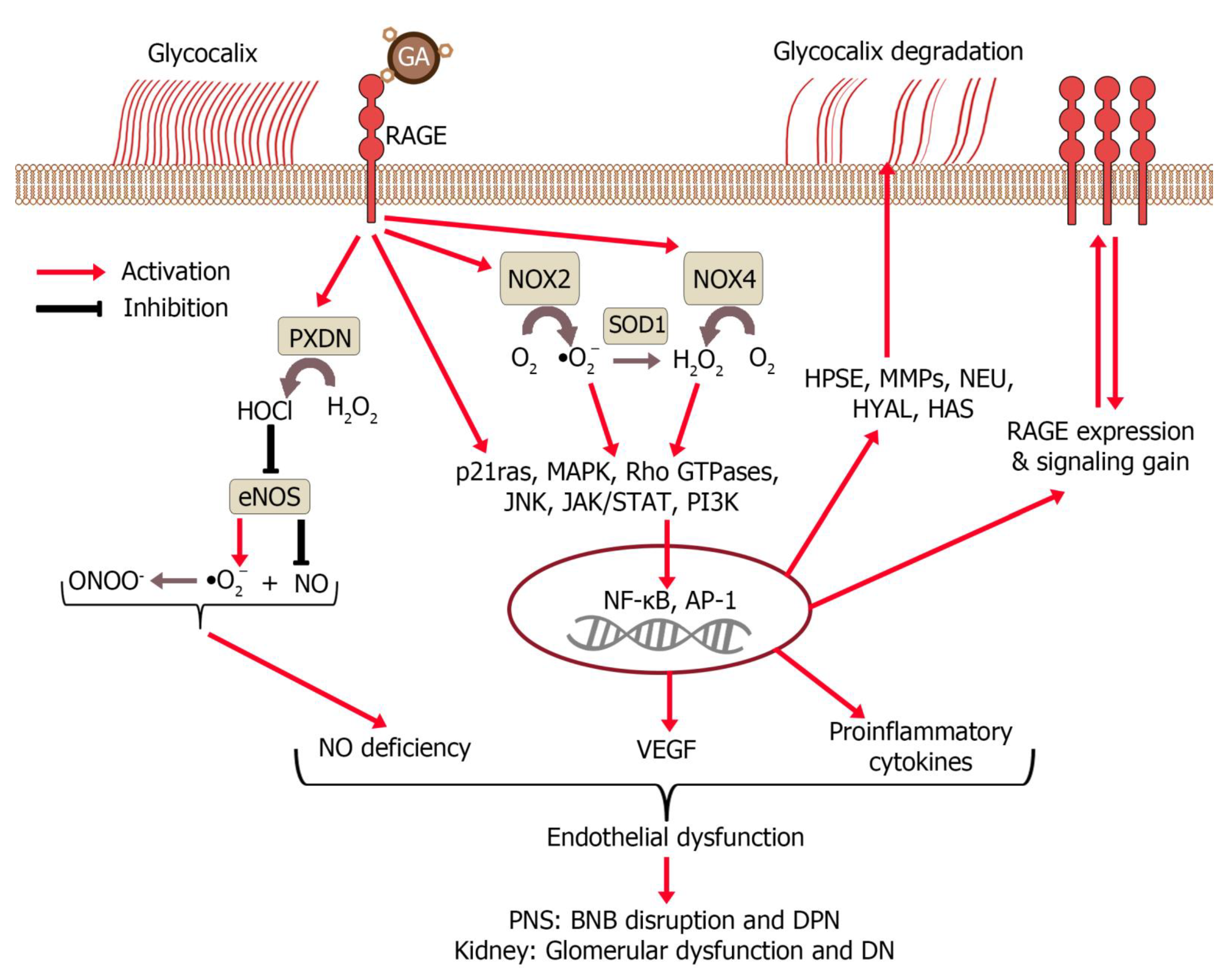
5.1. The Role of AGEs and GA in DM Pathophysiology
5.2. GA as a Diagnostic Tool for DM
5.3. Species Differences in Glycation Properties of Albumin
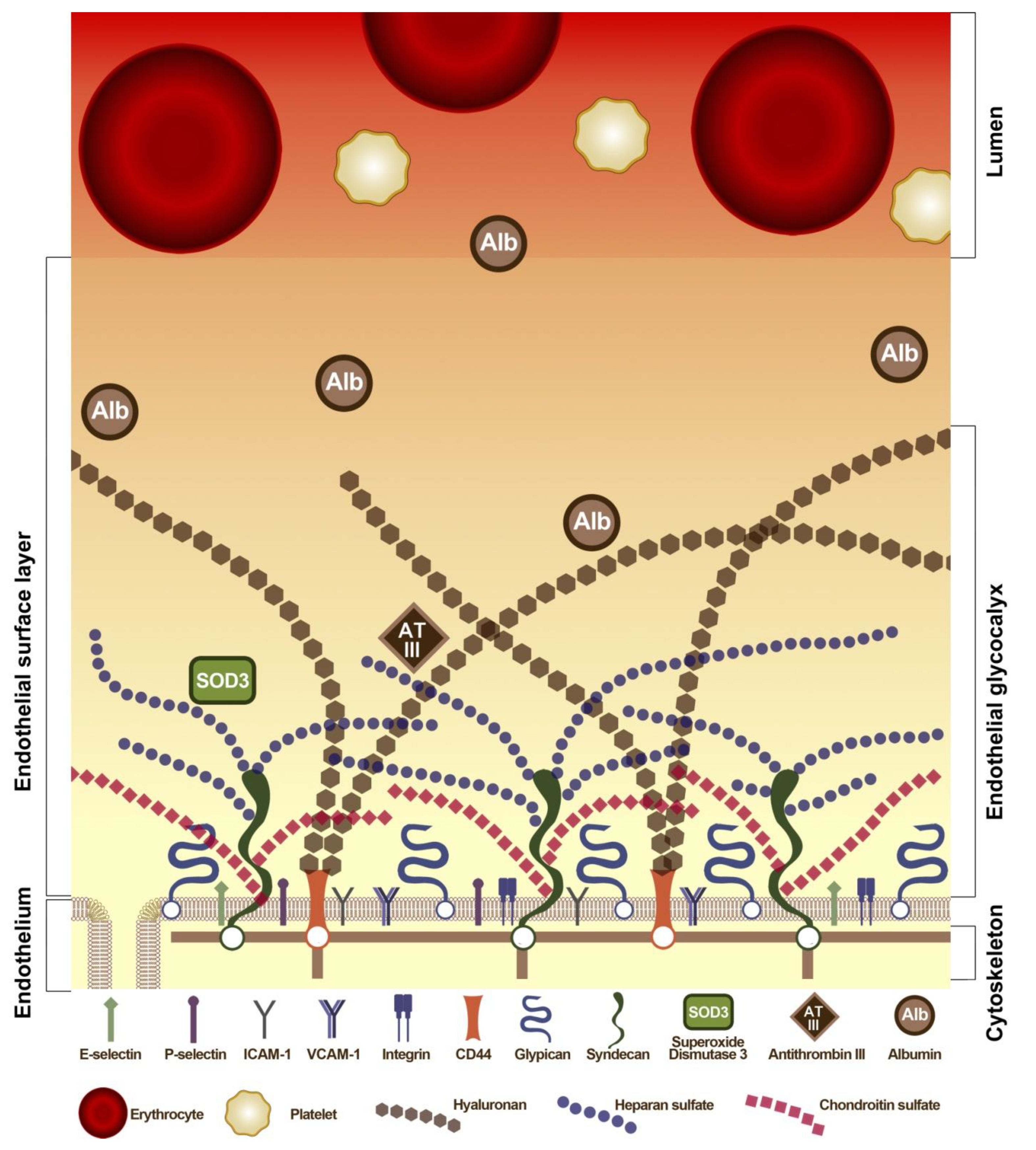
6. Interaction of Albumin with EC: Glycocalyx, Transcytosis and Glycoprotein CD36
7. Interaction of Glycated Albumin with the Endothelium
8. Role of Modified Albumin in Pathogenesis of Diseases
8.1. Obesity
8.2. Diabetic Polyneuropathy
8.3. Diabetic Nephropathy
8.4. COVID-19
8.5. The Role of Albumin in Epileptogenesis
9. Integrative Properties of Albumin in Diagnostics and Therapy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Peters, T., Jr. All about albumin. In Biochemistry, Genetics, and Medical Applications; Academic Press Ltd.: London, UK, 1996. [Google Scholar]
- Raoufinia, R.; Mota, A.; Keyhanvar, N.; Safari, F.; Shamekhi, S.; Abdolalizadeh, J. Overview of Albumin and Its Purification Methods. Adv. Pharm. Bull. 2016, 6, 495–507. [Google Scholar] [CrossRef]
- Mozzi, A.; Forni, D.; Cagliani, R.; Pozzoli, U.; Vertemara, J.; Bresolin, N.; Sironi, M. Albuminoid genes: Evolving at the interface of dispensability and selection. Genome Biol. Evol. 2014, 6, 2983–2997. [Google Scholar] [CrossRef]
- Li, S.; Cao, Y.; Geng, F. Genome-Wide Identification and Comparative Analysis of Albumin Family in Vertebrates. Evol. Bioinform. Online 2017, 3, 1176934317716089. [Google Scholar] [CrossRef]
- Albumin. Available online: http://albumin.org/ (accessed on 22 August 2021).
- Haefliger, D.N.; Moskaitis, J.E.; Schoenberg, D.R.; Wahli, W. Amphibian albumins as members of the albumin, alpha-fetoprotein, vitamin D-binding protein multigene family. J. Mol. Evol. 1989, 29, 344–354. [Google Scholar] [CrossRef]
- Lichenstein, H.S.; Lyons, D.E.; Wurfel, M.M.; Johnson, D.A.; McGinley, M.D.; Leidli, J.C.; Trollinger, D.B.; Mayer, J.P.; Wright, S.D.; Zukowski, M.M. Afamin is a new member of the albumin, alpha-fetoprotein, and vitamin D-binding protein gene family. J. Biol. Chem. 1994, 269, 18149–18154. [Google Scholar] [CrossRef]
- Doolittle, R.F. Stein and Moore Award address. Reconstructing history with amino acid sequences. Protein Sci. 1992, 1, 191–200. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 10, 1278–1289. [Google Scholar] [CrossRef]
- Metcalf, V.; Brennan, S.; George, P. Using serum albumin to infer vertebrate phylogenies. Appl. Bioinf. 2003, 2, S97–S107. [Google Scholar]
- Sarich, V.M.; Wilson, A.C. Rates of albumin evolution in primates. Proc. Natl. Acad. Sci. USA 1967, 58, 142–148. [Google Scholar] [CrossRef]
- Harper, M.E.; Dugaiczyk, A. Linkage of the evolutionarily-related serum albumin and alpha-fetoprotein genes within q11-22 of human chromosome 4. Am. J. Hum. Genet. 1983, 35, 565–572. [Google Scholar]
- Gray, J.E.; Doolittle, R.F. Characterization, primary structure, and evolution of lamprey plasma albumin. Protein Sci. 1992, 1, 289–302. [Google Scholar] [CrossRef]
- Nishio, H.; Heiskanen, M.; Palotie, A.; Bélanger, L.; Dugaiczyk, A. Tandem arrangement of the human serum albumin multigene family in the sub-centromeric region of 4q: Evolution and chromosomal direction of transcription. J. Mol. Biol. 1996, 259, 113–119. [Google Scholar] [CrossRef]
- Kragh-Hansen, U.; Brennan, S.O.; Galliano, M.; Sugita, O. Binding of warfarin, salicylate, and diazepam to genetic variants of human serum albumin with known mutations. Mol. Pharmacol. 1990, 37, 238–242. [Google Scholar]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Pietrangelo, A.; Pandurò, A.; Chowdhury, J.R.; Shafritz, D.A. Albumin gene expression is down-regulated by albumin or macromolecule infusion in the rat. J. Clin. Investig. 1992, 89, 1755–1760. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Fieux, M.; Le Quellec, S.; Bartier, S.; Coste, A.; Louis, B.; Giroudon, C.; Nourredine, M.; Bequignon, E. FcRn as a Transporter for Nasal Delivery of Biologics: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 6475. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef]
- Vetter, S.W. Glycated Serum Albumin and AGE Receptors. Adv. Clin. Chem. 2015, 72, 205–275. [Google Scholar] [CrossRef]
- Nakashima, F.; Shibata, T.; Kamiya, K.; Yoshitake, J.; Kikuchi, R.; Matsushita, T.; Ishii, I.; Giménez-Bastida, J.A.; Schneider, C.; Uchida, K. Structural and functional insights into S-thiolation of human serum albumins. Sci. Rep. 2018, 8, 932. [Google Scholar] [CrossRef]
- Hein, K.L.; Kragh-Hansen, U.; Morth, J.P.; Jeppesen, M.D.; Otzen, D.; Møller, J.V.; Nissen, P. Crystallographic analysis reveals a unique lidocaine binding site on human serum albumin. J. Struct. Biol. 2010, 171, 353–360. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Bteich, M. An overview of albumin and alpha-1-acid glycoprotein main characteristics: Highlighting the roles of amino acids in binding kinetics and molecular interactions. Heliyon 2019, 5, 02879. [Google Scholar] [CrossRef]
- Yamasaki, K.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Curry, S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab. Pharmacokinet. 2009, 24, 342–357. [Google Scholar] [CrossRef]
- Itoh, T.; Saura, Y.; Tsuda, Y.; Yamada, H. Stereoselectivity and enantiomer–enantiomer interactions in the binding of ibuprofen to human serum albumin. Chirality 1997, 9, 643–649. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Sjöholm, I.; Ekman, B.; Kober, A.; Ljungstedt-Påhlman, I.; Seiving, B.; Sjödin, T. Binding of drugs to human serum albumin: XI. The specificity of three binding sites as studied with albumin immobilized in microparticles. Mol. Pharmacol. 1979, 16, 767–777. [Google Scholar]
- Kragh-Hansen, U. Relations between high-affinity binding sites of markers for binding regions on human serum albumin. Biochem. J. 1985, 225, 629–638. [Google Scholar] [CrossRef]
- Dasgupta, A.; Havlik, D. Elevated free fosphenytoin concentrations in uremic sera: Uremic toxins hippuric acid and indoxyl sulfate do not account for the impaired protein binding of fosphenytoin. Ther. Drug Monit. 1998, 20, 658–662. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef]
- Sakai, T.; Takadate, A.; Otagiri, M. Characterization of binding site of uremic toxins on human serum albumin. Biol. Pharm. Bull. 1995, 18, 1755–1761. [Google Scholar] [CrossRef]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bolli, A.; Gullotta, F.; Fanali, G.; Fasano, M. Drug binding to Sudlow’s site I impairs allosterically human serum heme-albumin-catalyzed peroxynitrite detoxification. IUBMB Life 2010, 62, 776–780. [Google Scholar] [CrossRef]
- Bolli, A.; Marino, M.; Rimbach, G.; Fanali, G.; Fasano, M.; Ascenzi, P. Flavonoid binding to human serum albumin. Biochem. Biophys. Res. Commun. 2010, 398, 444–449. [Google Scholar] [CrossRef]
- Batalova, A.A.; Belinskaia, D.A.; Goncharov, N.V. Testing of polyphenols and fatty acids as modulators of albumin esterase activity towards organophosphates. In Proceedings of the Seventh Scientific Conference “Modern Trends in the Development of Health Care Technologies”, Moscow, Russia, 12–13 December 2019; Panin, V.P., Ed.; All-Russian Scientific Research Institute of Medicinal and Aromatic Plants: Moscow, Russia, 2019; pp. 428–434. (In Russian). [Google Scholar]
- Voronina, P.A.; Shmurak, V.I.; Batalova, A.A.; Belinskaia, D.A.; Goncharov, N.V. The effect of intramolecular disulfide bonds on the enzymatic activity of bovine serum albumin in vitro. In Modern Achievements of Chemical and Biological Sciences in Preventive and Clinical Medicine; Silina, A.V., Gaykova, L.B., Eds.; Mechnikov North-Western State Medical University: Saint Petersburg, Russia, 2020; pp. 53–60. (In Russian) [Google Scholar]
- Goncharov, N.V.; Terpilowski, M.A.; Shmurak, V.I.; Belinskaia, D.A.; Avdonin, P.V. The Rat (Rattus norvegicus) as a Model Object for Acute Organophosphate Poisoning. 1. Biochemical Aspects. J. Evol. Biochem. Physiol. 2019, 55, 112–123. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Terpilowski, M.A.; Kudryavtsev, I.V.; Serebryakova, M.K.; Belinskaia, D.A.; Sobolev, V.E.; Shmurak, V.I.; Korf, E.A.; Avdonin, P.V. The Rat (Rattus norvegicus) as a Model Object for Acute Organophosphate Poisoning. 2. A System Analysis of the Efficacy of Green Tea Extract in Preventing Delayed Effects of Poisoning. J. Evol. Biochem. Physiol. 2019, 55, 208–221. [Google Scholar] [CrossRef]
- Kuznetsov, S.V.; Goncharov, N.V. The Rat (Rattus norvegicus) as a Model Object for Acute Organophosphate Poisoning. 3. Cardiorespiratory Indices. J. Evol. Biochem. Physiol. 2019, 55, 239–243. [Google Scholar] [CrossRef]
- Sobolev, V.E.; Shmurak, V.I.; Goncharov, N.V. The Rat (Rattus norvegicus) as a Model Object for Acute Organophosphate Poisoning. 4. M1-Cholinoreceptors and Esterase Activity in Brain Homogenates. J. Evol. Biochem. Physiol. 2019, 55, 244–248. [Google Scholar] [CrossRef]
- Sobolev, V.E.; Korf, E.A.; Goncharov, N.V. The Rat (Rattus norvegicus) as a Model Object for Acute Organophosphate Poisoning. 5. Morphofunctional Alterations in Kidneys. J. Evol. Biochem. Physiol. 2019, 55, 302–312. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaia, D.A.; Shmurak, V.I. Probing of Albumin Esterase Activity Modulators as Components of Therapy for Acute Organophosphate Poisoning. In Actual Problems of Organic Chemistry and Biotechnology: The Materials of the International Scientific Conference (Written Reports); Glukhareva, T.V., Nein, Y.I., Pospelova, T.A., Bakulev, V.A., Eds.; AMB Publishing House: Ekaterinburg, Russia, 2020; pp. 305–307. Available online: https://orgchembiotech2020.urfu.ru/fileadmin/user_upload/site_22613/programm/Tom_2_s_korrekturoi_i_verstkoi__1__compressed.pdf (accessed on 22 August 2021). (In Russian)
- Lu, R.; Li, W.W.; Katzir, A.; Raichlin, Y.; Yu, H.Q.; Mizaikoff, B. Probing the secondary structure of bovine serum albumin during heat-induced denaturation using mid-infrared fiberoptic sensors. Analyst 2015, 140, 765–770. [Google Scholar] [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol. 2012, 52, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef]
- Taborskaya, K.I.; Belinskaya, D.A.; Goncharov, N.V.; Avdonin, P.V. Building a three-dimensional model of rat albumin molecule by homology modeling. J. Evol. Biochem. Physiol. 2017, 53, 384–393. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Batalova, A.A.; Goncharov, N.V. The Effect of Resveratrol on Binding and Esterase Activity of Human and Rat Albumin. J. Evol. Biochem. Physiol. 2019, 55, 174–183. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric modulation of drug binding to human serum albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Belinskaya, D.A.; Goncharov, N.V.; Shmurak, V.I.; Prokofieva, D.S. Serum albumin: Search for new sites of interaction with organophosphorus compounds by the example of soman. Russ. J. Bioorgan. Chem. 2014, 40, 499–506. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Taborskaya, K.I.; Avdonin, P.V.; Goncharov, N.V. Modulation of the albumin–paraoxon interaction sites by fatty acids: Analysis by the molecular modeling methods. Russ. J. Bioorgan. Chem. 2017, 43, 359–367. [Google Scholar] [CrossRef]
- Belinskaya, D.A.; Taborskaya, K.I.; Goncharov, N.V.; Shmurak, V.I.; Avdonin, P.P.; Avdonin, P.V. In silico analysis of paraoxon binding by human and bovine serum albumin. J. Evol. Biochem. Physiol. 2017, 53, 191–199. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaia, M.A.; Shmurak, V.I.; Terpilowski, M.A.; Jenkins, R.O.; Avdonin, P.V. Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates. Molecules 2017, 22, 1201. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Terpilovskii, M.A.; Batalova, A.A.; Goncharov, N.V. Effect of Cys34 oxidation state of albumin on its interaction with paraoxon according to molecular modeling data. Russ. J. Bioorgan. Chem. 2019, 45, 535–544. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Batalova, A.A.; Goncharov, N.V. Effect of Bovine Serum Albumin Redox Status on Its Interaction with Paraoxon as Determined by Molecular Modeling. J. Evol. Biochem. Physiol. 2020, 56, 434–438. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Terpilovskii, M.A.; Belinskaya, D.A.; Shmurak, V.I.; Avdonin, P.V. Comparative analysis of esterase and paraoxonase activities of different serum albumin species. J. Evol. Biochem. Physiol. 2017, 53, 271–281. [Google Scholar] [CrossRef]
- Bujacz, A.; Talaj, J.A.; Zielinski, K.; Pietrzyk, A.J.; Neumann, P. Crystal structures of serum albumins from domesticated ruminants and their complexes with 3,5-diiodosalicylic acid. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Yokomaku, K.; Kureishi, M.; Akiyama, M.; Kihira, K.; Komatsu, T. Artificial Blood for Dogs. Sci. Rep. 2016, 6, 36782. [Google Scholar] [CrossRef]
- Yokomaku, K.; Akiyama, M.; Morita, Y.; Kihira, K.; Komatsu, T. Core–shell protein clusters comprising haemoglobin and recombinant feline serum albumin as an artificial O2 carrier for cats. J. Mater. Chem. B 2018, 6, 2417–2425. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaya, D.A.; Razygraev, A.V.; Ukolov, A.I. On the enzymatic activity of albumin. Russ. J. Bioorgan. Chem. 2015, 41, 113–124. [Google Scholar] [CrossRef]
- Lee, W.Q.; Affandi, I.S.; Feroz, S.R.; Mohamad, S.B.; Tayyab, S. Evaluation of pendimethalin binding to human serum albumin: Insights from spectroscopic and molecular modeling approach. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef]
- Želonková, K.; Havadej, S.; Verebová, V.; Holečková, B.; Uličný, J.; Staničová, J. Fungicide Tebuconazole Influences the Structure of Human Serum Albumin Molecule. Molecules 2019, 24, 3190. [Google Scholar] [CrossRef]
- Lockridge, O.; Xue, W.; Gaydess, A.; Grigoryan, H.; Ding, S.J.; Schopfer, L.M.; Hinrichs, S.H.; Masson, P. Pseudo-esterase activity of human albumin: Slow turnover on tyrosine 411 and stable acetylation of 82 residues including 59 lysines. J. Biol. Chem. 2008, 283, 22582–22590. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Maddison, K.; LoCastro, L.; Borch, R.F. Accelerated decomposition of 4-hydroxycyclophosphamide by human serum albumin. Cancer Res. 1987, 47, 1505–1508. [Google Scholar]
- Gerasimova, Y.V.; Bobik, T.V.; Ponomarenko, N.A.; Shakirov, M.M.; Zenkova, M.A.; Tamkovich, N.V.; Popova, T.V.; Knorre, D.G.; Godovikova, T.S. RNA-hydrolyzing activity of human serum albumin and its recombinant analogue. Bioorgan. Med. Chem. Lett. 2010, 20, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Nachon, F.; Froment, M.T.; Verdier, L.; Debouzy, J.C.; Brasme, B.; Gillon, E.; Schopfer, L.M.; Lockridge, O.; Masson, P. Binding and hydrolysis of soman by human serum albumin. Chem. Res. Toxicol. 2008, 21, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; García-Argüelles, S.; Carrera, V.; Vilanova, E. Serum albumin is as efficient as paraxonase in the detoxication of paraoxon at toxicologically relevant concentrations. Chem. Res. Toxicol. 2008, 21, 1524–1529. [Google Scholar] [CrossRef]
- Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). The Enzyme List, Class 3—Hydrolases. Generated from the ExplorEnz Database. 2010. Available online: http://www.enzyme-database.org/downloads/ec3.pdf (accessed on 22 August 2021).
- Schomburg, D.; Schomburg, I. Springer Handbook of Enzymes; EC Number Index; Springer: Heidelberg/Berlin, Germany; New York, NY, USA, 2013. [Google Scholar]
- Kurdyukov, I.D.; Shmurak, V.I.; Nadeev, A.D.; Voitenko, N.G.; Prokofieva, D.S.; Goncharov, N.V. “Esterase status” of an organism at exposure by toxic substances and pharmaceuticals. Toksikol. Vestnik Toxicol. Bull. 2012, 6, 6–13. [Google Scholar]
- Sogorb, M.A.; Vilanova, E. Serum albumins and detoxication of anti-cholinesterase agents. Chem. Biol. Interact. 2010, 187, 325–329. [Google Scholar] [CrossRef]
- Fitzpatrick, F.A.; Wynalda, M.A. Albumin-catalyzed metabolism of prostaglandin D2. Identification of products formed in vitro. J. Biol. Chem. 1983, 258, 11713–11718. [Google Scholar] [CrossRef]
- Kimzey, M.J.; Yassine, H.N.; Riepel, B.M.; Tsaprailis, G.; Monks, T.J.; Lau, S.S. New site(s) of methylglyoxal-modified human serum albumin, identified by multiple reaction monitoring, alter warfarin binding and prostaglandin metabolism. Chem. Biol. Interact. 2011, 192, 122–128. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Aldini, G.; Ito, S.; Morishita, N.; Shibata, T.; Vistoli, G.; Carini, M.; Uchida, K. Delta12-prostaglandin J2 as a product and ligand of human serum albumin: Formation of an unusual covalent adduct at His146. J. Am. Chem. Soc. 2010, 132, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Presle, N.; Lapicque, F.; Maurice, M.H.; Fournel-Gigleux, S.; Magdalou, J.; Abiteboul, M.; Siest, G.; Netter, P. Stereoselective esterase activity of human serum albumin toward ketoprofen glucuronide. Mol. Pharmacol. 1995, 47, 647–653. [Google Scholar] [PubMed]
- Georges, H.; Presle, N.; Buronfosse, T.; Fournel-Gigleux, S.; Netter, P.; Magdalou, J.; Lapicque, F. In Vitro stereoselective degradation of carprofen glucuronide by human serum albumin. Characterization of sites and reactive amino acids. Chirality 2000, 12, 53–62. [Google Scholar] [CrossRef]
- Drmanovic, Z.; Voyatzi, S.; Kouretas, D.; Sahpazidou, D.; Papageorgiou, A.; Antonoglou, O. Albumin possesses intrinsic enolase activity towards dihydrotestosterone which can differentiate benign from malignant breast tumors. Anticancer Res. 1999, 19, 4113–4124. [Google Scholar]
- Benedetti, F.; Berti, F.; Bidoggia, S. Aldolase activity of serum albumins. Org. Biomol. Chem. 2011, 9, 4417–4420. [Google Scholar] [CrossRef]
- Luisi, I.; Pavan, S.; Fontanive, G.; Tossi, A.; Benedetti, F.; Savoini, A.; Maurizio, E.; Sgarra, R.; Sblattero, D.; Berti, F. An albumin-derived peptide scaffold capable of binding and catalysis. PLoS ONE 2013, 8, 56469. [Google Scholar] [CrossRef]
- Kirby, A.J.; Hollfelder, F.; Tawfik, D.S. Nonspecific catalysis by protein surfaces. Appl. Biochem. Biotechnol. 2000, 83, 173–180. [Google Scholar] [CrossRef]
- James, L.C.; Tawfik, D.S. Catalytic and binding poly-reactivities shared by two unrelated proteins: The potential role of promiscuity in enzyme evolution. Protein Sci. 2001, 10, 2600–2607. [Google Scholar] [CrossRef]
- Carbonell, P.; Faulon, J.L. Molecular signatures-based prediction of enzyme promiscuity. Bioinformatics 2010, 26, 2012–2019. [Google Scholar] [CrossRef]
- Moyon, N.S.; Islam, M.M.; Phukan, S.; Mitra, S. Fluorescence modulation and associative behavior of lumazine in hydrophobic domain of micelles and bovine serum albumin. J. Photochem. Photobiol. B 2013, 121, 37–45. [Google Scholar] [CrossRef]
- Copley, S.D. Shining a light on enzyme promiscuity. Curr. Opin. Struct. Biol. 2017, 47, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Miton, C.M.; Tokuriki, N. A mechanistic view of enzyme evolution. Protein Sci. 2020, 29, 1724–1747. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Lu, S.; Reed, C.; Schmidt, J.; Forconi, M. Kemp Elimination in Cationic Micelles: Designed Enzyme-Like Rates Achieved through the Addition of Long-Chain Bases. J. Phys. Org. Chem. 2016, 29, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Komatsu, T.; Ueno, T.; Hanaoka, K.; Urano, Y. Fluorescence detection of serum albumin with a turnover-based sensor utilizing Kemp elimination reaction. Bioorgan. Med. Chem. Lett. 2017, 27, 3464–3467. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Silva, C.; Bertran, J.; Branchadell, V.; Oliva, A. Kemp Elimination Reaction Catalyzed by Electric Fields. Chemphyschem. 2020, 21, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Röthlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Zanghellini, A.; Dym, O.; et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar] [CrossRef]
- Pen, J.; Beintema, J.J. Nomenclature of esterases. Biochem. J. 1986, 240, 933. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Huang, M. Structure and enzymatic activities of human serum albumin. Curr. Pharm. Des. 2015, 21, 1831–1836. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–230140. [Google Scholar] [CrossRef]
- Ota, C.; Takano, K. Behavior of Bovine Serum Albumin Molecules in Molecular Crowding Environments Investigated by Raman Spectroscopy. Langmuir 2016, 32, 7372–7382. [Google Scholar] [CrossRef]
- Zhu, T.T.; Zhang, Y.; Luo, X.A.; Wang, S.Z.; Jia, M.Q.; Chen, Z.X. Difference in Binding of Long- and Medium-Chain Fatty Acids with Serum Albumin: The Role of Macromolecular Crowding Effect. J. Agric. Food Chem. 2018, 66, 1242–1250. [Google Scholar] [CrossRef]
- Tabata, F.; Wada, Y.; Kawakami, S.; Miyaji, K. Serum Albumin Redox States: More Than Oxidative Stress Biomarker. Antioxidants 2021, 10, 503. [Google Scholar] [CrossRef]
- Oettl, K.; Marsche, G. Redox State of Human Serum Albumin in Terms of Cysteine-34 in Health and Disease. Methods Enzymol. 2010, 474, 181–195. [Google Scholar] [CrossRef]
- Michelis, R.; Sela, S.; Zeitun, T.; Geron, R.; Kristal, B. Unexpected Normal Colloid Osmotic Pressure in Clinical States with Low Serum Albumin. PLoS ONE 2016, 11, 0159839. [Google Scholar] [CrossRef]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-Cysteinylated Human Serum Albumin Is a Sensitive Plasma Marker in Oxidative Stress-Related Chronic Diseases. PLoS ONE 2014, 9, 85216. [Google Scholar] [CrossRef]
- Kurano, M.; Yasukawa, K.; Ikeda, H.; Aoki, J.; Yatomi, Y. Redox state of albumin affects its lipid mediator binding characteristics. Free Radic. Res. 2019, 53, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, S.M.; Araos, P.; Reyes, J.; Gravez, B.; Barrera-Chimal, J.; Amador, C.A. Oxidized Albumin as a Mediator of Kidney Disease. Antioxidants 2021, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.L.; Mira, J.P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Gryzunov, Y.A.; Arroyo, A.; Vigne, J.L.; Zhao, Q.; Tyurin, V.A.; Hubel, C.A.; Gandley, R.E.; Vladimirov, Y.A.; Taylor, R.N.; Kagan, V.E. Binding of fatty acids facilitates oxidation of cysteine-34 and converts copper-albumin complexes from antioxidants to prooxidants. Arch. Biochem. Biophys. 2003, 413, 53–66. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Ghuman, J.; McDonagh, A.F.; Curry, S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J. Mol. Biol. 2008, 381, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.D. Copper transport: An overview. Proc. Soc. Exp. Biol. Med. 1991, 196, 130–140. [Google Scholar] [CrossRef]
- Karpenko, M.N.; Ilyicheva, E.Y.; Muruzheva, Z.M.; Milyukhina, I.V.; Puchkova, L.V.; Orlov, Y.A. Role of copper dyshomeostasis in the pathogenesis of parkinson’s disease. Bull. Exp. Biol. Med. 2018, 164, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Laussac, J.P.; Sarkar, B. Characterization of the copper(II)- and nickel(II)-transport site of human serum albumin. Studies of copper(II) and nickel(II) binding to peptide 1–24 of human serum albumin by 13C and 1H NMR spectroscopy. Biochemistry 1984, 23, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Sendzik, M.; Pushie, M.J.; Stefaniak, E.; Haas, K.L. Structure and Affinity of Cu(I) Bound to Human Serum Albumin. Inorg. Chem. 2017, 56, 15057–15065. [Google Scholar] [CrossRef]
- Bar-Or, D.; Rael, L.T.; Lau, E.P.; Rao, N.K.; Thomas, G.W.; Winkler, J.V.; Yukl, R.L.; Kingston, R.G.; Curtis, C.G. An analog of the human albumin N-terminus (Asp-Ala-His-Lys) prevents formation of copper-induced reactive oxygen species. Biochem. Biophys. Res. Commun. 2001, 284, 856–862. [Google Scholar] [CrossRef]
- Pedersen, A.O.; Jacobsen, J. Reactivity of the thiol group in human and bovine albumin at pH 3–9, as measured by exchange with 2,2′-dithiodipyridine. Eur. J. Biochem. 1980, 106, 291–295. [Google Scholar] [CrossRef]
- Agarwal, R.P.; Phillips, M.; McPherson, R.A.; Hensley, P. Serum albumin and the metabolism of disulfiram. Biochem. Pharmacol. 1986, 35, 3341–3347. [Google Scholar] [CrossRef]
- Hurst, R.; Bao, Y.; Ridley, S.; Williamson, G. Phospholipid hydroperoxide cysteine peroxidase activity of human serum albumin. Biochem. J. 1999, 338, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.K.; Kim, I.H. Glutathione-linked thiol peroxidase activity of human serum albumin: A possible antioxidant role of serum albumin in blood plasma. Biochem. Biophys. Res. Commun. 1996, 222, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, I.H. Thioredoxin-linked lipid hydroperoxide peroxidase activity of human serum albumin in the presence of palmitoyl coenzyme A. Free Radic. Biol. Med. 2001, 30, 327–333. [Google Scholar] [CrossRef]
- Iwao, Y.; Ishima, Y.; Yamada, J.; Noguchi, T.; Kragh-Hansen, U.; Mera, K.; Honda, D.; Suenaga, A.; Maruyama, T.; Otagiri, M. Quantitative evaluation of the role of cysteine and methionine residues in the antioxidant activity of human serum albumin using recombinant mutants. IUBMB Life 2012, 64, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Costa, M.; Horrillo, R.; Ortiz, A.M.; Pérez, A.; Mestre, A.; Ruiz, A.; Boada, M.; Grancha, S. Increased Albumin Oxidation in Cerebrospinal Fluid and Plasma from Alzheimer’s Disease Patients. J. Alzheimers Dis. 2018, 63, 1395–1404. [Google Scholar] [CrossRef]
- Ueno, S.I.; Hatano, T.; Okuzumi, A.; Saiki, S.; Oji, Y.; Mori, A.; Koinuma, T.; Fujimaki, M.; Takeshige-Amano, H.; Kondo, A.; et al. Nonmercaptalbumin as an oxidative stress marker in Parkinson’s and PARK2 disease. Ann. Clin. Transl. Neurol. 2020, 7, 307–317. [Google Scholar] [CrossRef]
- Nasif, W.A.; Mukhtar, M.H.; El-Emshaty, H.M.; Alwazna, A.H. Redox State of Human Serum Albumin and Inflammatory Biomarkers in Hemodialysis Patients with Secondary Hyperparathyroidism During Oral Calcitriol Supplementation for Vitamin, D. Open Med. Chem. J. 2018, 12, 98–110. [Google Scholar] [CrossRef]
- Rael, L.T.; Leonard, J.; Salottolo, K.; Bar-Or, R.; Bartt, R.E.; Wagner, J.C.; Bar-Or, D. Plasma Oxidized Albumin in Acute Ischemic Stroke Is Associated With Better Outcomes. Front. Neurol. 2019, 10, 709. [Google Scholar] [CrossRef]
- Fujii, R.; Ueyama, J.; Aoi, A.; Ichino, N.; Osakabe, K.; Sugimoto, K.; Suzuki, K.; Hamajima, N.; Wakai, K.; Kondo, T. Oxidized human serum albumin as a possible correlation factor for atherosclerosis in a rural Japanese population: The results of the Yakumo Study. Environ. Health Prev. Med. 2018, 23, 1. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Gounden, V.; Ngu, M.; Anastasopoulou, C.; Jialal, I. Fructosamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Belsare, S.; Coté, G. Development of a colorimetric paper fluidic dipstick assay for measurement of glycated albumin to monitor gestational diabetes at the point-of-care. Talanta 2021, 223, 121728. [Google Scholar] [CrossRef]
- Bettiga, A.; Fiorio, F.; Di Marco, F.; Trevisani, F.; Romani, A.; Porrini, E.; Salonia, A.; Montorsi, F.; Vago, R. The Modern Western Diet Rich in Advanced Glycation End-Products (AGEs): An Overview of Its Impact on Obesity and Early Progression of Renal Pathology. Nutrients 2019, 11, 1748. [Google Scholar] [CrossRef]
- Bohlender, J.; Franke, S.; Stein, G.; Wolf, G. Advanced glycation end products and the kidney. Am. J. Physiol. Renal Physiol. 2005, 289, F645–F659. [Google Scholar] [CrossRef]
- Pötzsch, S.; Blankenhorn, A.; Navarrete Santos, A.; Silber, R.E.; Somoza, V.; Simm, A. The effect of an AGE-rich dietary extract on the activation of NF-κB depends on the cell model used. Food Funct. 2013, 4, 1023–1031. [Google Scholar] [CrossRef]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef]
- Frimat, M.; Daroux, M.; Litke, R.; Nevière, R.; Tessier, F.J.; Boulanger, E. Kidney, heart and brain: Three organs targeted by ageing and glycation. Clin. Sci. 2017, 131, 1069–1092. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.J. Advanced Glycation End Products (AGEs) May Be a Striking Link Between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K. Do advanced glycation end-products cause food allergy? Curr. Opin. Allergy Clin. Immunol. 2017, 17, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Sukkar, M.B.; Wood, L.G.; Tooze, M.; Simpson, J.L.; McDonald, V.M.; Gibson, P.G.; Wark, P.A. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur. Respir. J. 2012, 39, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.E.; Rojas, R.A.; Monasterio, A.V.; González, B.I.; Figueroa, C.I.; Manques, M.B.; Romero, E.J.; Llanos, L.J.; Valdés, M.E.; Cofré, L.C. Lesiones gástricas en pacientes infectados con Helicobacter pylori: Expresión de RAGE (receptor de productos de glicosilización avanzada) y otros inmunomarcadores. Expression of RAGE in Helicobacter pylori infested gastric biopsies. Rev. Med. Chil. 2013, 141, 1240–1248. [Google Scholar] [CrossRef]
- Kumar, P.A.; Chitra, P.S.; Reddy, G.B. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol. Concepts 2016, 7, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.N.; Oczypok, E.A.; Milutinovic, P.S.; Dutz, R.E.; Oury, T.D. RAGE-dependent VCAM-1 expression in the lung endothelium mediates IL-33-induced allergic airway inflammation. Allergy 2019, 74, 89–99. [Google Scholar] [CrossRef]
- Sotokawauchi, A.; Matsui, T.; Higashimoto, Y.; Yamagishi, S.I. Fructose causes endothelial cell damage via activation of advanced glycation end products-receptor system. Diabetes Vasc. Dis. Res. 2019, 16, 556–561. [Google Scholar] [CrossRef]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.-L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef]
- Li, L.-M.; Hou, D.-X.; Guo, Y.-L.; Yang, J.-W.; Liu, Y.; Zhang, C.-Y.; Zen, K. Role of MicroRNA-214–Targeting Phosphatase and Tensin Homolog in Advanced Glycation End Product-Induced Apoptosis Delay in Monocytes. J. Immunol. 2011, 186, 2552–2560. [Google Scholar] [CrossRef]
- Lyons, T.J.; Jenkins, A.J. Glycation, oxidation, and lipoxidation in the development of the complications of diabetes: A carbonyl stress hypothesis. Diabetes Rev. 1997, 5, 365–391. [Google Scholar]
- Neviere, R.; Yu, Y.; Wang, L.; Tessier, F.; Boulanger, E. Implication of advanced glycation end products (Ages) and their receptor (Rage) on myocardial contractile and mitochondrial functions. Glycoconj. J. 2016, 33, 607–617. [Google Scholar] [CrossRef]
- Ahmad, S.; Siddiqui, Z.; Rehman, S.; Khan, M.Y.; Khan, H.; Khanum, S.; Alouffi, S.; Saeed, M. A Glycation Angle to Look into the Diabetic Vasculopathy: Cause and Cure. Curr. Vasc. Pharmacol. 2017, 15, 352–364. [Google Scholar] [CrossRef]
- Copur, S.; Siriopol, D.; Afsar, B.; Comert, M.C.; Uzunkopru, G.; Sag, A.A.; Ortiz, A.; Covic, A.; van Raalte, D.H.; Cherney, D.Z.; et al. Serum glycated albumin predicts all-cause mortality in dialysis patients with diabetes mellitus: Meta-analysis and systematic review of a predictive biomarker. Acta Diabetol. 2021, 58, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, B.; Padmanabhan, G.; Ramanathan, K. Determination of serum glycated albumin and high sensitivity C—Reactive protein in the insight of cardiovascular complications in diabetic chronic kidney disease patients. Afr. Health Sci. 2020, 20, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Giglio, R.V.; Lo Sasso, B.; Agnello, L.; Bivona, G.; Maniscalco, R.; Ligi, D.; Mannello, F.; Ciaccio, M. Recent Updates and Advances in the Use of Glycated Albumin for the Diagnosis and Monitoring of Diabetes and Renal, Cerebro- and Cardio-Metabolic Diseases. J. Clin. Med. 2020, 9, 3634. [Google Scholar] [CrossRef]
- Sarmah, S.; Das, S.; Roy, A.S. Protective actions of bioactive flavonoids chrysin and luteolin on the glyoxal induced formation of advanced glycation end products and aggregation of human serum albumin: In vitro and molecular docking analysis. Int. J. Biol. Macromol. 2020, 165, 2275–2285. [Google Scholar] [CrossRef]
- Rondeau, P.; Bourdon, E. The glycation of albumin: Structural and functional impacts. Biochimie 2011, 93, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Basak, P.; Pramanik, A.; Majumder, R.; Ghosh, A.; Hazra, S.; Guria, M.; Bhattacharyya, M.; Banik, S.P. Ribosylation induced structural changes in Bovine Serum Albumin: Understanding high dietary sugar induced protein aggregation and amyloid formation. Heliyon 2020, 6, 05053. [Google Scholar] [CrossRef]
- Vlassopoulos, A.; Lean, M.E.; Combet, E. Role of oxidative stress in physiological albumin glycation: A neglected interaction. Free Radic. Biol. Med. 2013, 60, 318–324. [Google Scholar] [CrossRef]
- Ledesma-Osuna, A.I.; Ramos-Clamont, G.; Vázquez-Moreno, L. Characterization of bovine serum albumin glycated with glucose, galactose and lactose. Acta Biochim. Pol. 2008, 55, 491–497. [Google Scholar] [CrossRef]
- Soboleva, A.; Mavropulo-Stolyarenko, G.; Karonova, T.; Thieme, D.; Hoehenwarter, W.; Ihling, C.; Stefanov, V.; Grishina, T.; Frolov, A. Multiple Glycation Sites in Blood Plasma Proteins as an Integrated Biomarker of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 2329. [Google Scholar] [CrossRef]
- Qiu, H.; Jin, L.; Chen, J.; Shi, M.; Shi, F.; Wang, M.; Li, D.; Xu, X.; Su, X.; Yin, X.; et al. Comprehensive Glycomic Analysis Reveals That Human Serum Albumin Glycation Specifically Affects the Pharmacokinetics and Efficacy of Different Anticoagulant Drugs in Diabetes. Diabetes 2020, 69, 760–770. [Google Scholar] [CrossRef]
- Korça, E.; Piskovatska, V.; Börgermann, J.; Navarrete Santos, A.; Simm, A. Circulating antibodies against age-modified proteins in patients with coronary atherosclerosis. Sci. Rep. 2020, 10, 17105. [Google Scholar] [CrossRef]
- Qian, H.; Cao, Y.; Sun, J.; Zu, J.; Ma, L.; Zhou, H.; Tang, X.; Li, Y.; Yu, H.; Zhang, M.; et al. Anti-human serum albumin autoantibody may be involved in the pathogenesis of autoimmune bullous skin diseases. FASEB J. 2020, 34, 8574–8595. [Google Scholar] [CrossRef]
- Nass, N.; Bayreuther, K.; Simm, A. Systemic activation of NF-κB driven luciferase activity in transgenic mice fed advanced glycation end products modified albumin. Glycoconj. J. 2017, 34, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Freitas, P.A.C.; Ehlert, L.R.; Camargo, J.L. Glycated albumin: A potential biomarker in diabetes. Arch. Endocrinol. Metab. 2017, 61, 296–304. [Google Scholar] [CrossRef]
- Roohk, H.V.; Zaidi, A.R.; Patel, D. Glycated albumin (GA) and inflammation: Role of GA as a potential marker of inflammation. Inflamm. Res. 2018, 67, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, G.; Jing, P. Polyphenol binding disassembles glycation-modified bovine serum albumin amyloid fibrils. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 246, 119001. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, E.; Loreau, N.; Blache, D. Glucose and free radicals impair the antioxidant properties of serum albumin. FASEB J. 1999, 13, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Chesne, S.; Rondeau, P.; Armenta, S.; Bourdon, E. Effects of oxidative modifications induced by the glycation of bovine serum albumin on its structure and on cultured adipose cells. Biochimie 2006, 88, 1467–1477. [Google Scholar] [CrossRef]
- Rondeau, P.; Singh, N.R.; Caillens, H.; Tallet, F.; Bourdon, E. Oxidative stresses induced by glycoxidized human or bovine serum albumin on human monocytes. Free Radic. Biol. Med. 2008, 45, 799–812. [Google Scholar] [CrossRef]
- Martinez Fernandez, A.; Regazzoni, L.; Brioschi, M.; Gianazza, E.; Agostoni, P.; Aldini, G.; Banfi, C. Pro-oxidant and pro-inflammatory effects of glycated albumin on cardiomyocytes. Free Radic. Biol. Med. 2019, 144, 245–255. [Google Scholar] [CrossRef]
- Anthony-Regnitz, C.M.; Wilson, A.E.; Sweazea, K.L.; Braun, E.J. Fewer Exposed Lysine Residues May Explain Relative Resistance of Chicken Serum Albumin to In Vitro Protein Glycation in Comparison to Bovine Serum Albumin. J. Mol. Evol. 2020, 88, 653–661. [Google Scholar] [CrossRef]
- Martinez Del Rio, C.; Gutiérrez-Guerrero, Y.T. An Evolutionary Remedy for an Abominable Physiological Mystery: Benign Hyperglycemia in Birds. J. Mol. Evol. 2020, 88, 715–719. [Google Scholar] [CrossRef]
- Myers, G.J.; Wegner, J. Endothelial Glycocalyx and Cardiopulmonary Bypass. J. Extra Corpor. Technol. 2017, 49, 174–181. [Google Scholar]
- Perrin, R.M.; Harper, S.J.; Bates, D.O. A role for the endothelial glycocalyx in regulating microvascular permeability in diabetes mellitus. Cell Biochem. Biophys. 2007, 49, 65–72. [Google Scholar] [CrossRef]
- Bruegger, D.; Rehm, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Conzen, P.; Becker, B.F. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit. Care 2008, 12, R73. [Google Scholar] [CrossRef] [PubMed]
- Bundgaard, M. The three-dimensional organization of tight junctions in a capillary endothelium revealed by serial-section electron microscopy. J. Ultrastruct. Res. 1984, 88, 1–17. [Google Scholar] [CrossRef]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proc. Online 2009, 11, 32–51. [Google Scholar] [CrossRef]
- Aukland, K.; Kramer, G.C.; Renkin, E.M. Protein concentration of lymph and interstitial fluid in the rat tail. Am. J. Physiol. 1984, 247, H74–H79. [Google Scholar] [CrossRef] [PubMed]
- Rutili, G.; Arfors, K.E. Protein concentration in interstitial and lymphatic fluids from the subcutaneous tissue. Acta Physiol. Scand. 1997, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Milici, A.J.; Watrous, N.E.; Stukenbrok, H.; Palade, G.E. Transcytosis of albumin in capillary endothelium. J. Cell Biol. 1987, 105, 2603–2612. [Google Scholar] [CrossRef]
- Miyawaki-Shimizu, K.; Predescu, D.; Shimizu, J.; Broman, M.; Predescu, S.; Malik, A.B. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L405–L413. [Google Scholar] [CrossRef]
- Schubert, W.; Frank, P.G.; Razani, B.; Park, D.S.; Chow, C.W.; Lisanti, M.P. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 2001, 276, 48619–48622. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Batalova, A.A.; Goncharov, N.V. Serum Albumin. Encyclopedia 2021, 1, 65–75. [Google Scholar] [CrossRef]
- Armstrong, S.M.; Khajoee, V.; Wang, C.; Wang, T.; Tigdi, J.; Yin, J.; Kuebler, W.M.; Gillrie, M.; Davis, S.P.; Ho, M.; et al. Co-regulation of transcellular and paracellular leak across microvascular endothelium by dynamin and Rac. Am. J. Pathol. 2011, 180, 1308–1323. [Google Scholar] [CrossRef]
- Schubert, W.; Frank, P.G.; Woodman, S.E.; Hyogo, H.; Cohen, D.E.; Chow, C.W.; Lisanti, M.P. Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J. Biol. Chem. 2002, 277, 40091–40098. [Google Scholar] [CrossRef]
- Berde, C.B.; Hudson, B.S.; Simoni, R.D.; Sklar, L.A. Human serum albumin. Spectroscopic studies of binding and proximity relationships for fatty acids and bilirubin. J. Biol. Chem. 1979, 254, 391–400. [Google Scholar] [CrossRef]
- Baldo, G.; Fellin, R.; Manzatoa, E.; Baiocchi, M.R.; Ongaro, G.; Baggio, G.; Fabiani, F.; Pauluzzi, S.; Crepaldi, G. Characterization of hyperlipidemia in two patients with analbuminemia. Clin. Chim. Acta 1983, 128, 307–319. [Google Scholar] [CrossRef]
- Cormode, E.J.; Lyster, D.M.; Israels, S. Analbuminemia in a neonate. J. Pediatr. 1975, 86, 862–867. [Google Scholar] [CrossRef]
- Schnitzer, J.E.; Carley, W.W.; Palade, G.E. Albumin interacts specifically with a 60-kDa microvascular endothelial glycoprotein. Proc. Natl. Acad. Sci. USA 1988, 85, 6773–6777. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Song, W.; Bergenfeldt, M.; Sass, P.; Malik, A.B. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J. Biol. Chem. 1997, 272, 25968–25975. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, W.M.; Wittenberg, C.; Lee, W.L.; Reppien, E.; Goldenberg, N.M.; Lindner, K.; Gao, Y.; Winoto-Morbach, S.; Drab, M.; Mühlfeld, C.; et al. Thrombin stimulates albumin transcytosis in lung microvascular endothelial cells via activation of acid sphingomyelinase. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L720–L732. [Google Scholar] [CrossRef] [PubMed]
- John, T.A.; Vogel, S.M.; Tiruppathi, C.; Malik, A.B.; Minshall, R.D. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L187–L196. [Google Scholar] [CrossRef] [PubMed]
- Sarapultsev, A.P.; Rempel, S.V.; Kuznetsova, J.V.; Sarapultsev, G.P. Nanoparticle’s interactions with biological objects (The Review). J. Ural Med. Acad. Sci. 2016, 3, 97–111. (In Russian) [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef]
- Raheel, H.; Ghaffari, S.; Khosraviani, N.; Mintsopoulos, V.; Auyeung, D.; Wang, C.; Kim, Y.H.; Mullen, B.; Sung, H.K.; Ho, M.; et al. CD36 mediates albumin transcytosis by dermal but not lung microvascular endothelial cells: Role in fatty acid delivery. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L740–L750. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Popova, P.I.; Avdonin, P.P.; Kudryavtsev, I.V.; Serebryakova, M.K.; Korf, E.A.; Avdonin, P.V. Markers of Endothelial Cells in Normal and Pathological Conditions. Biochem. Moscow Suppl. Ser. A 2020, 14, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-β-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Undem, C.; Zaldumbide, J.; Rentsendorj, O.; Modekurty, S.; Dodd-O, J.M.; Scott, A.; Pearse, D.B.; et al. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L143–L153. [Google Scholar] [CrossRef]
- Hawighorst, T.; Oura, H.; Streit, M.; Janes, L.; Nguyen, L.; Brown, L.F.; Oliver, G.; Jackson, D.G.; Detmar, M. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene 2002, 21, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, A.; Abumrad, N.A. Role of CD36 in membrane transport of long-chain fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 139–145. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef]
- Peters, T., Jr. Serum albumin: Recent progress in the understanding of its structure and biosynthesis. Clin. Chem. 1977, 23, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug. Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Nergiz-Unal, R.; Rademakers, T.; Cosemans, J.M.; Heemskerk, J.W. CD36 as a multiple-ligand signaling receptor in atherothrombosis. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 42–55. [Google Scholar] [CrossRef]
- Nagase, S.; Shimamune, K.; Shumiya, S. Albumin-deficient rat mutant. Science 1979, 205, 590–591. [Google Scholar] [CrossRef]
- Lin, M.H.; Khnykin, D. Fatty acid transporters in skin development, function and disease. Biochim. Biophys. Acta 2014, 1841, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Colombo, E.S.; Lucas, S.N.; Hall, P.R.; Febbraio, M.; Paffett, M.L.; Campen, M.J. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol. Sci. 2013, 134, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Traoré, B.; Muanza, K.; Looareesuwan, S.; Supavej, S.; Khusmith, S.; Danis, M.; Viriyavejakul, P.; Gay, F. Cytoadherence characteristics of Plasmodium falciparum isolates in Thailand using an in vitro human lung endothelial cells model. Am. J. Trop. Med. Hyg. 2000, 62, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Azizi, P.M.; Li, Y.E.; Liu, J.; Wang, C.; Chan, K.L.; Hopperton, K.E.; Bazinet, R.P.; Heit, B.; Bilan, P.J.; et al. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E35–E44. [Google Scholar] [CrossRef]
- Amore, A.; Cirina, P.; Conti, G.; Cerutti, F.; Bagheri, N.; Emancipator, S.N.; Coppo, R. Amadori-configurated albumin induces nitric oxide-dependent apoptosis of endothelial cells: A possible mechanism of diabetic vasculopathy. Nephrol. Dial. Transplant. 2004, 19, 53–60. [Google Scholar] [CrossRef][Green Version]
- Moon, J.H.; Chae, M.K.; Kim, K.J.; Kim, H.M.; Cha, B.S.; Lee, H.C.; Kim, Y.J.; Lee, B.W. Decreased endothelial progenitor cells and increased serum glycated albumin are independently correlated with plaque-forming carotid artery atherosclerosis in type 2 diabetes patients without documented ischemic disease. Circ. J. 2012, 76, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.; Simionescu, M. Cellular mechanisms and signalling pathways activated by high glucose and AGE-albumin in the aortic endothelium. Arch. Physiol. Biochem. 2006, 112, 265–273. [Google Scholar] [CrossRef]
- Rubenstein, D.A.; Maria, Z.; Yin, W. Combined incubation of platelets and endothelial cells with glycated albumin: Altered thrombogenic and inflammatory responses. Diab. Vasc. Dis. Res. 2014, 11, 235–242. [Google Scholar] [CrossRef]
- Rubenstein, D.A.; Maria, Z.; Yin, W. Glycated albumin modulates endothelial cell thrombogenic and inflammatory responses. J. Diabetes Sci. Technol. 2011, 5, 703–713. [Google Scholar] [CrossRef]
- Nizheradze, K. Concanavalin A, but not glycated albumin, increases subendothelial deposition of von Willebrand factor in vitro. Endothelium 2006, 13, 245–248. [Google Scholar] [CrossRef]
- Wang, S.; Hirschberg, R. Diabetes-relevant regulation of cultured blood outgrowth endothelial cells. Microvasc. Res. 2009, 78, 174–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rashid, G.; Benchetrit, S.; Fishman, D.; Bernheim, J. Effect of advanced glycation end-products on gene expression and synthesis of TNF-alpha and endothelial nitric oxide synthase by endothelial cells. Kidney Int. 2004, 66, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Lo, W.Y.; Lin, L.J. Angiotensin-(1-7) decreases glycated albumin-induced endothelial interleukin-6 expression via modulation of miR-146a. Biochem. Biophys. Res. Commun. 2013, 430, 1157–1163. [Google Scholar] [CrossRef]
- Bala, K.; Gomes, J.; Gohil, N.K. Interaction of glycated human serum albumin with endothelial cells in a hemodynamic environment: Structural and functional correlates. Mol. Biosyst. 2011, 7, 3036–3041. [Google Scholar] [CrossRef]
- Kunt, T.; Forst, T.; Harzer, O.; Buchert, G.; Pfützner, A.; Löbig, M.; Zschäbitz, A.; Stofft, E.; Engelbach, M.; Beyer, J. The influence of advanced glycation endproducts (AGE) on the expression of human endothelial adhesion molecules. Exp. Clin. Endocrinol. Diabetes 1998, 106, 183–188. [Google Scholar] [CrossRef]
- Higai, K.; Shimamura, A.; Matsumoto, K. Amadori-modified glycated albumin predominantly induces E-selectin expression on human umbilical vein endothelial cells through NADPH oxidase activation. Clin. Chim. Acta 2006, 367, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Paradela-Dobarro, B.; Bravo, S.B.; Rozados-Luís, A.; González-Peteiro, M.; Varela-Román, A.; González-Juanatey, J.R.; García-Seara, J.; Alvarez, E. Inflammatory effects of in vivo glycated albumin from cardiovascular patients. Biomed. Pharmacother. 2019, 113, 108763. [Google Scholar] [CrossRef]
- Rodiño-Janeiro, B.K.; González-Peteiro, M.; Ucieda-Somoza, R.; González-Juanatey, J.R.; Alvarez, E. Glycated albumin, a precursor of advanced glycation end-products, up-regulates NADPH oxidase and enhances oxidative stress in human endothelial cells: Molecular correlate of diabetic vasculopathy. Diabetes Metab. Res. Rev. 2010, 26, 550–558. [Google Scholar] [CrossRef]
- Nagasu, H.; Satoh, M.; Kiyokage, E.; Kidokoro, K.; Toida, K.; Channon, K.M.; Kanwar, Y.S.; Sasaki, T.; Kashihara, N. Activation of endothelial NAD(P)H oxidase accelerates early glomerular injury in di-abetic mice. Lab. Investig. 2016, 96, 25–36. [Google Scholar] [CrossRef]
- Rodiño-Janeiro, B.K.; Paradela-Dobarro, B.; Raposeiras-Roubín, S.; González-Peteiro, M.; González-Juanatey, J.R.; Álvarez, E. Glycated human serum albumin induces NF-κB activation and endothelial nitric oxide synthase uncoupling in human umbilical vein endothelial cells. J. Diabetes Complicat. 2015, 29, 984–992. [Google Scholar] [CrossRef]
- Amore, A.; Cirina, P.; Mitola, S.; Peruzzi, L.; Gianoglio, B.; Rabbone, I.; Sacchetti, C.; Cerutti, F.; Grillo, C.; Coppo, R. Nonenzymatically glycated albumin (Amadori adducts) enhances nitric oxide synthase activity and gene expression in endothelial cells. Kidney Int. 1997, 51, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Hayes, R.G.; Stitt, A.W.; McAuley, E.; Archer, D.B. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is suppressed by high glucose and advanced glycation end products. Diabetes 1998, 47, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Dobi, A.; Bravo, S.B.; Veeren, B.; Paradela-Dobarro, B.; Álvarez, E.; Meilhac, O.; Viranaicken, W.; Baret, P.; Devin, A.; Rondeau, P. Advanced glycation end-products disrupt human endothelial cells redox homeostasis: New insights into reactive oxygen species production. Free Radic. Res. 2019, 53, 150–169. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, G.; Liu, Z.; Xu, Q.; Li, C.; Cheng, G.; Shi, R. Peroxidasin promotes diabetic vascular endothelial dysfunction induced by advanced glycation end products via NOX2/HOCl/Akt/eNOS pathway. Redox Biol. 2021, 45, 102031. [Google Scholar] [CrossRef]
- Ravi, R.; Ragavachetty Nagaraj, N.; Subramaniam Rajesh, B. Effect of advanced glycation end product on paraoxonase 2 expression: Its impact on endoplasmic reticulum stress and inflammation in HUVECs. Life Sci. 2020, 246, 117397. [Google Scholar] [CrossRef]
- Soaita, I.; Yin, W.; Rubenstein, D.A. Glycated albumin modifies platelet adhesion and aggregation responses. Platelets 2017, 28, 682–690. [Google Scholar] [CrossRef]
- Son, K.H.; Son, M.; Ahn, H.; Oh, S.; Yum, Y.; Choi, C.H.; Park, K.Y.; Byun, K. Age-related accumulation of advanced glycation end-products-albumin, S100beta, and the expressions of advanced glycation end product receptor differ in visceral and subcutaneous fat. Biochem. Biophys. Res. Commun. 2016, 477, 271–276. [Google Scholar] [CrossRef]
- Takeshita, Y.; Sato, R.; Kanda, T. Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. Int. J. Mol. Sci. 2021, 22, 62. [Google Scholar] [CrossRef]
- Goldsammler, M.; Merhi, Z.; Buyuk, E. Role of hormonal and inflammatory alterations in obesity-related reproductive dysfunction at the level of the hypothalamic-pituitary-ovarian axis. Reprod. Biol. Endocrinol. 2018, 16, 45. [Google Scholar] [CrossRef]
- Kuniyasu, A.; Ohgami, N.; Hayashi, S.; Miyazaki, A.; Horiuchi, S.; Nakayama, H. CD36-mediated endocytic uptake of advanced glycation end products (AGE) in mouse 3T3-L1 and human subcutaneous adipocytes. FEBS Lett. 2003, 537, 85–90. [Google Scholar] [CrossRef]
- Harati, Y. Diabetic neuropathies: Unanswered questions. Neurol. Clin. 2007, 25, 303–317. [Google Scholar] [CrossRef]
- Linn, T.; Ortac, K.; Laube, H.; Federlin, K. Intensive therapy in adult insulin-dependent diabetes mellitus is associated with improved insulin sensitivity and reserve: A randomized controlled prospective study over 5 years in newly diagnosed patients. Metabolism 1996, 45, 1508–1513. [Google Scholar] [CrossRef]
- Ismail-Beigi, F.; Craven, T.; Banerji, M.A.; Basile, J.; Calles, J.; Cohen, R.M.; Cuddihy, R.; Cushman, W.C.; Genuth, S.; Grimm, R.H., Jr.; et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: An analysis of the ACCORD randomised trial. Lancet 2010, 376, 419–430. [Google Scholar] [CrossRef]
- Ang, L.; Jaiswal, M.; Martin, C.; Pop-Busui, R. Glucose control and diabetic neuropathy: Lessons from recent large clinical trials. Curr. Diabetes Rep. 2014, 14, 528. [Google Scholar] [CrossRef]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.; van den Born, J.; van den Heuvel, M.C.; van der Vlag, J.; Krenning, G.; Hillebrands, J.L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.; Duan, J.L.; Xu, H.; Tao, K.S.; Han, H.; Dou, G.R.; Wang, L. Capillarized Liver Sinusoidal Endothelial Cells Undergo Partial Endothelial-Mesenchymal Transition to Actively Deposit Sinusoidal ECM in Liver Fibrosis. Front. Cell. Dev. Biol. 2021, 9, 671081. [Google Scholar] [CrossRef] [PubMed]
- Adjuto-Saccone, M.; Soubeyran, P.; Garcia, J.; Audebert, S.; Camoin, L.; Rubis, M.; Roques, J.; Binétruy, B.; Iovanna, J.L.; Tournaire, R. TNF-α induces endothelial-mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death Dis. 2021, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Li, Y.; Wang, C.; Zhang, J.; Chen, Y.; Chen, W.; Cao, J.; Wang, Y.; Hu, Z.; Lou, T. ROCK1 Induces Endothelial-to-Mesenchymal Transition in Glomeruli to Aggravate Albuminuria in Diabetic Nephropathy. Sci. Rep. 2016, 6, 20304. [Google Scholar] [CrossRef]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Welsh, G.I.; Raghu, G.; Menon, R.K.; Saleem, M.A.; Reddy, G.B. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch. Biochem. Biophys. 2016, 590, 10–19. [Google Scholar] [CrossRef][Green Version]
- Chen, S.; Cohen, M.P.; Ziyadeh, F.N. Amadori-glycated albumin in diabetic nephropathy: Pathophysiologic connections. Kidney Int. Suppl. 2000, 77, S40–S44. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cohen, M.P.; Lautenslager, G.T.; Shearman, C.W.; Ziyadeh, F.N. Glycated albumin stimulates TGF-beta 1 production and protein kinase C activity in glomerular endothelial cells. Kidney Int. 2001, 59, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Patschan, D.; Schwarze, K.; Henze, E.; Patschanm, S.; Muller, G.A. Endothelial autophagy and Endothelial-to-Mesenchymal Transition (EndoMT) in eEPC treatment of ischemic AKI. J. Nephrol. 2016, 29, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Feng, Y.; Wang, Y.; Xiang, D.; Zhang, X.; Yuan, F. Autophagy regulates Endothelial-Mesenchymal transition by decreasing the phosphorylation level of Smad3. Biochem. Biophys. Res. Commun. 2017, 487, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutiérrez-Ocampo, E.; Villamizar-Peña, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef] [PubMed]
- Rahmani-Kukia, N.; Abbasi, A.; Pakravan, N.; Hassan, Z.M. Measurement of oxidized albumin: An opportunity for diagnoses or treatment of COVID-19. Bioorgan. Chem. 2020, 105, 104429. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Salvadori, L.; Vukasinovic, A.; Donato, R.; Sorci, G.; Riuzzi, F. Targeting RAGE to prevent SARS-CoV-2-mediated multiple organ failure: Hypotheses and perspectives. Life Sci. 2021, 272, 119251. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Salvadori, L.; Donato, R.; Riuzzi, F.; Sorci, G. Hyperactivated RAGE in Comorbidities as a Risk Factor for Severe COVID-19-The Role of RAGE-RAS Crosstalk. Biomolecules 2021, 11, 876. [Google Scholar] [CrossRef]
- Dobi, A.; Rosanaly, S.; Devin, A.; Baret, P.; Meilhac, O.; Harry, G.J.; d’Hellencourt, C.L.; Rondeau, P. Advanced glycation end-products disrupt brain microvascular endothelial cell barrier: The role of mitochondria and oxidative stress. Microvasc. Res. 2021, 133, 104098. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Popova, P.I.; Golovkin, A.S.; Zalutskaya, N.M.; Palchikova, E.I.; Zanin, K.V.; Avdonin, P.V. Vascular endotelial dysfunction is a pathogenetic factor in the development of neurodegenerative diseases and cognitive impairment. Bekhterev Rev. Psychiatry Med. Psychol. 2020, 2020, 11–26. (In Russian) [Google Scholar] [CrossRef]
- Swissa, E.; Serlin, Y.; Vazana, U.; Prager, O.; Friedman, A. Blood-brain barrier dysfunction in status epileptics: Mechanisms and role in epileptogenesis. Epilepsy Behav. 2019, 101, 106285. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Ivens, S.; Kaufer, D.; Flores, L.P.; Bechmann, I.; Zumsteg, D.; Tomkins, O.; Seiffert, E.; Heinemann, U.; Friedman, A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007, 130, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Senatorov, V.V., Jr.; Morrissey, C.S.; Lippmann, K.; Vazquez, O.; Milikovsky, D.Z.; Gu, F.; Parada, I.; Prince, D.A.; Becker, A.J.; et al. TGFβ signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci. Rep. 2017, 7, 7711. [Google Scholar] [CrossRef]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11, eaaw8283. [Google Scholar] [CrossRef]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Vega-Zelaya, L.; Ortega, G.J.; Sola, R.G.; Pastor, J. Plasma albumin induces cytosolic calcium oscilations and DNA synthesis in human cultured astrocytes. Biomed. Res. Int. 2014, 2014, 539140. [Google Scholar] [CrossRef] [PubMed]
- Gatta, A.; Verardo, A.; Bolognesi, M. Hypoalbuminemia. Intern. Emerg. Med. 2012, 7, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Kettunen, J.; Würtz, P.; Haller, T.; Havulinna, A.S.; Kangas, A.J.; Soininen, P.; Esko, T.; Tammesoo, M.L.; Mägi, R.; et al. Biomarker profiling by nuclear magnetic resonance spectroscopy for the prediction of all-cause mortality: An observational study of 17,345 persons. PLoS Med. 2014, 11, 1001606. [Google Scholar] [CrossRef]
- Deelen, J.; Kettunen, J.; Fischer, K.; van der Spek, A.; Trompet, S.; Kastenmüller, G.; Boyd, A.; Zierer, J.; van den Akker, E.B.; Ala-Korpela, M.; et al. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nat. Commun. 2019, 10, 3346. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Kaur, N.; Sarangal, V. A study to evaluate the correlation of serum albumin levels with chronic periodontitis. Indian J. Dent. Res. 2015, 26, 11–14. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Z.; Tian, M.; Xu, P.; Li, B.; Yang, Q.; Yang, Y. Relationship Between Serum Albumin Levels and Infections in Newborn Late Preterm Infants. Med. Sci. Monit. 2016, 22, 92–98. [Google Scholar] [CrossRef]
- Merriel, S.W.; Carroll, R.; Hamilton, F.; Hamilton, W. Association between unexplained hypoalbuminaemia and new cancer diagnoses in UK primary care patients. Fam. Pract. 2016, 33, 449–452. [Google Scholar] [CrossRef]
- Ikeda, S.; Yoshioka, H.; Ikeo, S.; Morita, M.; Sone, N.; Niwa, T.; Nishiyama, A.; Yokoyama, T.; Sekine, A.; Ogura, T.; et al. Serum albumin level as a potential marker for deciding chemotherapy or best supportive care in elderly, advanced non-small cell lung cancer patients with poor performance status. BMC Cancer 2017, 17, 797. [Google Scholar] [CrossRef]
- Poudel-Tandukar, K.; Jacelon, C.S.; Bertone-Johnson, E.R.; Palmer, P.H.; Poudel, K.C. Serum albumin levels and depression in people living with Human Immunodeficiency Virus infection: A cross-sectional study. J. Psychosom. Res. 2017, 101, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Tsirpanlis, G.; Bagos, P.; Ioannou, D.; Bleta, A.; Marinou, I.; Lagouranis, A.; Chatzipanagiotou, S.; Nicolaou, C. Serum albumin: A late-reacting negative acute-phase protein in clinically evident inflammation in dialysis patients. Nephrol. Dial. Transplant. 2005, 20, 658–660. [Google Scholar] [CrossRef]
- Thi, T.N.D.; Gia, B.N.; Thi, H.L.L.; Thi, T.N.C.; Thanh, H.P. Evaluation of urinary L-FABP as an early marker for diabetic nephropathy in type 2 diabetic patients. J. Med. Biochem. 2020, 39, 224–230. [Google Scholar] [CrossRef]
- Chen, L.; Jin, C.; Chen, L.; Li, M.; Zhong, Y.; Xu, Y. Value of microalbuminuria in the diagnosis of heart failure with preserved ejection fraction. Herz 2020. [Google Scholar] [CrossRef]
- Arogundade, F.A. Detection of Early Renal Disease in Children With Sickle Cell Anaemia Using Microalbuminuria As A Surrogate Marker. West. Afr. J. Med. 2020, 37, 327. [Google Scholar]
- Hwang, J.C.; Jiang, M.Y.; Lu, Y.H.; Wang, C.T. Precedent fluctuation of serum hs-CRP to albumin ratios and mortality risk of clinically stable hemodialysis patients. PLoS ONE 2015, 10, 0120266. [Google Scholar] [CrossRef]
- Minigalin, A.D.; Voitenko, N.G.; Vorobyov, A.A.; Korf, E.A.; Novozhilov, A.V.; Petukhova, O.V.; Baranova, T.I.; Goncharov, N.V. Investigation of relations between physiological and biochemical parameters of human beings in dynamics after performing a maximal workload. Physiother. Sport Med. 2015, 132, 14–18. (In Russian) [Google Scholar]
- Suominen, A.; Jahnukainen, T.; Ojala, T.H.; Sarkola, T.; Turanlahti, M.; Saarinen-Pihkala, U.M.; Jahnukainen, K. Long-term renal prognosis and risk for hypertension after myeloablative therapies in survivors of childhood high-risk neuroblastoma: A nationwide study. Pediatr. Blood Cancer 2020, 67, e28209. [Google Scholar] [CrossRef]
- Violi, F.; Cangemi, R.; Romiti, G.F.; Ceccarelli, G.; Oliva, A.; Alessandri, F.; Pirro, M.; Pignatelli, P.; Lichtner, M.; Carraro, A.; et al. Is Albumin Predictor of Mortality in COVID-19? Antioxid. Redox Signal 2021, 35, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Yasukawa, K.; Sato, M.; Ikeda, H.; Inoguchi, Y.; Etoh, T.; Masakado, M.; Umeda, F.; Yatomi, Y.; Yamauchi, T.; et al. Clinical usefulness of human serum nonmercaptalbumin to mercaptalbumin ratio as a biomarker for diabetic complications and disability in activities of daily living in elderly patients with diabetes. Metabolism 2020, 103, 153995. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; He, Y.; Shi, B.; Yang, D. Human serum albumin from recombinant DNA technology: Challenges and strategies. Biochim. Biophys. Acta 2013, 1830, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Liumbruno, G.M.; Bennardello, F.; Lattanzio, A.; Piccoli, P.; Rossettias, G. Recommendations for the use of albumin and immunoglobulins. Blood Transfus. 2009, 7, 216–234. [Google Scholar] [CrossRef]
- Tullis, J.L. Albumin: 1. Background and Use. JAMA 1977, 237, 355. [Google Scholar] [CrossRef]
- Melia, D.; Post, B. Human albumin solutions in intensive care: A review. J. Intensive. Care Soc. 2021, 22, 248–254. [Google Scholar] [CrossRef]
- Schneider, F.; Dureau, A.F.; Hellé, S.; Betscha, C.; Senger, B.; Cremel, G.; Boulmedais, F.; Strub, J.M.; Corti, A.; Meyer, N.; et al. A Pilot Study on Continuous Infusion of 4% Albumin in Critically Ill Patients: Impact on Nosocomial Infection via a Reduction Mechanism for Oxidized Substrates. Crit. Care Explor. 2019, 1, 0044. [Google Scholar] [CrossRef]
- Ikeda, M.; Ishima, Y.; Kinoshita, R.; Chuang, V.T.G.; Tasaka, N.; Matsuo, N.; Watanabe, H.; Shimizu, T.; Ishida, T.; Otagiri, M.; et al. A novel S-sulfhydrated human serum albumin preparation suppresses melanin synthesis. Redox Biol. 2018, 14, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Sebe, A.; Ay, M.O.; Gumusay, U.; Topal, M.; Atli, M.; Icme, F.; Satar, S. Effectiveness of therapeutic plasma exchange in patients with intermediate syndrome due to organophosphate intoxication. Am. J. Emerg. Med. 2013, 31, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Tarin Remohi, M.J.; Sanchez Arcos, A.; Santos Ramos, B.; Bautista Paloma, J.; Guerrero Aznar, M.D. Costs related to inappropriate use of albumin in Spain. Ann. Pharmacother. 2000, 34, 1198–1205. [Google Scholar] [CrossRef]
- Arola-Arnal, A.; López de Las Hazas, M.C.; Iglesias-Carres, L.; Mantilla-Escalante, D.C.; Suárez, M.; Busto, R.; Visioli, F.; Bladé, C.; Dávalos, A. Exosomes transport trace amounts of (poly)phenols. Food Funct. 2020, 11, 7784–7792. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Kato, T.; Tanabe, H.; Taguchi-Atarashi, N.; Sato, Y.; Eguchi, A.; Watanabe, M.; Ohno, H.; Mori, C. Association between gut microbiota composition and glycoalbumin level during pregnancy in Japanese women: Pilot study from Chiba Study of Mother and Child Health. J. Diabetes Investig. 2020, 11, 699–706. [Google Scholar] [CrossRef]
- Hong, M.; Zhang, R.; Liu, Y.; Wu, Z.; Weng, P. The interaction effect between tea polyphenols and intestinal microbiota: Role in ameliorating neurological diseases [published online ahead of print, 2021 Jul 19]. J. Food. Biochem. 2021. [Google Scholar] [CrossRef]
- Vlassopoulos, A.; Lean, M.E.; Combet, E. Protein-phenolic interactions and inhibition of glycation—Combining a systematic review and experimental models for enhanced physiological relevance. Food Funct. 2014, 5, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.K.; Qian, Y.; Leonard, S.S.; Sbarra, D.C.; Shi, X. Luteolin and chrysin differentially inhibit cyclooxygenase-2 expression and scavenge reactive oxygen species but similarly inhibit prostaglandin-E2 formation in RAW 264.7 cells. J. Nutr. 2006, 136, 1517–1521. [Google Scholar] [CrossRef]
- Khan, M.W.A.; Al Otaibi, A.; Sherwani, S.; Khan, W.A.; Alshammari, E.M.; Al-Zahrani, S.A.; Saleem, M.; Khan, S.N.; Alouffi, S. Glycation and Oxidative Stress Increase Autoantibodies in the Elderly. Molecules 2020, 25, 3675. [Google Scholar] [CrossRef]
- Anwar, S.; Khan, S.; Almatroudi, A.; Khan, A.A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H. A review on mechanism of inhibition of advanced glycation end products formation by plant derived polyphenolic compounds. Mol. Biol. Rep. 2021, 48, 787–805. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. https://doi.org/10.3390/ijms221910318
Belinskaia DA, Voronina PA, Shmurak VI, Jenkins RO, Goncharov NV. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. International Journal of Molecular Sciences. 2021; 22(19):10318. https://doi.org/10.3390/ijms221910318
Chicago/Turabian StyleBelinskaia, Daria A., Polina A. Voronina, Vladimir I. Shmurak, Richard O. Jenkins, and Nikolay V. Goncharov. 2021. "Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties" International Journal of Molecular Sciences 22, no. 19: 10318. https://doi.org/10.3390/ijms221910318
APA StyleBelinskaia, D. A., Voronina, P. A., Shmurak, V. I., Jenkins, R. O., & Goncharov, N. V. (2021). Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. International Journal of Molecular Sciences, 22(19), 10318. https://doi.org/10.3390/ijms221910318