
Circular RNAs Repertoire and Expression Profile during Brassica rapa Pollen Development


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Growth Conditions, and Sample Collection
2.2. RNA Extraction and Sequencing
2.3. Identification and Differential Expression of Circular RNAs
2.4. Conservation Analysis of CircRNAs
2.5. Functional Enrichment Analysis
2.6. Prediction of Potential circRNA–miRNA Interactions and Visualization
2.7. Circular RNA Validation
3. Results
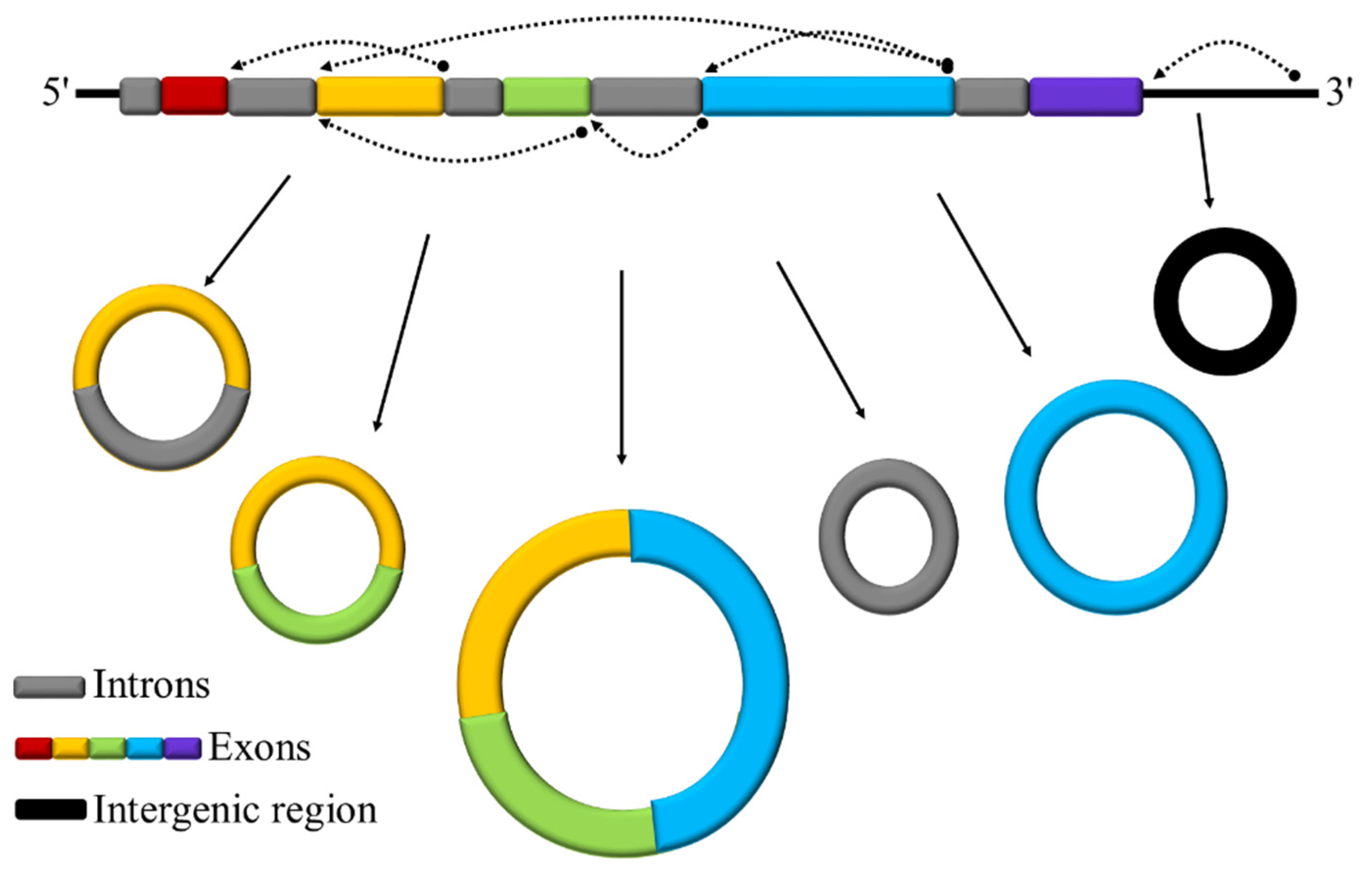
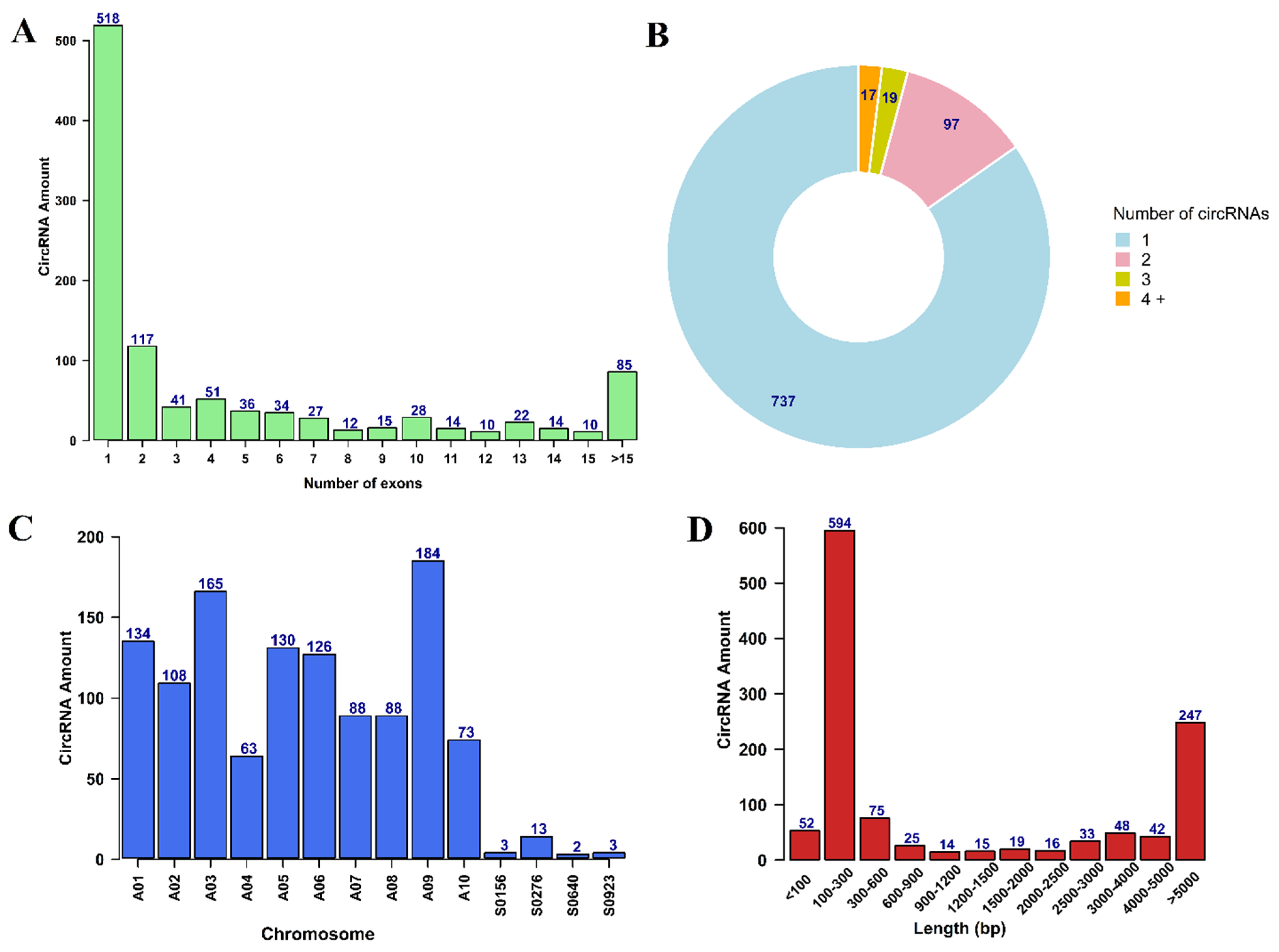
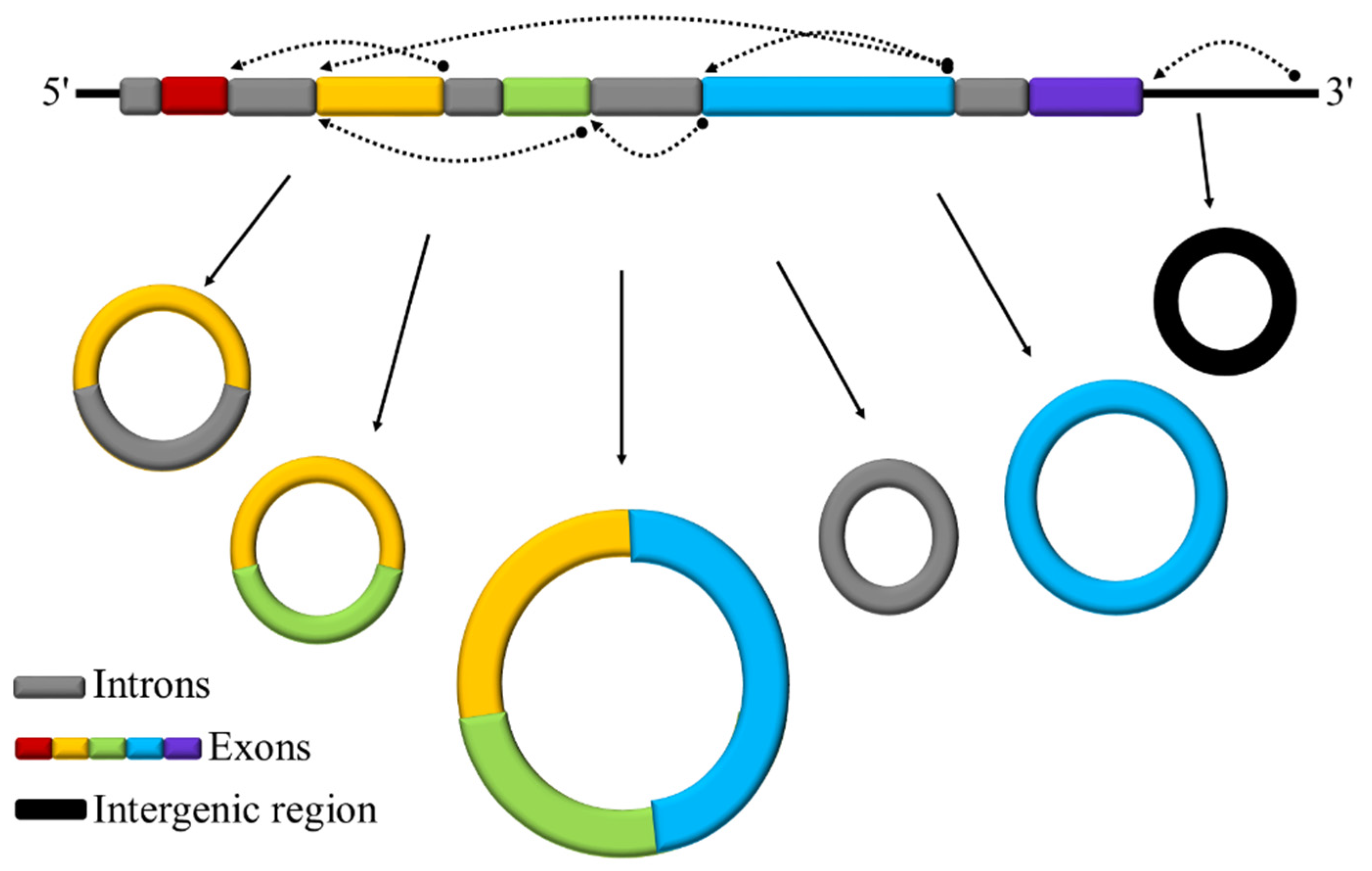
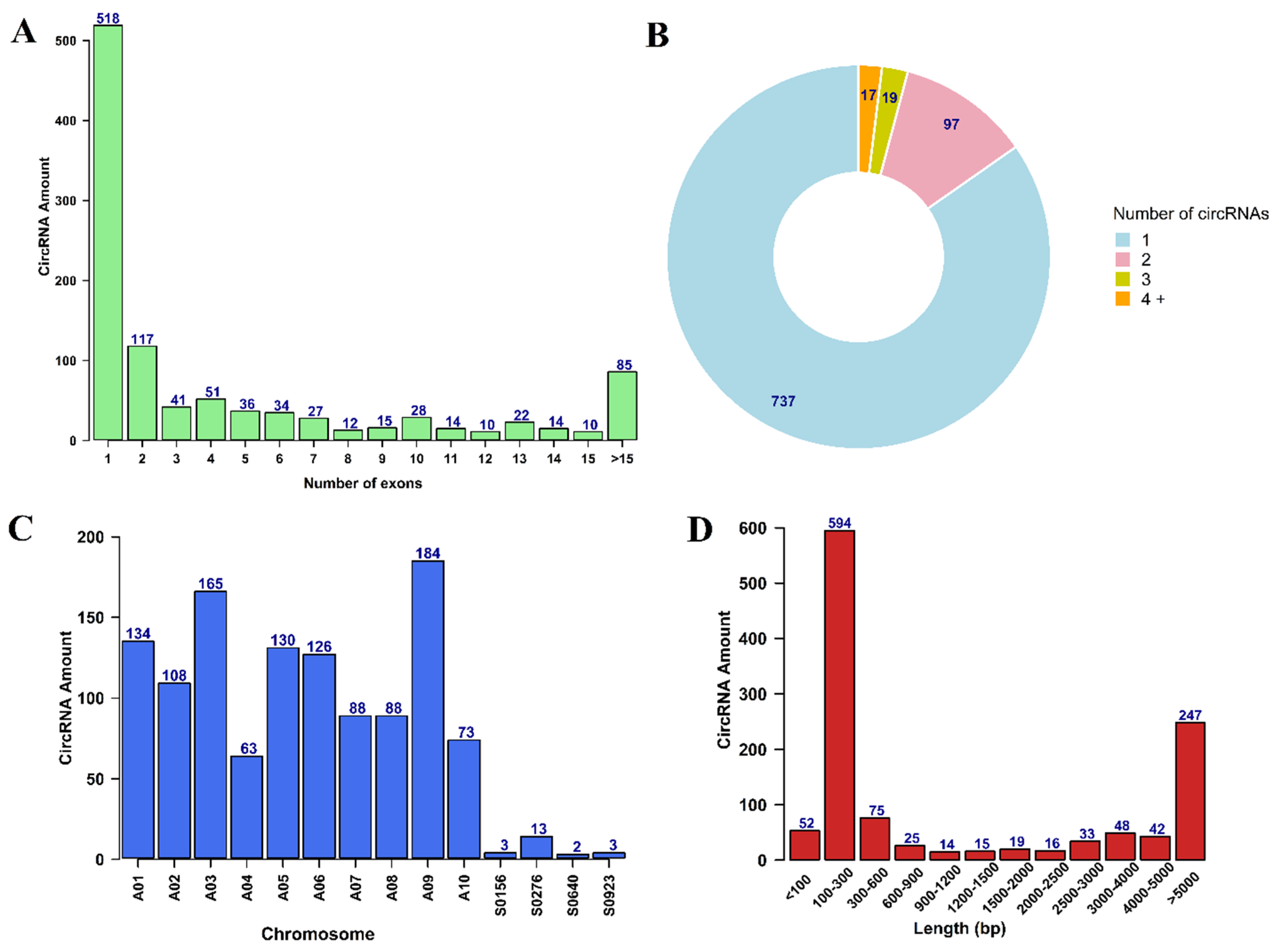
3.1. Identification and Characterization of circRNAs
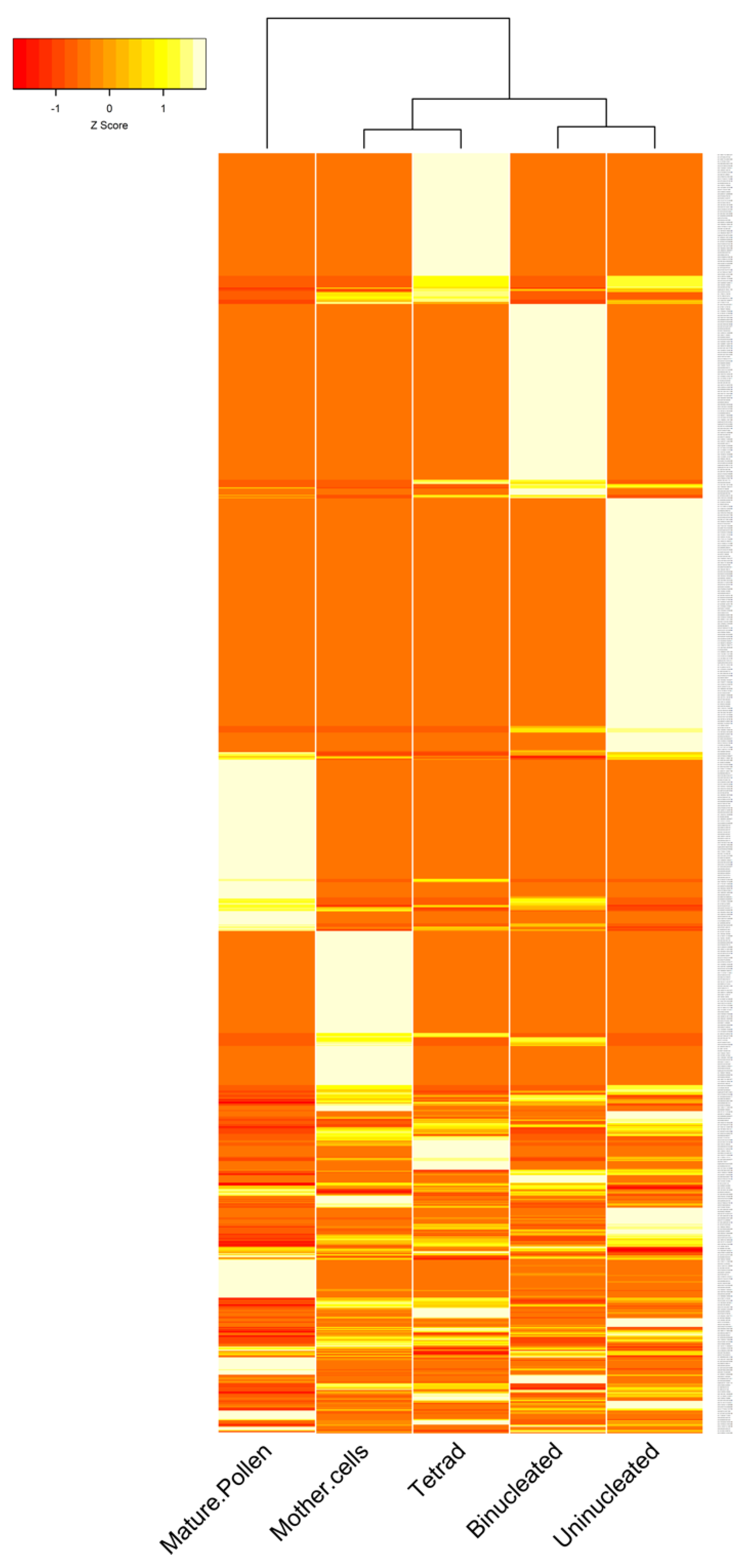
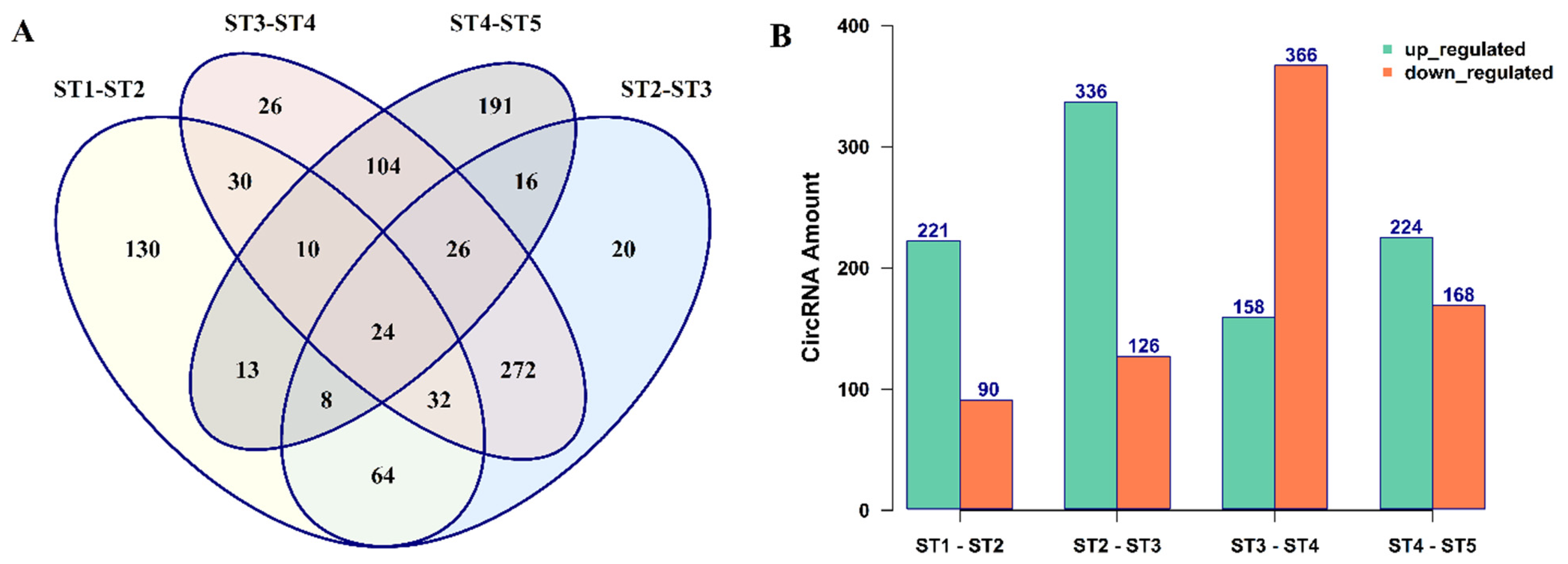
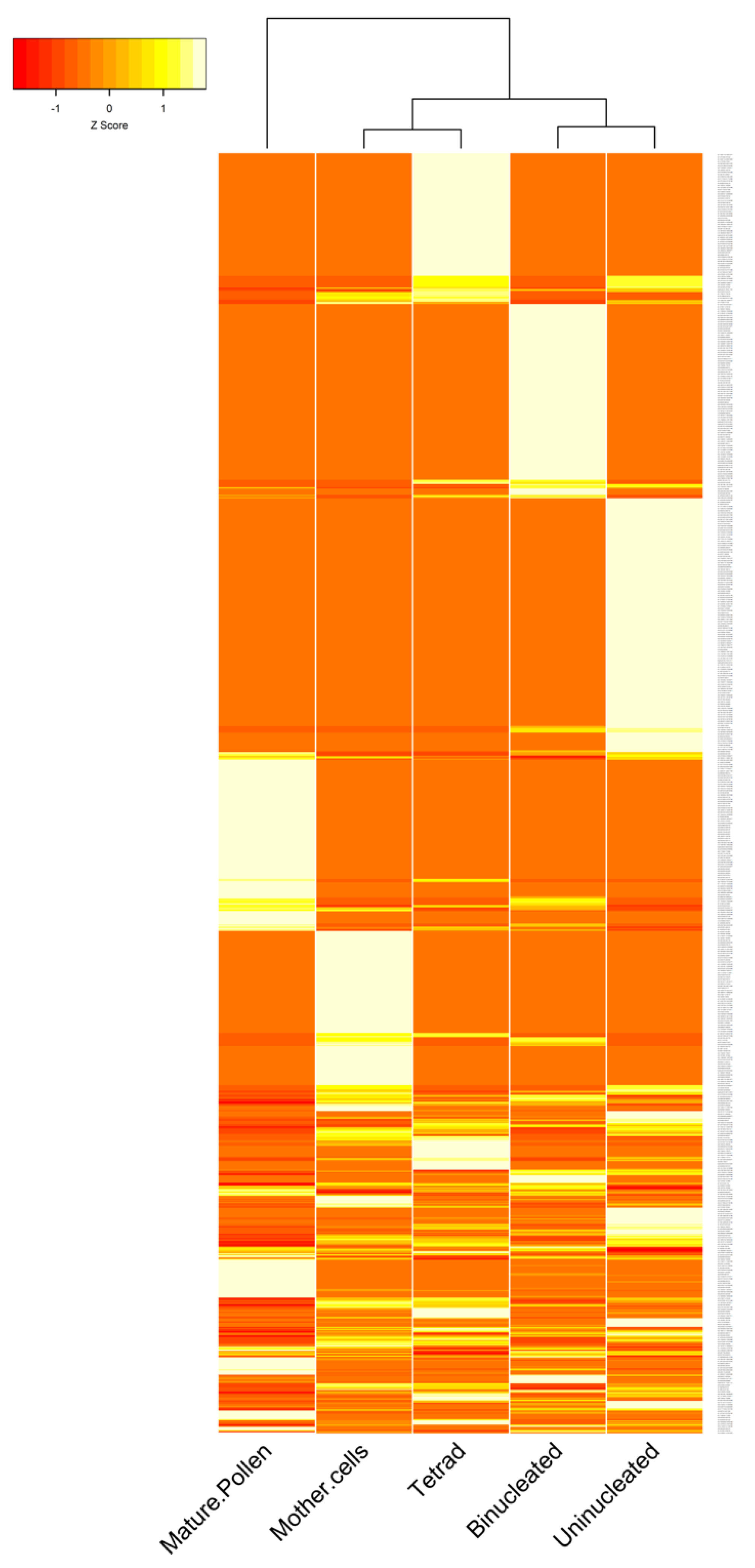
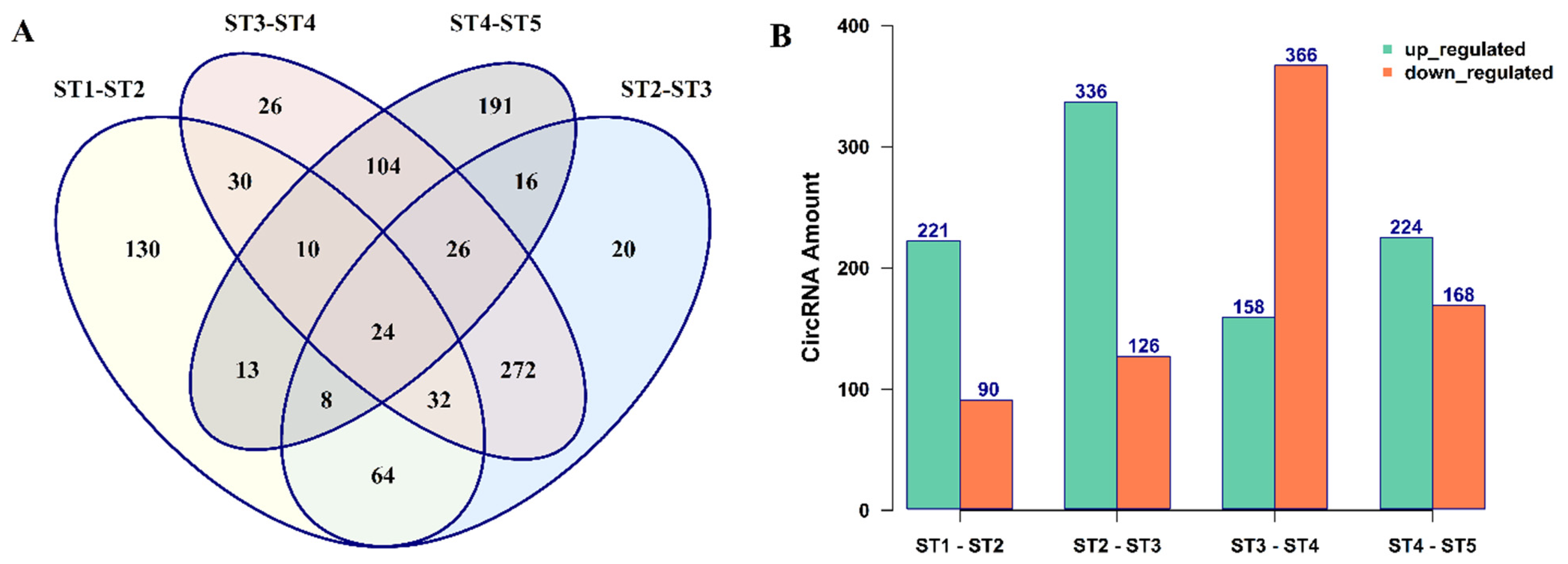
3.2. The Expression Patterns of circRNAs in B. rapa Pollen Development
3.3. Conservation Analysis of circRNAs between B. rapa and other Plant Species
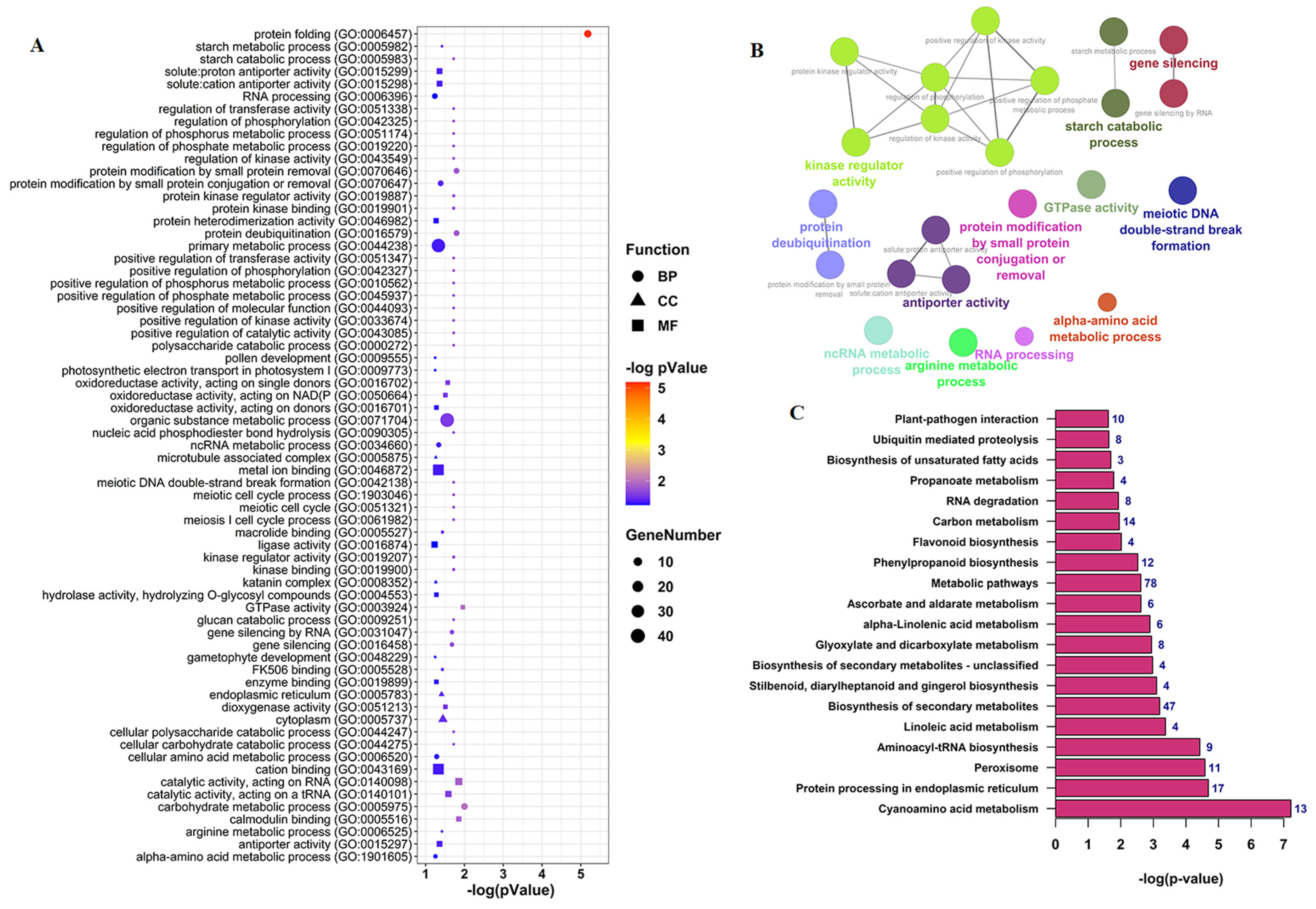
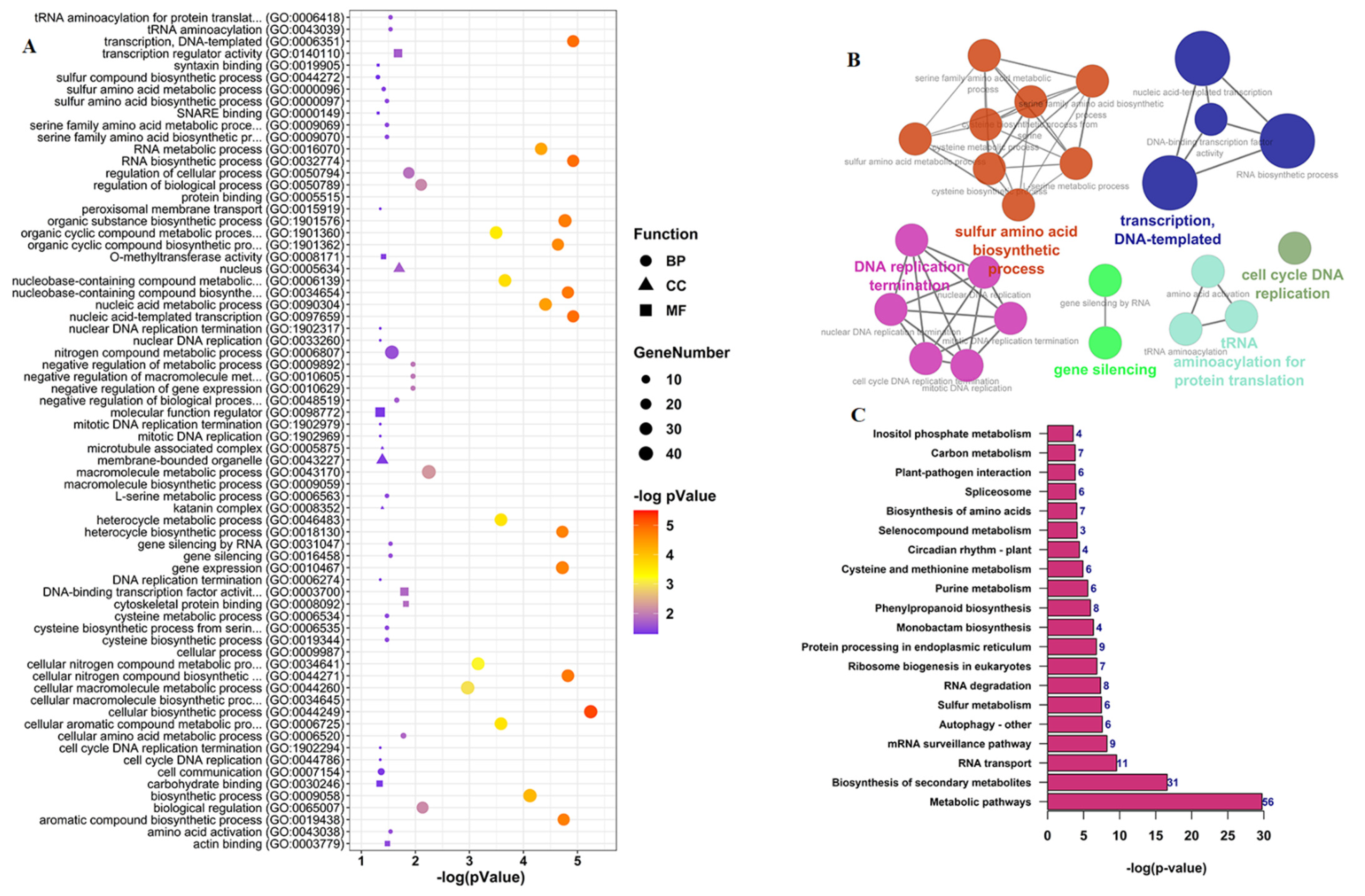
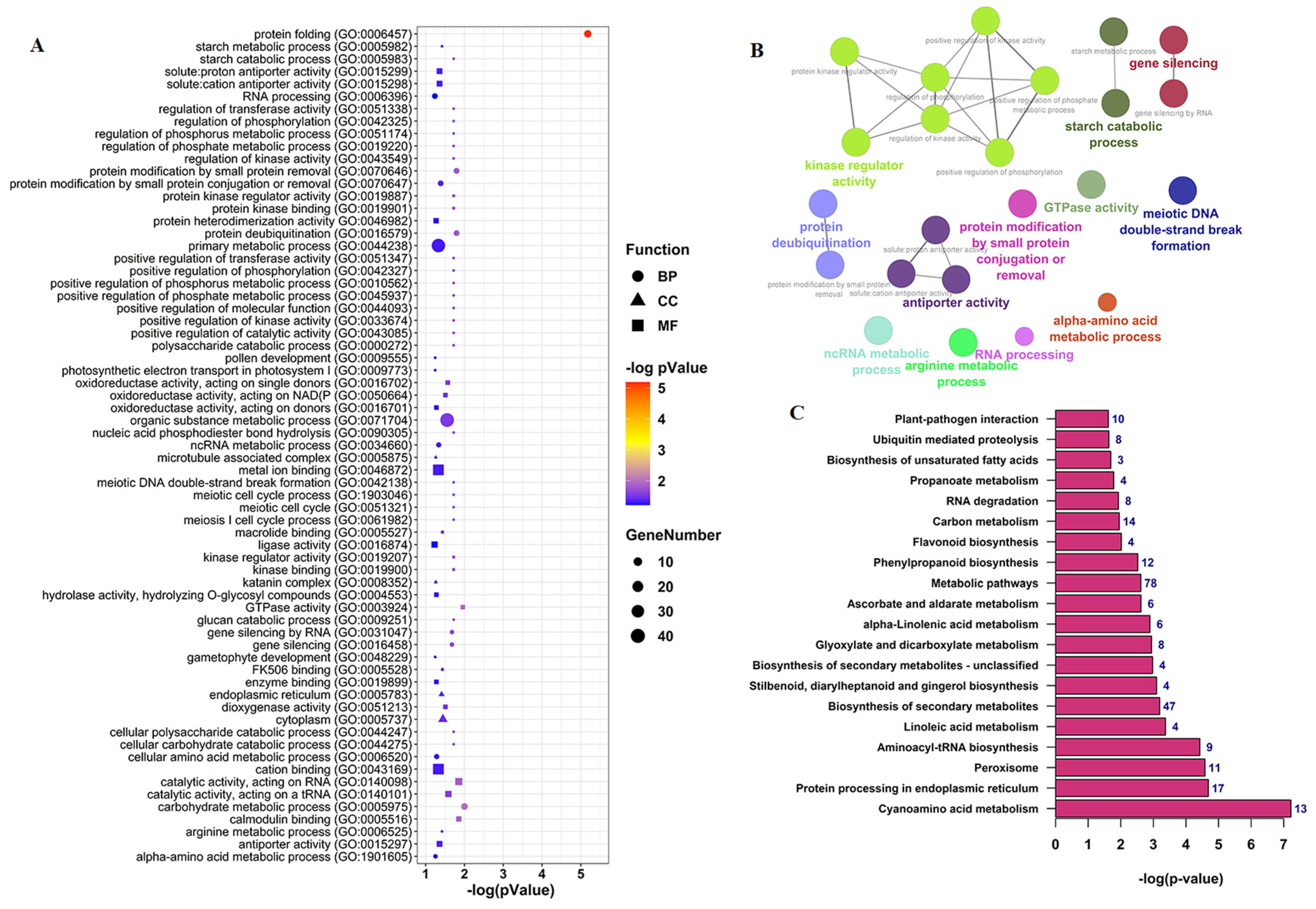
3.4. Functional Annotation Analysis of CircRNAs Parent Genes
3.5. Prediction of miRNA Target Sites in circRNAs
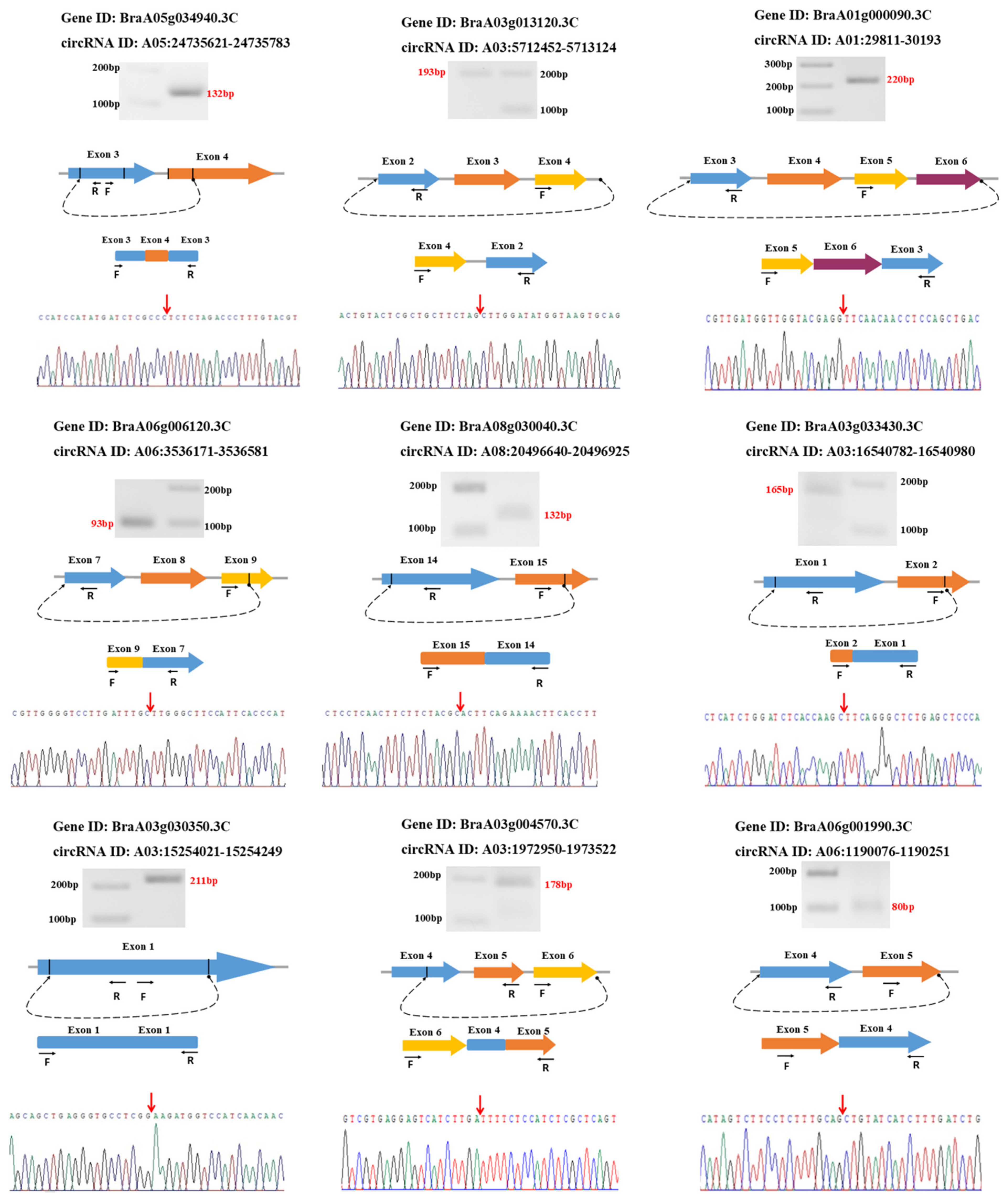
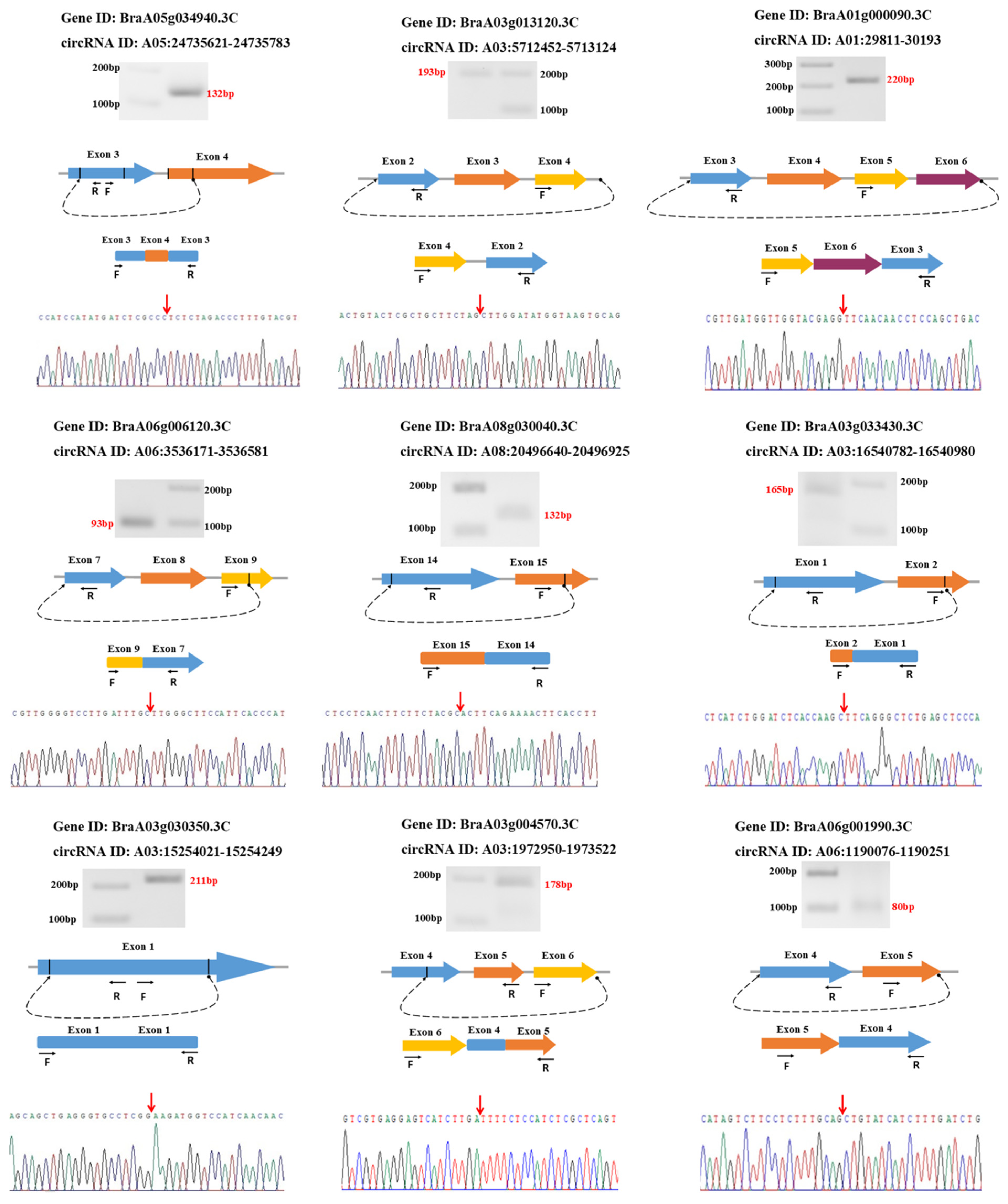
3.6. Validation of circRNAs Using Divergent Primers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Klerk, E.; AC‘t Hoen, P. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—Trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Li, X.; Zhang, P.; Wang, J.; Zhou, Y.; Chen, M. Circular RNA: An emerging key player in RNA world. Brief. Bioinform. 2017, 18, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Panigrahi, C.; Das, D.; Chandra Panda, A. Circular RNA translation, a path to hidden proteome. Wiley Interdiscip. Rev. RNA 2021, e1685. [Google Scholar] [CrossRef]
- Zhou, M.; Xiao, M.-S.; Li, Z.; Huang, C. New progresses of circular RNA biology: From nuclear export to degradation. RNA Biol. 2020, 1–9. [Google Scholar] [CrossRef]
- Xiao, M.-S.; Ai, Y.; Wilusz, J.E. Biogenesis and functions of circular RNAs come into focus. Trends Cell Biol. 2020, 30, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, S.; Chen, M. Characterization and function of circular RNAs in plants. Front. Mol. Biosci. 2020, 7, 91. [Google Scholar] [CrossRef]
- Guria, A.; Sharma, P.; Natesan, S.; Pandi, G. Circular RNAs—The road less traveled. Front. Mol. Biosci. 2020, 6, 146. [Google Scholar] [CrossRef]
- Wang, P.L.; Bao, Y.; Yee, M.-C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar]
- Golicz, A.A.; Bhalla, P.L.; Singh, M.B. lncRNAs in plant and animal sexual reproduction. Trends Plant Sci. 2018, 23, 195–205. [Google Scholar] [CrossRef]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [Green Version]
- Komiya, R.; Ohyanagi, H.; Niihama, M.; Watanabe, T.; Nakano, M.; Kurata, N.; Nonomura, K.I. Rice germline-specific A rgonaute MEL 1 protein binds to phasi RNA s generated from more than 700 linc RNA s. Plant J. 2014, 78, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.B.; Bhalla, P.L. Control of male germ-cell development in flowering plants. Bioessays 2007, 29, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Haerizadeh, F.; Wong, C.E.; Bhalla, P.L.; Gresshoff, P.M.; Singh, M.B. Genomic expression profiling of mature soybean (Glycine max) pollen. BMC Plant Biol. 2009, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.D.; Gou, X.; Wong, C.E.; Wang, X.; Yuan, T.; Wei, X.; Bhalla, P.L.; Singh, M.B. Genomic profiling of rice sperm cell transcripts reveals conserved and distinct elements in the flowering plant male germ lineage. New Phytol. 2012, 195, 560–573. [Google Scholar] [CrossRef]
- Golicz, A.A.; Allu, A.D.; Li, W.; Lohani, N.; Singh, M.B.; Bhalla, P.L. A dynamic intron retention program regulates the expression of several hundred genes during pollen meiosis. Plant Reprod. 2021, 34, 225–242. [Google Scholar] [CrossRef]
- Janousek, B.; Zluvova, J.; Vyskot, B. Histone H4 acetylation and DNA methylation dynamics during pollen development. Protoplasma 2000, 211, 116–122. [Google Scholar] [CrossRef]
- Sharma, N.; Russell, S.D.; Bhalla, P.L.; Singh, M.B. Putative cis-regulatory elements in genes highly expressed in rice sperm cells. BMC Res. Notes 2011, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Singh, M.B.; Bhalla, P.L. Histone H3 variants in male gametic cells of lily and H3 methylation in mature pollen. Plant Mol. Biol. 2006, 62, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Bhalla, P.L.; Xu, H.; Singh, M.B. Isolation and characterization of a flowering plant male gametic cell-specific promoter. FEBS Lett. 2003, 542, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Swoboda, I.; Bhalla, P.L.; Singh, M.B. Male gametic cell-specific expression of H2A and H3 histone genes. Plant Mol. Biol. 1999, 39, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yan, B.; Qu, Y.; Qin, F.; Yang, Y.; Hao, X.; Yu, J.; Zhao, Q.; Zhu, D.; Ao, G. Zm401, a short-open reading-frame mRNA or noncoding RNA, is essential for tapetum and microspore development and can regulate the floret formation in maize. J. Cell. Biochem. 2008, 105, 136–146. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Liu, Y.; Zhang, F.; Feng, Y.; Yu, D.; Lin, S.; Cao, J. Systematic identification of long non-coding RNA s during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiong, Z.; Li, Q.; Sun, Y.; Jin, J.; Chen, H.; Zou, Y.; Huang, X.; Ding, Y. Circular RNA profiling of the rice photo-thermosensitive genic male sterile line Wuxiang S reveals circRNA involved in the fertility transition. BMC Plant Biol. 2019, 19, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ding, X.; Zhang, H.; He, T.; Li, Y.; Wang, T.; Li, X.; Jin, L.; Song, Q.; Yang, S. Comparative analysis of circular RNAs between soybean cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B by high-throughput sequencing. BMC Genom. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, Y.; Xu, L.; Zhou, D.; Jin, Z.; Zhou, H.; Lin, S.; Cao, J.; Huang, L. CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris. Int. J. Mol. Sci. 2019, 20, 4808. [Google Scholar] [CrossRef] [Green Version]
- Lohani, N.; Singh, M.B.; Bhalla, P. RNA-seq highlights molecular events associated with impaired pollen-pistil interactions following short-term heat stress in Brassica napus. Front. Plant Sci. 2020, 11, 2150. [Google Scholar]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Chen, L.; Wang, C.; Sun, H.; Wang, J.; Liang, Y.; Wang, Y.; Wong, G. The bioinformatics toolbox for circRNA discovery and analysis. Brief. Bioinform. 2021, 22, 1706–1728. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Zhang, X.-O.; Dong, R.; Zhang, Y.; Zhang, J.-L.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R Programming Tools for Plotting Data. In R Package Vers 3.1.1.; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. In R Package Vers 2.42.0; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E. A 360 view of circular RNAs: From biogenesis to functions. Wiley Interdiscip. Rev. RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [Green Version]
- Ebbesen, K.K.; Kjems, J.; Hansen, T.B. Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 163–168. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [Green Version]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.; Zhang, X.; Zhu, X.; Liu, C.; Mao, L.; Ye, C.; Zhu, Q.-H.; Fan, L. PlantcircBase: A database for plant circular RNAs. Mol. Plant 2017, 10, 1126–1128. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Chen, G.; Shi, T. Identifying and characterizing the circular RNAs during the lifespan of Arabidopsis leaves. Front. Plant Sci. 2017, 8, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Yang, T.; Tang, Y.; Aisimutuola, P.; Zhang, G.; Wang, B.; Li, N.; Wang, J.; Yu, Q. Transcriptomic profile analysis of non-coding RNAs involved in Capsicum chinense Jacq. fruit ripening. Sci. Hortic. 2020, 264, 109158. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, Y.; Li, Z.; Wang, T.; Zhang, X.; Zheng, B. A lariat-derived circular RNA is required for plant development in Arabidopsis. Sci. China Life Sci. 2018, 61, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef]
- Jakobi, T.; Dieterich, C. Computational approaches for circular RNA analysis. Wiley Interdiscip. Rev. RNA 2019, 10, e1528. [Google Scholar] [CrossRef]
- Hansen, T.B. Improved circRNA identification by combining prediction algorithms. Front. Cell Dev. Biol. 2018, 6, 20. [Google Scholar] [CrossRef]
- Sun, X.; Wang, L.; Ding, J.; Wang, Y.; Wang, J.; Zhang, X.; Che, Y.; Liu, Z.; Zhang, X.; Ye, J. Integrative analysis of Arabidopsis thaliana transcriptomics reveals intuitive splicing mechanism for circular RNA. FEBS Lett. 2016, 590, 3510–3516. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wang, L.; Li, S.; Xu, M.; Guan, X.; Zhou, B. Characterization of conserved circular RNA in polyploid Gossypium species and their ancestors. FEBS Lett. 2017, 591, 3660–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Cui, J.; Wang, L.; Zhu, Y.; Lu, Z.; Jin, B. Genome-wide identification of circular RNAs in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Wang, W.; Wang, J.; Wei, Q.; Li, B.; Zhong, X.; Hu, T.; Hu, H.; Bao, C. Transcriptome-wide identification and characterization of circular RNAs in leaves of Chinese cabbage (Brassica rapa L. ssp. pekinensis) in response to calcium deficiency-induced tip-burn. Sci. Rep. 2019, 9, 1–9. [Google Scholar]
- Zhang, X.; Ma, X.; Ning, L.; Li, Z.; Zhao, K.; Li, K.; He, J.; Yin, D. Genome-wide identification of circular RNAs in peanut (Arachis hypogaea L.). BMC Genom. 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cui, L.; Zhou, Y.; Zhu, C.; Fan, D.; Gong, H.; Zhao, Q.; Zhou, C.; Zhao, Y.; Lu, D. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Yu, K.; Lü, H.; Zhang, X.; Liu, X.; Sun, C.; Xu, H.; Zhang, J.; He, X.; Zhang, D. Transcriptome-wide identification of novel circular RNAs in soybean in response to low-phosphorus stress. PLoS ONE 2020, 15, e0227243. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.Y.; Chen, L.; Liu, C.; Zhu, Q.H.; Fan, L. Widespread noncoding circular RNA s in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, J.; Luo, M.; Li, H.; Chen, Q.; Wang, L.; Song, S.; Zhao, L.; Xu, W.; Zhang, C. Characterization and cloning of grape circular RNAs identified the cold resistance-related Vv-circATS1. Plant Physiol. 2019, 180, 966–985. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Yu, J.; Hou, Y.; Li, F.; Zhou, Q.; Wei, C.; Bennetzen, J.L. Circular RNA architecture and differentiation during leaf bud to young leaf development in tea (Camellia sinensis). Planta 2018, 248, 1417–1429. [Google Scholar] [CrossRef]
- Rutter, M.T.; Cross, K.V.; Van Woert, P.A. Birth, death and subfunctionalization in the Arabidopsis genome. Trends Plant Sci. 2012, 17, 204–212. [Google Scholar] [CrossRef]
- Liu, Y.; Su, H.; Zhang, J.; Liu, Y.; Feng, C.; Han, F. Back-spliced RNA from retrotransposon binds to centromere and regulates centromeric chromatin loops in maize. PLoS Biol. 2020, 18, e3000582. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256. [Google Scholar] [CrossRef]
- Conn, V.M.; Hugouvieux, V.; Nayak, A.; Conos, S.A.; Capovilla, G.; Cildir, G.; Jourdain, A.; Tergaonkar, V.; Schmid, M.; Zubieta, C. A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat. Plants 2017, 3, 1–5. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [Green Version]
- Rutley, N.; Twell, D. A decade of pollen transcriptomics. Plant Reprod. 2015, 28, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragkostefanakis, S.; Mesihovic, A.; Hu, Y.; Schleiff, E. Unfolded protein response in pollen development and heat stress tolerance. Plant Reprod. 2016, 29, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.B.; Lohani, N.; Bhalla, P.L. The Role of Endoplasmic Reticulum Stress Response in Pollen Development and Heat Stress Tolerance. Front. Plant Sci. 2021, 12, 661062. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, Z.; Cao, J. Pollen wall development: The associated enzymes and metabolic pathways. Plant Biol. 2013, 15, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, M.B.; Bhalla, P.L. Ultrastructure of microsporogenesis and microgametogenesis in Brachypodium distachyon. Protoplasma 2015, 252, 1575–1586. [Google Scholar] [CrossRef]
- Dong, X.; Feng, H.; Xu, M.; Lee, J.; Kim, Y.K.; Lim, Y.P.; Piao, Z.; Park, Y.D.; Ma, H.; Hur, Y. Comprehensive analysis of genic male sterility-related genes in Brassica rapa using a newly developed Br300K oligomeric chip. PLoS ONE 2013, 8, e72178. [Google Scholar]
- Waititu, J.K.; Zhang, C.; Liu, J.; Wang, H. Plant Non-Coding RNAs: Origin, Biogenesis, Mode of Action and Their Roles in Abiotic Stress. Int. J. Mol. Sci. 2020, 21, 8401. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Chen, L.-L. The biogenesis, functions, and challenges of circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 2018, 174, 350–362.e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Jara, C.A.C.; Fenske, P. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 2017, 357, 6357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.; Bai, P.; Zhu, X.; Zhang, X.; Mao, L.; Zhu, Q.-H.; Fan, L.; Ye, C.-Y. Characteristics of plant circular RNAs. Brief. Bioinform. 2020, 21, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Frydrych Capelari, É.; da Fonseca, G.C.; Guzman, F.; Margis, R. Circular and micro RNAs from Arabidopsis thaliana flowers are simultaneously isolated from AGO-IP libraries. Plants 2019, 8, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wu, L.; Qi, H.; Xu, M. LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of Populus. Ind. Crop. Prod. 2019, 133, 333–347. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, Z.; Yin, S.; Li, G. GreenCircRNA: A database for plant circRNAs that act as miRNA decoys. Database 2020, 2020, baaa039. [Google Scholar] [CrossRef] [PubMed]
- Luján-Soto, E.; Dinkova, T.D. Time to Wake Up: Epigenetic and Small-RNA-Mediated Regulation during Seed Germination. Plants 2021, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. Plant microRNAs and development. J. Genet. Genom. 2013, 40, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, B. MicroRNAs in control of plant development. J. Cell. Physiol. 2016, 231, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.-H.; Lee, S.; Yun, J.; Lee, M.; Park, C.-M. The miR172 target TOE3 represses AGAMOUS expression during Arabidopsis floral patterning. Plant Sci. 2014, 215, 29–38. [Google Scholar] [CrossRef]
- Nair, S.K.; Wang, N.; Turuspekov, Y.; Pourkheirandish, M.; Sinsuwongwat, S.; Chen, G.; Sameri, M.; Tagiri, A.; Honda, I.; Watanabe, Y. Cleistogamous flowering in barley arises from the suppression of microRNA-guided HvAP2 mRNA cleavage. Proc. Natl. Acad. Sci. USA 2010, 107, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Wang, J.; Ju, Z.; Liu, Q.; Li, S.; Tian, H.; Fu, D.; Zhu, H.; Luo, Y.; Zhu, B. Regulations on growth and development in tomato cotyledon, flower and fruit via destruction of miR396 with short tandem target mimic. Plant Sci. 2016, 247, 1–12. [Google Scholar] [CrossRef]
- Wang, T.; Ping, X.; Cao, Y.; Jian, H.; Gao, Y.; Wang, J.; Tan, Y.; Xu, X.; Lu, K.; Li, J. Genome-wide exploration and characterization of miR172/euAP2 genes in Brassica napus L. for likely role in flower organ development. BMC Plant Biol. 2019, 19, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Wei, W.; Li, Y.; Kan, L.; Wang, F.; Zhang, X.; Li, F.; Liu, Z.; Kang, C. Conserved and novel roles of miR164-CUC 2 regulatory module in specifying leaf and floral organ morphology in strawberry. New Phytol. 2019, 224, 480–492. [Google Scholar] [CrossRef]
- Ma, Z.; Jiang, J.; Hu, Z.; Lyu, T.; Yang, Y.; Jiang, J.; Cao, J. Over-expression of miR158 causes pollen abortion in Brassica campestris ssp. chinensis. Plant Mol. Biol. 2017, 93, 313–326. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant Species | No. of CircRNAs | No. of Conserved CircRNAs | % Conservation |
|---|---|---|---|
| Arabidopsis thaliana | 52,393 | 421/1180 | 35.68 |
| Oryza sativa | 40,311 | 9/1180 | 0.76 |
| Glycine max | 8148 | 8/1180 | 0.68 |
| Gossypium hirsutum | 3944 | 5/1180 | 0.42 |
| Cucumis sativus | 4832 | 4/1180 | 0.34 |
| Solanum tuberosum | 1728 | 3/1180 | 0.25 |
| Solanum lycopersicum | 3727 | 3/1180 | 0.25 |
| Zea mays | 6434 | 3/1180 | 0.25 |
| Gossypium raimondii | 1478 | 1/1180 | 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babaei, S.; Singh, M.B.; Bhalla, P.L. Circular RNAs Repertoire and Expression Profile during Brassica rapa Pollen Development. Int. J. Mol. Sci. 2021, 22, 10297. https://doi.org/10.3390/ijms221910297
Babaei S, Singh MB, Bhalla PL. Circular RNAs Repertoire and Expression Profile during Brassica rapa Pollen Development. International Journal of Molecular Sciences. 2021; 22(19):10297. https://doi.org/10.3390/ijms221910297
Chicago/Turabian StyleBabaei, Saeid, Mohan B. Singh, and Prem L. Bhalla. 2021. "Circular RNAs Repertoire and Expression Profile during Brassica rapa Pollen Development" International Journal of Molecular Sciences 22, no. 19: 10297. https://doi.org/10.3390/ijms221910297
APA StyleBabaei, S., Singh, M. B., & Bhalla, P. L. (2021). Circular RNAs Repertoire and Expression Profile during Brassica rapa Pollen Development. International Journal of Molecular Sciences, 22(19), 10297. https://doi.org/10.3390/ijms221910297