
RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective



Abstract
1. Introduction
2. A Brief History of ALS
3. Face with ALS: Onset, Clinical Manifestation, and Diagnosis
4. RNA Biology of ALS
5. Noncoding RNA Landscape
5.1. MicroRNAs
5.1.1. MiRNA Biosynthesis Is Affected in ALS
5.1.2. Integrative miRNA-Omics Studies in ALS
5.1.3. MiR-9 and miR-124 in ALS MNs
5.1.4. Other miRNAs in ALS MNs
5.1.5. ALS miRNAs in Non-MN Cells: Microglia
5.1.6. ALS miRNAs in Non-MN Cells: Astrocytes
5.1.7. MyomiRs in ALS
5.2. Long Noncoding RNAs
5.2.1. C9ORF72-AS
5.2.2. ATXN2-AS
5.2.3. The Interplay between lncRNAs and ALS Genes
5.2.4. NEAT1
5.2.5. Other lncRNAs in ALS
5.3. Circular RNAs
Circular RNAs in ALS
5.4. Noncoding RNAs as ALS Biomarkers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ASO | antisense oligonucleotide |
| BS | brainstem |
| circRNA | circular RNA |
| CLIP | cross-linking immunoprecipitation |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DRP | dipeptide-repeat-containing protein |
| ER | endoplasmic reticulum |
| ESC | embryonic stem cell |
| fALS | familial ALS |
| FTD | frontotemporal dementia |
| iPSC | induced pluripotent stem cell |
| lncRNA | long noncoding RNA |
| MCx | motor cortex |
| MG | microglia |
| miRNA | microRNA |
| MN | motoneuron |
| mRNA | messenger RNA |
| ncRNA | noncoding RNA |
| Nf | neurofilament |
| NMJ | neuromuscular junction |
| NS | nervous system |
| pNfH | phosphorylated neurofilament heavy chain |
| RBP | RNA binding protein |
| RNP | ribonucleoprotein |
| sALS | sporadic ALS |
| SC | spinal cord |
| SG | stress granule |
| TF | transcription factor |
| UTR | untranslated region |
References
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef]
- Li, T.M.; Alberman, E.; Swash, M. Comparison of Sporadic and Familial Disease amongst 580 Cases of Motor Neuron Disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Brown, R.H. Molecular Biology of Amyotrophic Lateral Sclerosis: Insights from Genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Al-Chalabi, A. Clinical Genetics of Amyotrophic Lateral Sclerosis: What Do We Really Know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Chiò, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic Counselling in ALS: Facts, Uncertainties and Clinical Suggestions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 478–485. [Google Scholar] [CrossRef]
- Gamez, J.; Corbera-Bellalta, M.; Nogales, G.; Raguer, N.; García-Arumí, E.; Badia-Canto, M.; Lladó-Carbó, E.; Álvarez-Sabín, J. Mutational Analysis of the Cu/Zn Superoxide Dismutase Gene in a Catalan ALS Population: Should All Sporadic ALS Cases Also Be Screened for SOD1? J. Neurol. Sci. 2006, 247, 21–28. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-Pathological Features in Amyotrophic Lateral Sclerosis with Expansions in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long Pre-MRNA Depletion and RNA Missplicing Contribute to Neuronal Vulnerability from Loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-Linked TDP-43 Mutations Produce Aberrant RNA Splicing and Adult-Onset Motor Neuron Disease without Aggregation or Loss of Nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 10, 712. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor Complex Mediates the Genesis of MicroRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-Associated Mutations in TDP-43 Increase Its Stability and Promote TDP-43 Complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef]
- Charcot, J.-M.; Joffroy, A. Deux Cas d’atrophie Musculaire Progressive Avec Lésions de La Substance Grise et Des Faisceaux Antérolatéraux de La Moelle Épinière. Arch. Physiol. Norm. Pathol. 1869, 2, 744–760. [Google Scholar]
- Kurland, L.K.; Mulder, D.W. Epidemiologic Investigations of Amyotrophic Lateral Sclerosis: 1. Preliminary Report on Geographic Distribution, with Special Reference to the Mariana Islands, Including Clinical and Pathologic Observations. Neurology 1954, 4, 355–378. [Google Scholar] [CrossRef]
- Mulder, D.W. Epidemiologic Investigations of Amyotrophic Lateral Sclerosis: 2. Familial Aggregations Indicative of Dominant Inheritance Part II. Neurology 1955, 5, 249–268. [Google Scholar] [CrossRef]
- Ganesalingam, J.; Stahl, D.; Wijesekera, L.; Galtrey, C.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Latent Cluster Analysis of ALS Phenotypes Identifies Prognostically Differing Groups. PLoS ONE 2009, 4, e7107. [Google Scholar] [CrossRef]
- Finegan, E.; Chipika, R.H.; Li Hi Shing, S.; Hardiman, O.; Bede, P. Pathological Crying and Laughing in Motor Neuron Disease: Pathobiology, Screening, Intervention. Front. Neurol. 2019, 10, 260. [Google Scholar] [CrossRef]
- Pape, J.A.; Grose, J.H. The Effects of Diet and Sex in Amyotrophic Lateral Sclerosis. Rev. Neurol. (Paris) 2020, 176, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Trojsi, F.; D’Alvano, G.; Bonavita, S.; Tedeschi, G. Genetics and Sex in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS): Is There a Link? Int. J. Mol. Sci. 2020, 21, 3647. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H.; Mehal, J.M.; Holman, R.C.; Rowland, L.P.; Rowland, A.S.; Cheek, J.E. Incidence of Amyotrophic Lateral Sclerosis Among American Indians and Alaska Natives. JAMA Neurol. 2013, 70, 476–480. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; EURALS. Incidence of Amyotrophic Lateral Sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic Factors in ALS: A Critical Review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- del Aguila, M.A.; Longstreth, W.T.; McGuire, V.; Koepsell, T.D.; van Belle, G. Prognosis in Amyotrophic Lateral Sclerosis. Neurology 2003, 60, 813–819. [Google Scholar] [CrossRef]
- Turner, M.R.; Parton, M.J.; Shaw, C.E.; Leigh, P.N.; Al-Chalabi, A. Prolonged Survival in Motor Neuron Disease: A Descriptive Study of the King’s Database 1990–2002. J. Neurol. Neurosurg. Psychiatry 2003, 74, 995–997. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS Study Group. Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2009, 1, 293–299. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; ALS/Riulzole Study Group. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2010, 330, 585–591. [Google Scholar] [CrossRef]
- Gagliardi, D.; Meneri, M.; Saccomanno, D.; Bresolin, N.; Comi, G.P.; Corti, S. Diagnostic and Prognostic Role of Blood and Cerebrospinal Fluid and Blood Neurofilaments in Amyotrophic Lateral Sclerosis: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 4152. [Google Scholar] [CrossRef] [PubMed]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament Markers for ALS Correlate with Extent of Upper and Lower Motor Neuron Disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Falzone, Y.M.; Domi, T.; Agosta, F.; Pozzi, L.; Schito, P.; Fazio, R.; Del Carro, U.; Barbieri, A.; Comola, M.; Leocani, L.; et al. Serum Phosphorylated Neurofilament Heavy-Chain Levels Reflect Phenotypic Heterogeneity and Are an Independent Predictor of Survival in Motor Neuron Disease. J. Neurol. 2020, 267, 1. [Google Scholar] [CrossRef] [PubMed]
- De Schaepdryver, M.; Jeromin, A.; Gille, B.; Claeys, K.G.; Herbst, V.; Brix, B.; Van Damme, P.; Poesen, K. Comparison of Elevated Phosphorylated Neurofilament Heavy Chains in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 367–373. [Google Scholar] [CrossRef]
- Menzies, F.M.; Grierson, A.J.; Cookson, M.R.; Heath, P.R.; Tomkins, J.; Figlewicz, D.A.; Ince, P.G.; Shaw, P.J. Selective Loss of Neurofilament Expression in Cu/Zn Superoxide Dismutase (SOD1) Linked Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 82, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zheng, L.; Viera, L.; Suswam, E.; Li, Y.; Li, X.; Estévez, A.G.; King, P.H. Mutant Cu/Zn-Superoxide Dismutase Associated with Amyotrophic Lateral Sclerosis Destabilizes Vascular Endothelial Growth Factor MRNA and Downregulates Its Expression. J. Neurosci. 2007, 27, 7929–7938. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with IPSCs Reveals That Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef]
- Strong, M.J. The Evidence for Altered RNA Metabolism in Amyotrophic Lateral Sclerosis (ALS). J. Neurol. Sci. 2010, 288, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 Hexanucleotide Repeat Expansion in Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: A Cross-Sectional Study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense Transcripts of the Expanded C9ORF72 Hexanucleotide Repeat Form Nuclear RNA Foci and Undergo Repeat-Associated Non-ATG Translation in C9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Heusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted Degradation of Sense and Antisense C9orf72 RNA Foci as Therapy for ALS and Frontotemporal Degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Armstrong, G.A.B.; Drapeau, P. Calcium Channel Agonists Protect against Neuromuscular Dysfunction in a Genetic Model of TDP-43 Mutation in ALS. J. Neurosci. 2013, 33, 1741–1752. [Google Scholar] [CrossRef]
- Lu, L.; Wang, S.; Zheng, L.; Li, X.; Suswam, E.A.; Zhang, X.; Wheeler, C.G.; Nabors, L.B.; Filippova, N.; King, P.H. Amyotrophic Lateral Sclerosis-Linked Mutant SOD1 Sequesters Hu Antigen R (HuR) and TIA-1-Related Protein (TIAR). J. Biol. Chem. 2009, 284, 33989–33998. [Google Scholar] [CrossRef] [PubMed]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 Affects Drosophila Motoneurons Terminal Synapsis and Locomotive Behavior. FEBS Lett. 2009, 583, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.H.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of Neuronal RNA Targets of TDP-43-Containing Ribonucleoprotein Complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, V.; Grossi, E.; Laneve, P.; Morlando, M.; Dini Modigliani, S.; Ballarino, M.; Bozzoni, I.; Caffarelli, E. TDP-43 Regulates the Microprocessor Complex Activity during in Vitro Neuronal Differentiation. Mol. Neurobiol. 2013, 48, 952–963. [Google Scholar] [CrossRef]
- Herzog, J.J.; Deshpande, M.; Shapiro, L.; Rodal, A.A.; Paradis, S. TDP-43 Misexpression Causes Defects in Dendritic Growth. Sci. Rep. 2017, 7, 15656. [Google Scholar] [CrossRef]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse Dysfunction of Layer V Pyramidal Neurons Precedes Neurodegeneration in a Mouse Model of TDP-43 Proteinopathies. Cereb. Cortex 2017, 27, 3630–3647. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Buratti, E. Physiological Functions and Pathobiology of TDP-43 and FUS/TLS Proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear Factor TDP-43 Can Affect Selected MicroRNA Levels. FEBS J. 2010, 277, 2268–2281. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 Promotes MicroRNA Biogenesis as a Component of the Drosha and Dicer Complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef]
- Zhang, Z.; Almeida, S.; Lu, Y.; Nishimura, A.L.; Peng, L.; Sun, D.; Wu, B.; Karydas, A.M.; Tartaglia, M.C.; Fong, J.C.; et al. Downregulation of MicroRNA-9 in IPSC-Derived Neurons of FTD/ALS Patients with TDP-43 Mutations. PLoS ONE 2013, 8, e76055. [Google Scholar] [CrossRef] [PubMed]
- King, I.N.; Yartseva, V.; Salas, D.; Kumar, A.; Heidersbach, A.; Ando, D.M.; Stallings, N.R.; Elliott, J.L.; Srivastava, D.; Ivey, K.N. The RNA-Binding Protein TDP-43 Selectively Disrupts MicroRNA-1/206 Incorporation into the RNA-Induced Silencing Complex. J. Biol. Chem. 2014, 289, 14263–14271. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-Binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-Transcriptional Fate in Motoneuron-like Cells. J. Biol. Chem. 2012, 287, 15635. [Google Scholar] [CrossRef]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) Binds RNA in Vivo and Engages in Nucleo-Cytoplasmic Shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA Binding Protein TLS Is Translocated to Dendritic Spines by MGluR5 Activation and Regulates Spine Morphology. Curr. Biol. 2005, 15, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Takumi, T. TLS Facilitates Transport of MRNA Encoding an Actin-Stabilizing Protein to Dendritic Spines. J. Cell Sci. 2005, 118, 5755–5765. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Dormann, D.; Haass, C. Fused in Sarcoma (FUS): An Oncogene Goes Awry in Neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Daigle, G.G.; Lanson, N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. Rna-Binding Ability of FUS Regulates Neurodegeneration, Cytoplasmic Mislocalization and Incorporation into Stress Granules Associated with FUS Carrying ALS-Linked Mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long Non-Coding and Coding RNAs Characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis Patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef]
- Garofalo, M.; Pandini, C.; Bordoni, M.; Pansarasa, O.; Rey, F.; Costa, A.; Minafra, B.; Diamanti, L.; Zucca, S.; Carelli, S.; et al. Alzheimer’s, Parkinson’s Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement. Int. J. Mol. Sci. 2020, 21, 9500. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. Roles for MicroRNAs in Conferring Robustness to Biological Processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef]
- Cloutier, F.; Marrero, A.; O’Connell, C.; Morin, P.J. MicroRNAs as Potential Circulating Biomarkers for Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 2014, 56, 102–112. [Google Scholar] [CrossRef] [PubMed]
- de Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Åström, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An Integrated Expression Atlas of MiRNAs and Their Promoters in Human and Mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Zolboot, N.; Du, J.X.; Zampa, F.; Lippi, G. MicroRNAs Instruct and Maintain Cell Type Diversity in the Nervous System. Front. Mol. Neurosci. 2021, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-L.; Yoo, A.S. Mechanistic Insights into MicroRNA-Induced Neuronal Reprogramming of Human Adult Fibroblasts. Front. Neurosci. 2018, 12, 522. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered MicroRNA Expression Profile in Amyotrophic Lateral Sclerosis: A Role in the Regulation of NFL MRNA Levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef]
- Emde, A.; Eitan, C.; Liou, L.-L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated MiRNA Biogenesis Downstream of Cellular Stress and ALS-Causing Mutations: A New Mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of MicroRNAs in Human Post-Mortem Amyotrophic Lateral Sclerosis Spinal Cords Provides Insight into Disease Mechanisms. Mol. Cell. Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.P.; Barabino, S.; Nizzardo, M.; et al. MicroRNA Expression Analysis Identifies a Subset of Downregulated MiRNAs in ALS Motor Neuron Progenitors. Sci. Rep. 2018, 8, 10105. [Google Scholar] [CrossRef]
- Jawaid, A.; Woldemichael, B.T.; Kremer, E.A.; Laferriere, F.; Gaur, N.; Afroz, T.; Polymenidou, M.; Mansuy, I.M. Memory Decline and Its Reversal in Aging and Neurodegeneration Involve MiR-183/96/182 Biogenesis. Mol. Neurobiol. 2018, 56, 3451–3462. [Google Scholar] [CrossRef]
- Paez-Colasante, X.; Figueroa-Romero, C.; Rumora, A.E.; Hur, J.; Mendelson, F.E.; Hayes, J.M.; Backus, C.; Taubman, G.F.; Heinicke, L.; Walter, N.G.; et al. Cytoplasmic TDP43 Binds MicroRNAs: New Disease Targets in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2020, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Morlando, M.; Modigliani, S.D.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS Stimulates MicroRNA Biogenesis by Facilitating Co-Transcriptional Drosha Recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, Y.-C.; Mullane, P.; Ji, Y.J.; Liu, H.; He, L.; Arora, A.; Hwang, H.-Y.; Alessi, A.F.; Niaki, A.G.; et al. FUS Regulates Activity of MicroRNA-Mediated Gene Silencing. Mol. Cell 2018, 69, 787–801.e8. [Google Scholar] [CrossRef] [PubMed]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. Evidence of a Negative Feedback Network between TDP-43 and MiRNAs Dependent on TDP-43 Nuclear Localization. J. Mol. Biol. 2020, 432, 166695. [Google Scholar] [CrossRef]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. Novel MiR-B2122 Regulates Several ALS-Related RNA-Binding Proteins. Mol. Brain 2017, 10, 46. [Google Scholar] [CrossRef]
- Dini Modigliani, S.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-Associated Mutation in the FUS 3′-UTR Disrupts a MicroRNA–FUS Regulatory Circuitry. Nat. Commun. 2014, 5, 4335. [Google Scholar] [CrossRef]
- Eitan, C.; Hornstein, E. Vulnerability of MicroRNA Biogenesis in FTD–ALS. Brain Res. 2016, 1647, 105–111. [Google Scholar] [CrossRef]
- Shinde, S.; Arora, N.; Bhadra, U. A Complex Network of MicroRNAs Expressed in Brain and Genes Associated with Amyotrophic Lateral Sclerosis. Int. J. Genom. 2013, 2013, 383024. [Google Scholar] [CrossRef]
- Hamzeiy, H.; Suluyayla, R.; Brinkrolf, C.; Janowski, S.J.; Hofestädt, R.; Allmer, J. Visualization and Analysis of MiRNAs Implicated in Amyotrophic Lateral Sclerosis Within Gene Regulatory Pathways. Ger. Med. Data Sci. 2018, 253, 183–187. [Google Scholar] [CrossRef]
- Recabarren-Leiva, D.; Alarcón, M. New Insights into the Gene Expression Associated to Amyotrophic Lateral Sclerosis. Life Sci. 2018, 193, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zuo, X.; Zhang, P.; Zhao, R.; Lai, D.; Chen, K.; Han, Y.; Wan, G.; Zheng, Y.; Lu, C.; et al. The Novel Regulatory Role of LncRNA-MiRNA-MRNA Axis in Amyotrophic Lateral Sclerosis: An Integrated Bioinformatics Analysis. Comput. Math. Methods Med. 2021, 2021, 5526179. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, K.; Katsila, T.; Patrinos, G.P.; Pampalakis, G. Multi-Omics for Biomarker Discovery and Target Validation in Biofluids for Amyotrophic Lateral Sclerosis Diagnosis. Omics A J. Integr. Biol. 2018, 22, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Rotem, N.; Magen, I.; Ionescu, A.; Gershoni-Emek, N.; Altman, T.; Costa, C.J.; Gradus, T.; Pasmanik-Chor, M.; Willis, D.E.; Ben-Dov, I.Z.; et al. ALS Along the Axons–Expression of Coding and Noncoding RNA Differs in Axons of ALS Models. Sci. Rep. 2017, 7, 44500. [Google Scholar] [CrossRef]
- Helferich, A.M.; Brockmann, S.J.; Reinders, J.; Deshpande, D.; Holzmann, K.; Brenner, D.; Andersen, P.M.; Petri, S.; Thal, D.R.; Michaelis, J.; et al. Dysregulation of a Novel MiR-1825/TBCB/TUBA4A Pathway in Sporadic and Familial ALS. Cell. Mol. Life Sci. 2018, 75, 4301–4319. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kim, Y.R.; Choi, K.W.; Lee, M.; Lee, S.; Im, W.; Shin, J.Y.; Kim, J.Y.; Hong, Y.H.; Kim, M.; et al. Downregulated MiR-18b-5p Triggers Apoptosis by Inhibition of Calcium Signaling and Neuronal Cell Differentiation in Transgenic SOD1 (G93A) Mice and SOD1 (G17S and G86S) ALS Patients. Transl. Neurodegener. 2020, 9, 23. [Google Scholar] [CrossRef]
- Freischmidt, A.; Goswami, A.; Limm, K.; Zimyanin, V.L.; Demestre, M.; Glaß, H.; Holzmann, K.; Helferich, A.M.; Brockmann, S.J.; Tripathi, P.; et al. A Serum MicroRNA Sequence Reveals Fragile X Protein Pathology in Amyotrophic Lateral Sclerosis. Brain 2021, 144, 1214–1229. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Božič, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.F.; et al. Serum MicroRNAs in Patients with Genetic Amyotrophic Lateral Sclerosis and Pre-Manifest Mutation Carriers. Brain 2014, 137, 2938–2950. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Mayer, B.; von Arnim, C.A.F.; Hübers, A.; Dorst, J.; Otto, M.; Holzmann, K.; et al. Serum MicroRNAs in Sporadic Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2015, 36, 2660.e15–2660.e20. [Google Scholar] [CrossRef]
- De Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; de Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS Mutant Human Motoneurons Display Altered Transcriptome and MicroRNA Pathways with Implications for ALS Pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef]
- Bronicki, L.M.; Jasmin, B.J. Emerging Complexity of the HuD/ELAVl4 Gene; Implications for Neuronal Development, Function, and Dysfunction. RNA 2013, 19, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Duchen, L.W.; Strich, S.J. An Hereditary Motor Neurone Disease with Progressive Denervation of Muscle in the Mouse: The Mutant “Wobbler”. J. Neurol. Neurosurg. Psychiatry 1968, 31, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; May, C.; Marcus, K.; Steinbach, S.; Theis, V.; Theiss, C.; Matschke, V. The MicroRNA MiR-375-3p and the Tumor Suppressor NDRG2 Are Involved in Sporadic Amyotrophic Lateral Sclerosis. Cell. Physiol. Biochem. 2019, 52, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Capauto, D.; Colantoni, A.; Lu, L.; Santini, T.; Peruzzi, G.; Biscarini, S.; Morlando, M.; Shneider, N.A.; Caffarelli, E.; Laneve, P.; et al. A Regulatory Circuitry Between Gria2, MiR-409, and MiR-495 Is Affected by ALS FUS Mutation in ESC-Derived Motor Neurons. Mol. Neurobiol. 2018, 55, 7635–7651. [Google Scholar] [CrossRef]
- Li, Z.; Lu, Y.; Xu, X.-L.; Gao, F.-B. The FTD/ALS-Associated RNA-Binding Protein TDP-43 Regulates the Robustness of Neuronal Specification through MicroRNA-9a in Drosophila. Hum. Mol. Genet. 2013, 22, 218–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, F.; Guan, Y.; Chen, Y.; Zhang, C.; Yu, L.; Gao, H.; Du, H.; Liu, B.; Wang, X. MiRNA-9 Expression Is Upregulated in the Spinal Cord of G93A-SOD1 Transgenic Mice. Int. J. Clin. Exp. Pathol. 2013, 6, 1826. [Google Scholar] [PubMed]
- Bergeron, C.; Beric-Maskarel, K.; Muntasser, S.; Weyer, L.; Somerville, M.J.; Percy, M.E. Neurofilament Light and Polyadenylated MRNA Levels Are Decreased in Amyotrophic Lateral Sclerosis Motor Neurons. J. Neuropathol. Exp. Neurol. 1994, 53, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ishtiaq, M.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Analysis of Novel NEFL MRNA Targeting MicroRNAs in Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e85653. [Google Scholar] [CrossRef]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. MiR-105 and MiR-9 Regulate the MRNA Stability of Neuronal Intermediate Filaments. Implications for the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Brain Res. 2019, 1706, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.-M.; Wirguin, I.; et al. MiRNA Malfunction Causes Spinal Motor Neuron Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef]
- Cong, C.; Liang, W.; Zhang, C.; Wang, Y.; Yang, Y.; Wang, X.; Wang, S.; Huo, D.; Wang, H.; Wang, D.; et al. PAK4 Suppresses Motor Neuron Degeneration in HSOD1G93A-Linked Amyotrophic Lateral Sclerosis Cell and Rat Models. Cell Prolif. 2021, 54, e13003. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, C.; Guan, Y.; Chen, Y.; Lu, Q.; Jie, L.; Gao, H.; Du, H.; Zhang, H.; Liu, Y.; et al. Screening the Expression Characteristics of Several MiRNAs in G93A-SOD1 Transgenic Mouse: Altered Expression of MiRNA-124 Is Associated with Astrocyte Differentiation by Targeting Sox2 and Sox9. J. Neurochem. 2018, 145, 51–67. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Kapetis, D.; Mantegazza, R.; Baggi, F.; Bonanno, S.; Barzago, C.; Cavalcante, P.; de Rosbo, N.K.; Bernasconi, P. Altered MiRNA Expression Is Associated with Neuronal Fate in G93A-SOD1 Ependymal Stem Progenitor Cells. Exp. Neurol. 2014, 253, 91–101. [Google Scholar] [CrossRef]
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial Membrane Potential Is Regulated by Vimentin Intermediate Filaments. FASEB J. 2015, 29, 820–827. [Google Scholar] [CrossRef]
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin Intermediate Filaments Modulate the Motility of Mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, T.; Fine, R.; Joshi, Y.; Gradus-Pery, T.; Kozer, N.; Reichenstein, I.; Yanowski, E.; Nevo, S.; Weiss-Tishler, H.; Eisenberg-Bord, M.; et al. High Content Image Analysis Reveals Function of MiR-124 Upstream of Vimentin in Regulating Motor Neuron Mitochondria. Sci. Rep. 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Vizinha, D.; Morais, H.; Colaço, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of MiR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. Int. J. Mol. Sci. 2021, 22, 6128. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Bernardini, C.; Martini, M.; Baranzini, M.; Barba, M.; Musarò, A. Muscle Expression of SOD1G93A Modulates MicroRNA and MRNA Transcription Pattern Associated with the Myelination Process in the Spinal Cord of Transgenic Mice. Front. Cell. Neurosci. 2015, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef]
- Nolan, K.; Walter, F.; Tuffy, L.P.; Poeschel, S.; Gallagher, R.; Haunsberger, S.; Bray, I.; Stallings, R.L.; Concannon, C.G.; Prehn, J.H.M. Endoplasmic Reticulum Stress-Mediated Upregulation of MiR-29a Enhances Sensitivity to Neuronal Apoptosis. Eur. J. Neurosci. 2016, 43, 640–652. [Google Scholar] [CrossRef]
- Nolan, K.; Mitchem, M.R.; Jimenez-Mateos, E.M.; Henshall, D.C.; Concannon, C.G.; Prehn, J.H.M. Increased Expression of MicroRNA-29a in ALS Mice: Functional Analysis of Its Inhibition. J. Mol. Neurosci. 2014, 53, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Klatt, C.L.; Theis, V.; Hahn, S.; Theiss, C.; Matschke, V. Deregulated MiR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1077. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.; Chen, X.; Wei, Q.; Cao, B.; Shang, H. Downregulation of MicroRNA-193b-3p Promotes Autophagy and Cell Survival by Targeting TSC1/MTOR Signaling in NSC-34 Cells. Front. Mol. Neurosci. 2017, 10, 160. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, Q.; Chen, X.; Li, C.; Cao, B.; Ou, R.; Hadano, S.; Shang, H.-F. Aberration of MiRNAs Expression in Leukocytes from Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2016, 9, 69. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy Induction Enhances TDP43 Turnover and Survival in Neuronal ALS Models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kurita, H.; Okuda, R.; Yokoo, K.; Inden, M.; Hozumi, I. Protective Roles of SLC30A3 against Endoplasmic Reticulum Stress via ERK1/2 Activation. Biochem. Biophys. Res. Commun. 2016, 479, 853–859. [Google Scholar] [CrossRef]
- Patrushev, N.; Seidel-Rogol, B.; Salazar, G. Angiotensin II Requires Zinc and Downregulation of the Zinc Transporters ZnT3 and ZnT10 to Induce Senescence of Vascular Smooth Muscle Cells. PLoS ONE 2012, 7, e33211. [Google Scholar] [CrossRef]
- Kurita, H.; Yabe, S.; Ueda, T.; Inden, M.; Kakita, A.; Hozumi, I. MicroRNA-5572 Is a Novel MicroRNA-Regulating SLC30A3 in Sporadic Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 4482. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, Y.; Chen, X.; Wei, Q.; Ou, R.; Gu, X.; Cao, B.; Shang, H. MicroRNA-183-5p Is Stress-Inducible and Protects Neurons against Cell Death in Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2020, 24, 8614–8622. [Google Scholar] [CrossRef]
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of MiR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-T.; Peng, K.-C.; Chen, Y.-C.; Yen, Y.-P.; Chang, M.; Thams, S.; Chen, J.-A. Mir-17∼92 Confers Motor Neuron Subtype Differential Resistance to ALS-Associated Degeneration. Cell Stem Cell 2019, 25, 193–209.e7. [Google Scholar] [CrossRef]
- Li, C.; Wei, Q.; Gu, X.; Chen, Y.; Chen, X.; Cao, B.; Ou, R.; Shang, H. Decreased Glycogenolysis by MiR-338-3p Promotes Regional Glycogen Accumulation Within the Spinal Cord of Amyotrophic Lateral Sclerosis Mice. Front. Mol. Neurosci. 2019, 12, 114. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy Metabolism in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- D’amico, E.; Grosso, G.; Nieves, J.W.; Zanghì, A.; Factor-Litvak, P.; Mitsumoto, H. Metabolic Abnormalities, Dietary Risk Factors and Nutritional Management in Amyotrophic Lateral Sclerosis. Nutrients 2021, 13, 2273. [Google Scholar] [CrossRef]
- Caplliure-Llopis, J.; Peralta-Chamba, T.; Carrera-Juliá, S.; Cuerda-Ballester, M.; Drehmer-Rieger, E.; López-Rodriguez, M.M.; de la Rubia Ortí, J.E. Therapeutic Alternative of the Ketogenic Mediterranean Diet to Improve Mitochondrial Activity in Amyotrophic Lateral Sclerosis (ALS): A Comprehensive Review. Food Sci. Nutr. 2020, 8, 23–35. [Google Scholar] [CrossRef]
- Boumil, E.F.; Vohnoutka, R.B.; Liu, Y.; Lee, S.; Shea, T.B. Omega-3 Hastens and Omega-6 Delays the Progression of Neuropathology in a Murine Model of Familial ALS. Open Neurol. J. 2017, 11, 84–91. [Google Scholar] [CrossRef][Green Version]
- Mathers, J.C.; Strathdee, G.; Relton, C.L. Induction of Epigenetic Alterations by Dietary and Other Environmental Factors. Adv. Genet. 2010, 71, 3–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzo, S.; Bonanno, S.; Kapetis, D.; Barzago, C.; Cavalcante, P.; D’Alessandro, S.; Mantegazza, R.; Bernasconi, P. Up-Regulation of Neural and Cell Cycle-Related MicroRNAs in Brain of Amyotrophic Lateral Sclerosis Mice at Late Disease Stage. Mol. Brain 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonté, C. Dysregulated MicroRNAs in Amyotrophic Lateral Sclerosis Microglia Modulate Genes Linked to Neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef]
- Parisi, C.; Napoli, G.; Amadio, S.; Spalloni, A.; Apolloni, S.; Longone, P.; Volonte, C. MicroRNA-125b Regulates Microglia Activation and Motor Neuron Death in ALS. Cell Death Differ. 2016, 23, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating Inflammatory Monocytes with a Unique MicroRNA Gene Signature Ameliorates Murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.A.; Moreira, R.; Brites, D. Phenotypic Effects of Wild-Type and Mutant SOD1 Expression in N9 Murine Microglia at Steady State, Inflammatory and Immunomodulatory Conditions. Front. Cell. Neurosci. 2019, 13, 109. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting MiR-155 Restores Abnormal Microglia and Attenuates Disease in SOD1 Mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for Widespread MicroRNA-155 Inhibition Prolongs Survival in ALS-Model Mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastião, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased MiRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar] [CrossRef] [PubMed]
- Giunti, D.; Marini, C.; Parodi, B.; Usai, C.; Milanese, M.; Bonanno, G.; Kerlero de Rosbo, N.; Uccelli, A. Role of MiRNAs Shuttled by Mesenchymal Stem Cell-Derived Small Extracellular Vesicles in Modulating Neuroinflammation. Sci. Rep. 2021, 11, 1740. [Google Scholar] [CrossRef]
- Lasiene, J.; Yamanaka, K. Glial Cells in Amyotrophic Lateral Sclerosis. Neurol. Res. Int. 2011, 2011, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs Secreted through Astrocyte-Derived Extracellular Vesicles Cause Neuronal Network Degeneration in C9orf72 ALS. EBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef]
- Hoye, M.L.; Regan, M.R.; Jensen, L.A.; Lake, A.M.; Reddy, L.V.; Vidensky, S.; Richard, J.P.; Maragakis, N.J.; Rothstein, J.D.; Dougherty, J.D.; et al. Motor Neuron-Derived MicroRNAs Cause Astrocyte Dysfunction in Amyotrophic Lateral Sclerosis. Brain 2018, 141, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Reichenstein, I.; Eitan, C.; Diaz-Garcia, S.; Haim, G.; Magen, I.; Siany, A.; Hoye, M.L.; Rivkin, N.; Olender, T.; Toth, B.; et al. Human Genetics and Neuropathology Suggest a Link between MiR-218 and Amyotrophic Lateral Sclerosis Pathophysiology. Sci. Transl. Med. 2019, 11, eaav5264. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and MiR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Gonçalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of Depleted MiR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 2021, 9, 634355. [Google Scholar] [CrossRef]
- Greenway, M.J.; Andersen, P.M.; Russ, G.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG Mutations Segregate with Familial and “sporadic” Amyotrophic Lateral Sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Crivello, M.; Hogg, M.C.; Jirström, E.; Halang, L.; Woods, I.; Rayner, M.; Coughlan, K.S.; Lewandowski, S.A.; Prehn, J.H.M. Vascular Regression Precedes Motor Neuron Loss in the FUS (1–359) ALS Mouse Model. Dis. Model. Mech. 2019, 12, dmm040238. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-Specific MicroRNAs in Skeletal Muscle Development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 Delays ALS Progression and Promotes Regeneration of Neuromuscular Synapses in Mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A Potential Circulating Biomarker Candidate for Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Valdez, G.; Heyer, M.P.; Feng, G.; Sanes, J.R. The Role of Muscle MicroRNAs in Repairing the Neuromuscular Junction. PLoS ONE 2014, 9, e93140. [Google Scholar] [CrossRef]
- Bruneteau, G.; Simonet, T.; Bauché, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.-L.; Behin, A.; Khiami, F.; Sariali, E.; et al. Muscle Histone Deacetylase 4 Upregulation in Amyotrophic Lateral Sclerosis: Potential Role in Reinnervation Ability and Disease Progression. Brain 2013, 136, 2359–2368. [Google Scholar] [CrossRef]
- Pegoraro, V.; Marozzo, R.; Angelini, C. MicroRNAs and HDAC4 Protein Expression in the Skeletal Muscle of ALS Patients. Clin. Neuropathol. 2020, 39, 105–114. [Google Scholar] [CrossRef]
- Sobuś, A.; Baumert, B.; Litwińska, Z.; Gołąb-Janowska, M.; Stępniewski, J.; Kotowski, M.; Pius-Sadowska, E.; Kawa, M.P.; Gródecka-Szwajkiewicz, D.; Peregud-Pogorzelski, J.; et al. Safety and Feasibility of Lin- Cells Administration to ALS Patients: A Novel View on Humoral Factors and MiRNA Profiles. Int. J. Mol. Sci. 2018, 19, 1312. [Google Scholar] [CrossRef]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Léger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of Skeletal Muscle Mitochondrial Network Genes and MiRNAs in Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef]
- Kang, C.; Ji, L.L. Role of PGC-1α Signaling in Skeletal Muscle Health and Disease. Ann. N. Y. Acad. Sci. 2012, 1271, 110–117. [Google Scholar] [CrossRef]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. Biomed Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef]
- Martini, M.; Dobrowolny, G.; Aucello, M.; Musarò, A. Postmitotic Expression of SOD1G93A Gene Affects the Identity of Myogenic Cells and Inhibits Myoblasts Differentiation. Mediators Inflamm. 2015, 2015, 537853. [Google Scholar] [CrossRef] [PubMed]
- DI Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential Therapeutic Targets for ALS: MIR206, MIR208b and MIR499 Are Modulated during Disease Progression in the Skeletal Muscle of Patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, V.; Merico, A.; Angelini, C. Micro-RNAs in ALS Muscle: Differences in Gender, Age at Onset and Disease Duration. J. Neurol. Sci. 2017, 380, 58–63. [Google Scholar] [CrossRef]
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Ristic Kovacic, S.; Curk, T.; Rogelj, B. Differential Expression of MicroRNAs and Other Small RNAs in Muscle Tissue of Patients with ALS and Healthy Age-Matched Controls. Sci. Rep. 2018, 8, 5609. [Google Scholar] [CrossRef]
- Malacarne, C.; Galbiati, M.; Giagnorio, E.; Cavalcante, P.; Salerno, F.; Andreetta, F.; Cagnoli, C.; Taiana, M.; Nizzardo, M.; Corti, S.; et al. Dysregulation of Muscle-Specific Micrornas as Common Pathogenic Feature Associated with Muscle Atrophy in Als, Sma and Sbma: Evidence from Animal Models and Human Patients. Int. J. Mol. Sci. 2021, 22, 5673. [Google Scholar] [CrossRef] [PubMed]
- Maimon, R.; Ionescu, A.; Bonnie, A.; Sweetat, S.; Wald-Altman, S.; Inbar, S.; Gradus, T.; Trotti, D.; Weil, M.; Behar, O.; et al. MiR126-5p Downregulation Facilitates Axon Degeneration and NMJ Disruption via a Non–Cell-Autonomous Mechanism in ALS. J. Neurosci. 2018, 38, 5478–5494. [Google Scholar] [CrossRef]
- Fochi, S.; Giuriato, G.; De Simone, T.; Gomez-Lira, M.; Tamburin, S.; Del Piccolo, L.; Schena, F.; Venturelli, M.; Romanelli, M.G. Regulation of MicroRNAs in Satellite Cell Renewal, Muscle Function, Sarcopenia and the Role of Exercise. Int. J. Mol. Sci. 2020, 21, 6732. [Google Scholar] [CrossRef]
- Park, D.; Kwak, S.G.; Park, J.-S.; Choo, Y.J.; Chang, M.C. Can Therapeutic Exercise Slow Down Progressive Functional Decline in Patients With Amyotrophic Lateral Sclerosis? A Meta-Analysis. Front. Neurol. 2020, 11, 853. [Google Scholar] [CrossRef]
- Pegoraro, V.; Merico, A.; Angelini, C. MyomiRNAs Dysregulation in ALS Rehabilitation. Brain Sci. 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-Coding RNA: What Is Functional and What Is Junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Basu, S.; Müller, F.; Sanges, R. Examples of Sequence Conservation Analyses Capture a Subset of Mouse Long Non-Coding RNAs Sharing Homology with Fish Conserved Genomic Elements. BMC Bioinform. 2013, 14, S14. [Google Scholar] [CrossRef]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved Function of LincRNAs in Vertebrate Embryonic Development despite Rapid Sequence Evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative Annotation of Human Large Intergenic Noncoding RNAs Reveals Global Properties and Specific Subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. LincRNAs: Genomics, Evolution, and Mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The Multidimensional Mechanisms of Long Noncoding RNA Function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Laneve, P.; Rea, J.; Caffarelli, E. Long Noncoding RNAs: Emerging Players in Medulloblastoma. Front. Pediatr. 2019, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific Expression of Long Noncoding RNAs in the Mouse Brain. Proc. Natl. Acad. Sci. USA. 2008, 105, 716–721. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 Catalog of Human Long Noncoding RNAs: Analysis of Their Gene Structure, Evolution, and Expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-Coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef]
- Molyneaux, B.J.; Goff, L.A.; Brettler, A.C.; Chen, H.-H.; Brown, J.R.; Hrvatin, S.; Rinn, J.L.; Arlotta, P. DeCoN: Genome-Wide Analysis of In Vivo Transcriptional Dynamics during Pyramidal Neuron Fate Selection in Neocortex. Neuron 2015, 85, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Oliver, P.L.; Lunter, G.; Ponting, C.P. Genomic and Transcriptional Co-Localization of Protein-Coding and Long Non-Coding RNA Pairs in the Developing Brain. PLoS Genet. 2009, 5, e1000617. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Rea, J.; Menci, V.; Tollis, P.; Santini, T.; Armaos, A.; Garone, M.G.; Iberite, F.; Cipriano, A.; Tartaglia, G.G.; Rosa, A.; et al. HOTAIRM1 Regulates Neuronal Differentiation by Modulating NEUROGENIN 2 and the Downstream Neurogenic Cascade. Cell Death Dis. 2020, 11, 527. [Google Scholar] [CrossRef]
- Zimmer-Bensch, G. Emerging Roles of Long Non-Coding RNAs as Drivers of Brain Evolution. Cells 2019, 8, 1399. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.-L.; Corbier, C.J.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 Impairs Autophagy and Synergizes with PolyQ Ataxin-2 to Induce Motor Neuron Dysfunction and Cell Death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Heckman, M.G.; DeJesus-Hernandez, M.; Baker, M.C.; Soto-Ortolaza, A.I.; Rayaprolu, S.; Stewart, H.; Finger, E.; Volkening, K.; Seeley, W.W.; et al. Length of Normal Alleles of C9ORF72 GGGGCC Repeat Do Not Influence Disease Phenotype. Neurobiol. Aging 2012, 33, 2950.e5–2950.e7. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Lai, J.D.; Ichida, J.K. C9ORF72 Protein Function and Immune Dysregulation in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 713, 134523. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to C9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN Proteins and RNA Foci from Antisense Transcripts in C9ORF72 ALS and Frontotemporal Dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 Frontotemporal Lobar Degeneration Is Characterised by Frequent Neuronal Sense and Antisense RNA Foci. Acta Neuropathol. 2013, 126, 845–857. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Higginbottom, A.; Stopford, M.J.; Highley, J.R.; Ince, P.G.; Wharton, S.B.; Pickering-Brown, S.; Kirby, J.; Hautbergue, G.M.; Shaw, P.J. Antisense RNA Foci in the Motor Neurons of C9ORF72-ALS Patients Are Associated with TDP-43 Proteinopathy. Acta Neuropathol. 2015, 130, 63–75. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Frick, P.; Grässer, F.A.; Gendron, T.F.; Petrucelli, L.; Cashman, N.R.; Edbauer, D.; Kremmer, E.; Prudlo, J.; Troost, D.; et al. Quantitative Analysis and Clinico-Pathological Correlations of Different Dipeptide Repeat Protein Pathologies in C9ORF72 Mutation Carriers. Acta Neuropathol. 2015, 130, 845–861. [Google Scholar] [CrossRef]
- Tabet, R.; Schaeffer, L.; Freyermuth, F.; Jambeau, M.; Workman, M.; Lee, C.Z.; Lin, C.C.; Jiang, J.; Jansen-West, K.; Abou-Hamdan, H.; et al. CUG Initiation and Frameshifting Enable Production of Dipeptide Repeat Proteins from ALS/FTD C9ORF72 Transcripts. Nat. Commun. 2018, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 Repeat Expansion Disrupts Nucleocytoplasmic Transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of Multiple RNA Recognition Motif-Containing Proteins by C9orf72 Repeat Expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 Nucleotide Repeat Structures Initiate Molecular Cascades of Disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef]
- Moens, T.G.; Mizielinska, S.; Niccoli, T.; Mitchell, J.S.; Thoeng, A.; Ridler, C.E.; Grönke, S.; Esser, J.; Heslegrave, A.; Zetterberg, H.; et al. Sense and Antisense RNA Are Not Toxic in Drosophila Models of C9orf72-Associated ALS/FTD. Acta Neuropathol. 2018, 135, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 Repeat Expansions Cause Neurodegeneration in Drosophila through Arginine-Rich Proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A Zebrafish Model for C9orf72 ALS Reveals RNA Toxicity as a Pathogenic Mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Dodd, D.W.; Tomchick, D.R.; Corey, D.R.; Gagnon, K.T. Pathogenic C9ORF72 Antisense Repeat RNA Forms a Double Helix with Tandem C:C Mismatches. Biochemistry 2016, 55, 1283–1286. [Google Scholar] [CrossRef]
- Kovanda, A.; Zalar, M.; Šket, P.; Plavec, J.; Rogelj, B. Anti-Sense DNA d(GGCCCC)n Expansions in C9ORF72 Form i-Motifs and Protonated Hairpins. Sci. Rep. 2015, 5, 17944. [Google Scholar] [CrossRef]
- Ostrowski, L.A.; Hall, A.C.; Mekhail, K. Ataxin-2: From RNA Control to Human Health and Disease. Genes (Basel) 2017, 8, 2–21. [Google Scholar] [CrossRef]
- Pulst, S.M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate Expansion of a Normally Biallelic Trinucleotide Repeat in Spinooerebellar Ataxia Type. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.-M.; Weber, C.; Mandel, J.-L.; Cancel, G.; Abbas, N.; et al. Cloning of the Gene for Spinocerebellar Ataxia 2 Reveals a Locus with High Sensitivity to Expanded CAG/Glutamine Repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Yokoshi, M.; Li, Q.; Yamamoto, M.; Okada, H.; Suzuki, Y.; Kawahara, Y. Direct Binding of Ataxin-2 to Distinct Elements in 3′ UTRs Promotes MRNA Stability and Protein Expression. Mol. Cell 2014, 55, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Li, P.P.; Sun, X.; Xia, G.; Arbez, N.; Paul, S.; Zhu, S.; Peng, H.B.; Ross, C.A.; Koeppen, A.H.; Margolis, R.L.; et al. ATXN2-AS, a Gene Antisense to ATXN2, Is Associated with Spinocerebellar Ataxia Type 2 and Amyotrophic Lateral Sclerosis. Ann. Neurol. 2016, 80, 600–615. [Google Scholar] [CrossRef]
- Nalavade, R.; Griesche, N.; Ryan, D.P.; Hildebrand, S.; Krauß, S. Mechanisms of RNA-Induced Toxicity in CAG Repeat Disorders. Cell Death Dis. 2013, 4, e752. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, G.F.; Janitz, M.; Huang, Y.; Halliday, G.M. Long Noncoding RNAs in TDP-43 and FUS/TLS-Related Frontotemporal Lobar Degeneration (FTLD). Neurobiol. Dis. 2015, 82, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Sas-Nowosielska, H.; Magalska, A. Long Noncoding Rnas—Crucial Players Organizing the Landscape of the Neuronal Nucleus. Int. J. Mol. Sci. 2021, 22, 3478. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA Targets and Position-Dependent Splicing Regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced NcRNAs Allosterically Modify RNA-Binding Proteins in Cis to Inhibit Transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Hoell, J.I.; Larsson, E.; Runge, S.; Nusbaum, J.D.; Duggimpudi, S.; Farazi, T.A.; Hafner, M.; Borkhardt, A.; Sander, C.; Tuschl, T. RNA Targets of Wild-Type and Mutant FET Family Proteins. Nat. Struct. Mol. Biol. 2011, 18, 1428–1431. [Google Scholar] [CrossRef]
- Yen, Y.P.; Hsieh, W.F.; Tsai, Y.Y.; Lu, Y.L.; Liau, E.S.; Hsu, H.C.; Chen, Y.C.; Liu, T.C.; Chang, M.; Li, J.; et al. Dlk1-Dio3 Locus-Derived LncRNAs Perpetuate Postmitotic Motor Neuron Cell Fate and Subtype Identity. Elife 2018, 7, e38080. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.F.; Goshima, N.; Hirose, T. Alternative 3′-End Processing of Long Noncoding RNA Initiates Construction of Nuclear Paraspeckles. EMBO J. 2012, 31, 4020–4034. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Kukharsky, M.S.; An, H.; Dimasi, P.; Alexeeva, S.; Shabir, O.; Heath, P.R.; Buchman, V.L. Protective Paraspeckle Hyper-Assembly Downstream of TDP-43 Loss of Function in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2018, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.I.; Imai, T.; Murayama, S.; et al. The Long Non-Coding RNA Nuclear-Enriched Abundant Transcript 1-2 Induces Paraspeckle Formation in the Motor Neuron during the Early Phase of Amyotrophic Lateral Sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef]
- Banerjee, A.; Vest, K.E.; Pavlath, G.K.; Corbett, A.H. Nuclear Poly(A) Binding Protein 1 (PABPN1) and Matrin3 Interact in Muscle Cells and Regulate RNA Processing. Nucleic Acids Res. 2017, 45, 10706–10725. [Google Scholar] [CrossRef]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. C9-ALS/FTD-Linked Proline–Arginine Dipeptide Repeat Protein Associates with Paraspeckle Components and Increases Paraspeckle Formation. Cell Death Dis. 2019, 10, 746. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, K.; Kukharsky, M.S.; Park, S.K.; Park, S.; Watanabe, N.; Iwatsubo, T.; Hashimoto, T.; Liebman, S.W.; Shelkovnikova, T.A. Long Non-Coding RNA NEAT1_1 Ameliorates TDP-43 Toxicity in in Vivo Models of TDP-43 Proteinopathy. RNA Biol. 2021, 1–9. [Google Scholar] [CrossRef]
- Biscarini, S.; Capauto, D.; Peruzzi, G.; Lu, L.; Colantoni, A.; Santini, T.; Shneider, N.A.; Caffarelli, E.; Laneve, P.; Bozzoni, I. Characterization of the LncRNA Transcriptome in MESC-Derived Motor Neurons: Implications for FUS-ALS. Stem Cell Res. 2018, 27, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Lo Piccolo, L.; Bonaccorso, R.; Attardi, A.; Li Greci, L.; Romano, G.; Sollazzo, M.; Giurato, G.; Ingrassia, A.M.R.; Feiguin, F.; Corona, D.F.V.; et al. Loss of ISWI Function in Drosophila Nuclear Bodies Drives Cytoplasmic Redistribution of Drosophila TDP-43. Int. J. Mol. Sci. 2018, 19, 1082. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Berson, A.; Kennerdell, J.R.; Sartoris, A.; Unger, T.; Porta, S.; Kim, H.J.; Smith, E.R.; Shilatifard, A.; Van Deerlin, V.; et al. Aberrant Activation of Non-Coding RNA Targets of Transcriptional Elongation Complexes Contributes to TDP-43 Toxicity. Nat. Commun. 2018, 9, 4406. [Google Scholar] [CrossRef] [PubMed]
- Lo Piccolo, L.; Yamaguchi, M. RNAi of ArcRNA Hsrω Affects Sub-Cellular Localization of Drosophila FUS to Drive Neurodiseases. Exp. Neurol. 2017, 292, 125–134. [Google Scholar] [CrossRef]
- Rey, F.; Marcuzzo, S.; Bonanno, S.; Bordoni, M.; Giallongo, T.; Malacarne, C.; Cereda, C.; Zuccotti, G.V.; Carelli, S. Lncrnas Associated with Neuronal Development and Oncogenesis Are Deregulated in Sod1-G93a Murine Model of Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural Circular RNAs Are Derived from Synaptic Genes and Regulated by Development and Plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and Characterizing Circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- D’Ambra, E.; Capauto, D.; Morlando, M. Exploring the Regulatory Role of Circular RNAs in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 5477. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-Intron Circular RNAs Regulate Transcription in the Nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A Novel Protein Encoded by the Circular Form of the SHPRH Gene Suppresses Glioma Tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfò, R.; Peruzzi, G.; et al. FUS Affects Circular RNA Expression in Murine Embryonic Stem Cell-Derived Motor Neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef]
- Wu, L.S.; Cheng, W.C.; Chen, C.Y.; Wu, M.C.; Wang, Y.C.; Tseng, Y.H.; Chuang, T.J.; Shen, C.K.J. Transcriptomopathies of Pre- and Post-Symptomatic Frontotemporal Dementia-like Mice with TDP-43 Depletion in Forebrain Neurons. Acta Neuropathol. Commun. 2019, 7, 50. [Google Scholar] [CrossRef]
- Wang, S.; Latallo, M.J.; Zhang, Z.; Huang, B.; Bobrovnikov, D.G.; Dong, D.; Livingston, N.M.; Tjoeng, W.; Hayes, L.R.; Rothstein, J.D.; et al. Nuclear Export and Translation of Circular Repeat-Containing Intronic RNA in C9ORF72-ALS/FTD. Nat. Commun. 2021, 12, 4908. [Google Scholar] [CrossRef]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A MiRNA Signature in Leukocytes from Sporadic Amyotrophic Lateral Sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- Raheja, R.; Regev, K.; Healy, B.C.; Mazzola, M.A.; Beynon, V.; Von Glehn, F.; Paul, A.; Diaz-Cruz, C.; Gholipour, T.; Glanz, B.I.; et al. Correlating Serum Micrornas and Clinical Parameters in Amyotrophic Lateral Sclerosis. Muscle Nerve 2018, 58, 261–269. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Wang, H. Exploring MicroRNA Biomarker for Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 1318. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef]
- Takahashi, I.; Hama, Y.; Matsushima, M.; Hirotani, M.; Kano, T.; Hohzen, H.; Yabe, I.; Utsumi, J.; Sasaki, H. Identification of Plasma MicroRNAs as a Biomarker of Sporadic Amyotrophic Lateral Sclerosis. Mol. Brain 2015, 8, 67. [Google Scholar] [CrossRef]
- de Andrade, H.M.T.; de Albuquerque, M.; Avansini, S.H.; de S Rocha, C.; Dogini, D.B.; Nucci, A.; Carvalho, B.; Lopes-Cendes, I.; França, M.C. MicroRNAs-424 and 206 Are Potential Prognostic Markers in Spinal Onset Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2016, 368, 19–24. [Google Scholar] [CrossRef]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating MicroRNAs as Biomarkers of Muscle Differentiation and Atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kmetzsch, V.; Anquetil, V.; Saracino, D.; Rinaldi, D.; Camuzat, A.; Gareau, T.; Jornea, L.; Forlani, S.; Couratier, P.; Wallon, D.; et al. Plasma MicroRNA Signature in Presymptomatic and Symptomatic Subjects with C9orf72-Associated Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Soliman, R.; Mousa, N.O.; Rashed, H.R.; Moustafa, R.R.; Hamdi, N.; Osman, A.; Fahmy, N. Assessment of Diagnostic Potential of Some Circulating MicroRNAs in Amyotrophic Lateral Sclerosis Patients, an Egyptian Study. Clin. Neurol. Neurosurg. 2021, 208, 106883. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Mori, F.; Kakita, A.; Takahashi, H.; Utsumi, J.; Sasaki, H. Analysis of MicroRNA from Archived Formalin-Fixed Paraffin-Embedded Specimens of Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2014, 2, 173. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with HSOD1-G93A Are Enriched in Mir-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Jovičić, A.; Gitler, A.D. Distinct Repertoires of MicroRNAs Present in Mouse Astrocytes Compared to Astrocyte-Secreted Exosomes. PLoS ONE 2017, 12, e0171418. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, Y.; Zhou, X.; Luan, J.; Cui, Y.; Han, J. Comparison of the Extraction and Determination of Serum Exosome and MiRNA in Serum and the Detection of MiR-27a-3p in Serum Exosome of ALS Patients. Intractable Rare Dis. Res. 2018, 7, 13–18. [Google Scholar] [CrossRef]
- Saucier, D.; Wajnberg, G.; Roy, J.; Beauregard, A.P.; Chacko, S.; Crapoulet, N.; Fournier, S.; Ghosh, A.; Lewis, S.M.; Marrero, A.; et al. Identification of a Circulating MiRNA Signature in Extracellular Vesicles Collected from Amyotrophic Lateral Sclerosis Patients. Brain Res. 2019, 1708, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Katsu, M.; Hama, Y.; Utsumi, J.; Takashina, K.; Yasumatsu, H.; Mori, F.; Wakabayashi, K.; Shoji, M.; Sasaki, H. MicroRNA Expression Profiles of Neuron-Derived Extracellular Vesicles in Plasma from Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 708, 134176. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Cox, P.A. An MiRNA Fingerprint Using Neural-Enriched Extracellular Vesicles from Blood Plasma: Towards a Biomarker for Amyotrophic Lateral Sclerosis/Motor Neuron Disease. Open Biol. 2020, 10, 200116. [Google Scholar] [CrossRef]
- Pregnolato, F.; Cova, L.; Doretti, A.; Bardelli, D.; Silani, V.; Bossolasco, P. Exosome MicroRNAs in Amyotrophic Lateral Sclerosis: A Pilot Study. Biomolecules 2021, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Sproviero, D.; Gagliardi, S.; Zucca, S.; Arigoni, M.; Giannini, M.; Garofalo, M.; Olivero, M.; Dell’Orco, M.; Pansarasa, O.; Bernuzzi, S.; et al. Different MiRNA Profiles in Plasma Derived Small and Large Extracellular Vesicles from Patients with Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 2737. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H. Systemic Dysregulation of TDP-43 Binding MicroRNAs in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2013, 1, 42. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. MiR-338-3p Is over-Expressed in Blood, CFS, Serum and Spinal Cord from Sporadic Amyotrophic Lateral Sclerosis Patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Benigni, M.; Ricci, C.; Jones, A.R.; Giannini, F.; Al-Chalabi, A.; Battistini, S. Identification of MiRNAs as Potential Biomarkers in Cerebrospinal Fluid from Amyotrophic Lateral Sclerosis Patients. Neuro Mol. Med. 2016, 18, 551–560. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed MiRNAs Related to Neural and Glial Activity. Front. Neurosci. 2018, 11, 731. [Google Scholar] [CrossRef]
- Yelick, J.; Men, Y.; Jin, S.; Seo, S.; Espejo-Porras, F.; Yang, Y. Elevated Exosomal Secretion of MiR-124-3p from Spinal Neurons Positively Associates with Disease Severity in ALS. Exp. Neurol. 2020, 333, 113414. [Google Scholar] [CrossRef]
- Matamala, J.M.; Arias-Carrasco, R.; Sanchez, C.; Uhrig, M.; Bargsted, L.; Matus, S.; Maracaja-Coutinho, V.; Abarzua, S.; van Zundert, B.; Verdugo, R.; et al. Genome-Wide Circulating MicroRNA Expression Profiling Reveals Potential Biomarkers for Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2018, 64, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Cui, X.; Crossman, D.K.; Hao, J.; Kazamel, M.; Kwon, Y.; King, P.H. Muscle MicroRNA Signatures as Biomarkers of Disease Progression in Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2018, 114, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Martone, J.; Lepore, E.; Casola, I.; Petrucci, A.; Inghilleri, M.; Morlando, M.; Colantoni, A.; Scicchitano, B.M.; Calvo, A.; et al. A Longitudinal Study Defined Circulating MicroRNAs as Reliable Biomarkers for Disease Prognosis and Progression in ALS Human Patients. Cell Death Discov. 2021, 7, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating Brain-Enriched MicroRNAs as Novel Biomarkers for Detection and Differentiation of Neurodegenerative Diseases. Alzheimers Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum MiRNAs MiR-206, 143-3p and 374b-5p as Potential Biomarkers for Amyotrophic Lateral Sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Vrabec, K.; Boštjančič, E.; Koritnik, B.; Leonardis, L.; Dolenc Grošelj, L.; Zidar, J.; Rogelj, B.; Glavač, D.; Ravnik-Glavač, M. Differential Expression of Several MiRNAs and the Host Genes AATK and DNM2 in Leukocytes of Sporadic ALS Patients. Front. Mol. Neurosci. 2018, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Manfellotto, F.; Fiorentino, G.; Annunziata, A.; Biffali, E.; Pannone, R.; Federico, A. Wide-Ranging Analysis of MicroRNA Profiles in Sporadic Amyotrophic Lateral Sclerosis Using Next-Generation Sequencing. Front. Genet. 2018, 9, 310. [Google Scholar] [CrossRef]
- Dolinar, A.; Koritnik, B.; Glavač, D.; Ravnik-Glavač, M. Circular RNAs as Potential Blood Biomarkers in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 8052–8062. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Yamashita, T.; Teramoto, S.; Hirose, N.; Tamaoka, A.; Kwak, S. ADAR2-Dependent A-to-I RNA Editing in the Extracellular Linear and Circular RNAs. Neurosci. Res. 2019, 147, 48–57. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound Downregulation of the RNA Editing Enzyme ADAR2 in ALS Spinal Motor Neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef]
- Loffreda, A.; Nizzardo, M.; Arosio, A.; Ruepp, M.D.; Calogero, R.A.; Volinia, S.; Galasso, M.; Bendotti, C.; Ferrarese, C.; Lunetta, C.; et al. MiR-129-5p: A Key Factor and Therapeutic Target in Amyotrophic Lateral Sclerosis. Prog. Neurobiol. 2020, 190, 101803. [Google Scholar] [CrossRef] [PubMed]
- Dirren, E.; Aebischer, J.; Rochat, C.; Towne, C.; Schneider, B.L.; Aebischer, P. SOD1 Silencing in Motoneurons or Glia Rescues Neuromuscular Function in ALS Mice. Ann. Clin. Transl. Neurol. 2015, 2, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, B.; Qiu, L.; Yang, C.; Kramer, J.; Su, Q.; Guo, Y.; Brown, R.H.; Gao, G.; Xu, Z. Widespread Spinal Cord Transduction by Intrathecal Injection of RAAV Delivers Efficacious RNAi Therapy for Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2014, 23, 668–681. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabreja, G.C.; Song, L.; Su, Q.; Gao, G.P.; ElMallah, M.K.; Brown, R.H.; et al. Therapeutic RAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H.; Sena-Esteves, M. Adeno-Associated Virus–Delivered Artificial MicroRNA Extends Survival and Delays Paralysis in an Amyotrophic Lateral Sclerosis Mouse Model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Keeler, A.M.; Zieger, M.; Semple, C.; Pucci, L.; Veinbachs, A.; Brown, R.H.; Mueller, C.; ElMallah, M.K. Intralingual and Intrapleural AAV Gene Therapy Prolongs Survival in a SOD1 ALS Mouse Model. Mol. Ther.-Methods Clin. Dev. 2020, 17, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Martier, R.; Liefhebber, J.M.; Miniarikova, J.; van der Zon, T.; Snapper, J.; Kolder, I.; Petry, H.; van Deventer, S.J.; Evers, M.M.; Konstantinova, P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-Nuclear Transcripts in ALS and FTD Patients. Mol. Ther.-Nucleic Acids 2019, 14, 593–608. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef]



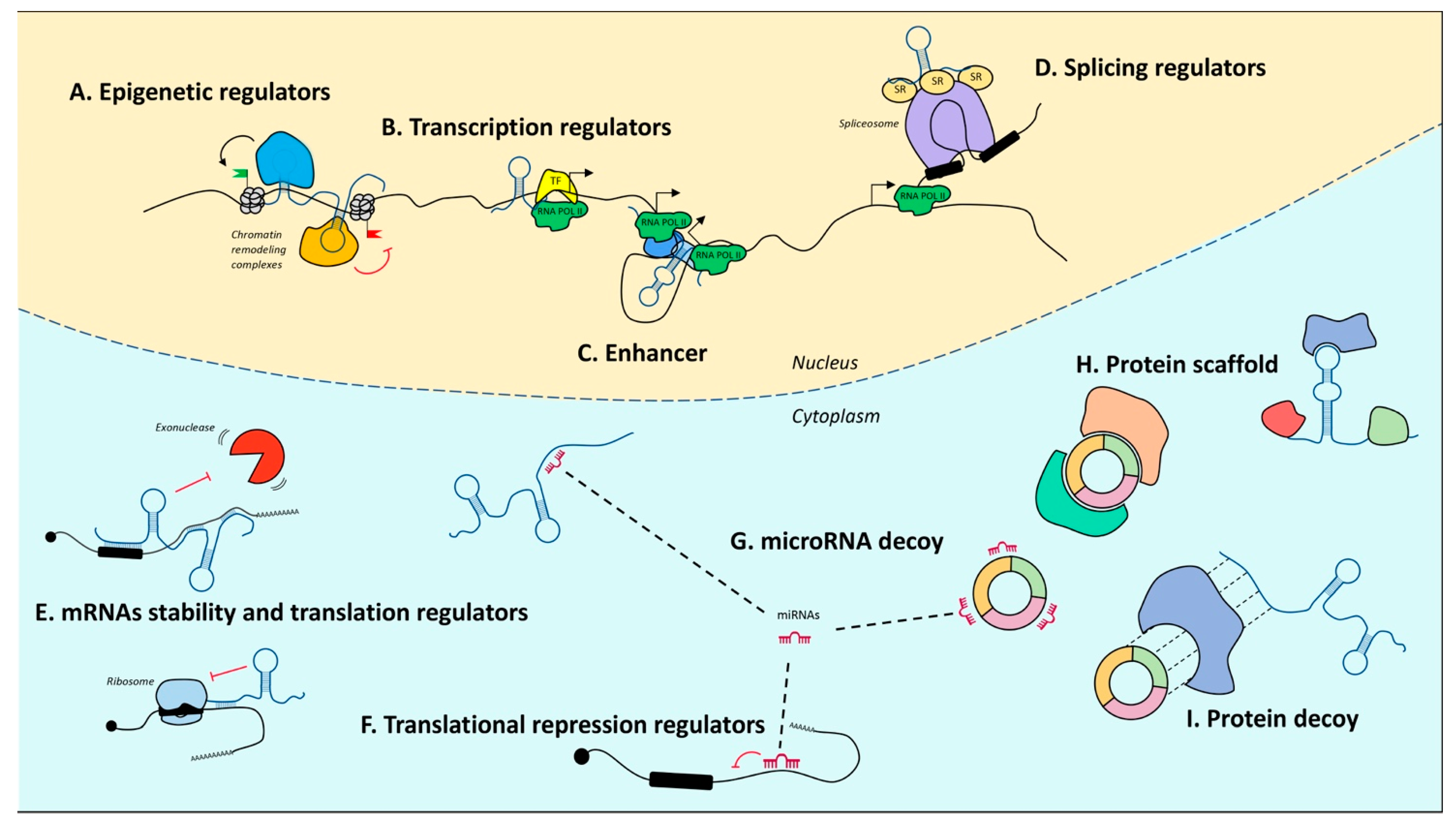{kind=link}
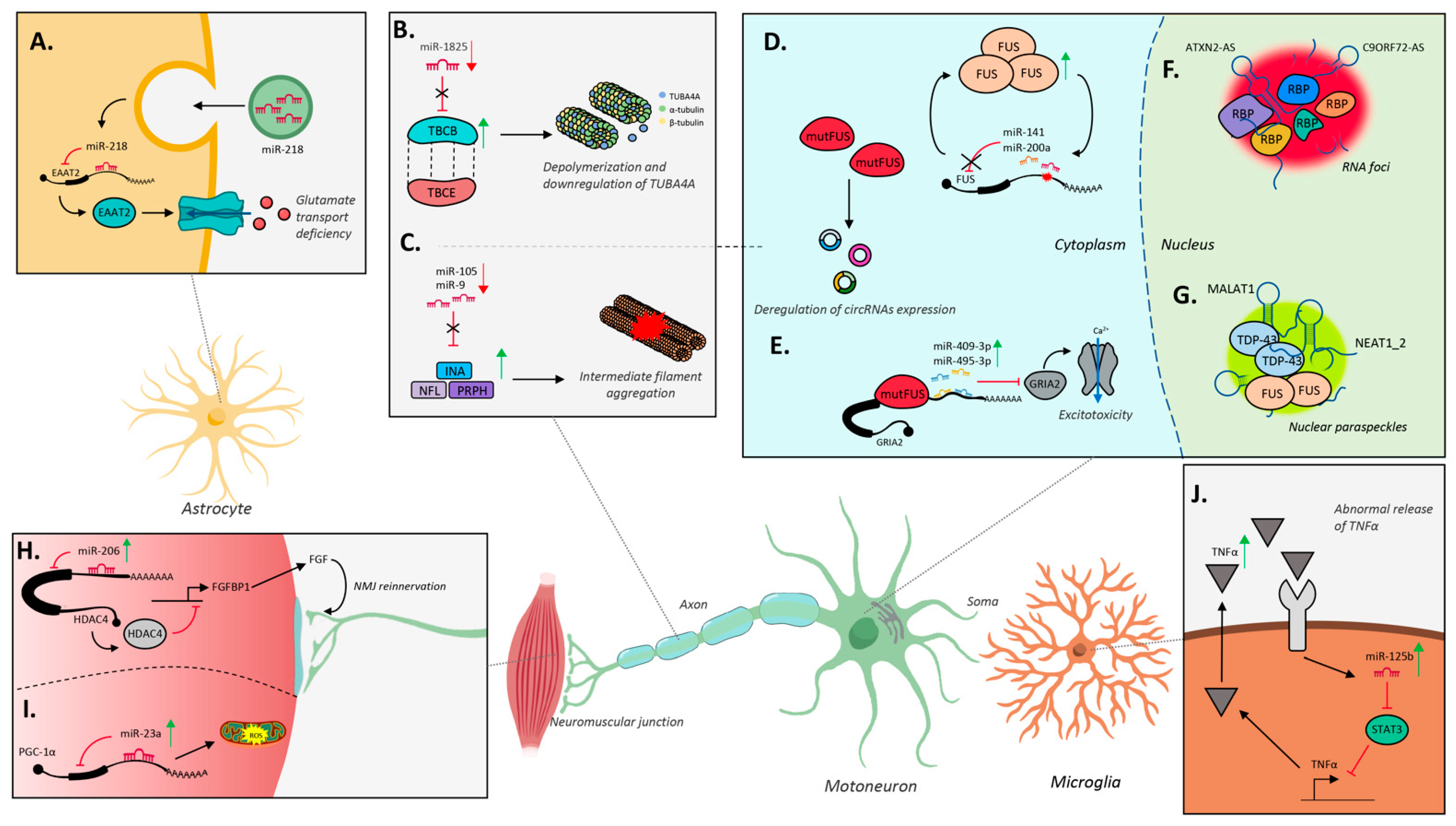{kind=link}
| Sample | Disease | Upregulated | Downregulated | Reference |
|---|---|---|---|---|
| Cortico-spinal tract tissue (EV) | C9orf72 ALS | miRNA-494-3p | [156] | |
| Cerebro-spinal fluid | sALS, fALS | miRNA-27b, miRNA-99b, miRNA-146a, miRNA-150, miRNA-328, miRNA-532-3p | [149] | |
| Cerebro-spinal fluid | sALS | miRNA-143-5p, miRNA-574-5p | miRNA-132-3p, miRNA-132-5p, miRNA-143-3p | [276] |
| Cerebro-spinal fluid | sALS | miRNA-181a-5p | let7a-5p, let7b-5p, let7f-5p, miRNA-15b-5p, miRNA-21-5p, miRNA-148-3p, miRNA-195-5p | [278] |
| Cerebro-spinal fluid | sALS | miRNA-9-5p, miRNA-23b-3p, miRNA-27b-3p, miRNA-99b-5p, miRNA-124-3p, miRNA-126-5p, miRNA-127-3p | let-7f-5p, miRNA-i50-5p, miRNA-142-5p, miRNA-378a-3p | [281] |
| Cerebro-spinal fluid | sALS | miRNA-9-5p, miRNA-27b-3p, miRNA-124-3p, miRNA-125b-2-3p, miRNA-127-3p, miRNA-143-3p | let7f-5p, miRNA-16-5p, miRNA-28-3p, miRNA-92a-5p, miRNA-142-5p, miRNA-146a-3p, miRNA-150-5p, miRNA-378a-3p, miRNA-486-5p | [279] |
| Peripheral blood mononuclear cells | ALS | miRNA-183, miRNA-193b, miRNA-451, miRNA-3935 | [130] | |
| Plasma | sALS | miRNA-4649-5p | miRNA-4299 | [261] |
| Plasma | sALS | miRNA-206, miRNA-424 | [262] | |
| Plasma | ALS | miRNA-9, miRNA-129-3p, miRNA-206, miRNA-335-5p, miRNA-338-3p | [282] | |
| Plasma | sALS | let-7a-5p, let-7d-5p, let-7f-5p, let-7g-5p, let-7i-5p, miRNA-15a-5p, miRNA-15b-5p, miRNA-16-5p, miRNA-22-3p, miRNA-23a-3p, miRNA-26a-5p, miRNA-26b-5p, miRNA-27b-3p, miRNA-28-3p, miRNA-30b-5p, miRNA-30c-5p, miRNA-93-5p, miRNA-103a-3p, miRNA-106b-3p, miRNA-128-3p, miRNA-130a-3p, miRNA-130b-3p, miRNA-144-5p, miRNA-148a-3p, miRNA-148b-3p, miRNA-151a-5p, miRNA-151b, miRNA-182-5p, miRNA-183-5p, miRNA-186-5p, miRNA-221-3p, miRNA-223-3p, miRNA-342-3p, miRNA-425-5p, miRNA-451a, miRNA-532-5p, miRNA-550a-3p, miRNA-584-5p | [260] | |
| Plasma | ALS | miRNA-532.3p, miRNA-144-3p, miRNA-15a-5p, miRNA-363-3p, miRNA-183-5p | let-7c-5p, miRNA-4454, miRNA-9-1-5p, miRNA-9-3-5p, miRNA-338-3p, miRNA-9-2-5p, miRNA-100-5p, miRNA-7977, miRNA-1246, miRNA-664a-5p, miRNA-7641-1, miRNA-1290, miRNA-4286, miRNA-181b-1-5p, miRNA-1260b, miRNA-181b-2-5p, miRNA-127-3p, miRNA-181a-2-5p, miRNA-181a-1-5p, miRNA-199b-3p, miRNA-199a-1-3p | [271] |
| Plasma | C9orf72 ALS | miRNA-34a-5p, miRNA-345-5p | miRNA-200c-3p, miRNA-10a-3p | [264] |
| Plasma | sALS, fALS | let7f-5p, miRNA-106, miRNA-142, miRNA-143, miRNA-206, miRNA-4516 | let7f-5p | [265] |
| Plasma (EV) | ALS | miRNA-24-3p, miRNA-149-3p, miRNA-371a-5p, miRNA-939-5p, miRNA-1207-5p, miRNA-3619-3p, miRNA-4298, miRNA-4484, miRNA-4505, miRNA-4688, miRNA-4700-5p, miRNA-4736, miRNA-4739 | miRNA-150-3p, miRNA-634, miRNA-1268a, miRNA-1913, miRNA-2861, miRNA-3176, miRNA-3177-3p, miRNA-3605-5p, miRNA-3911, miRNA-3940-3p, miRNA-4507, miRNA-4508, miRNA-4646-5p, miRNA-4674, miRNA-4687-5p, miRNA-4745-5p, miRNA-4788 | [272] |
| Plasma (EV) | ALS | miRNA-146a-5p, miRNA-151a-3p, miRNA-151a-5p, miRNA-199a-3p, miRNA-199a-5p | miRNA-10b-5p, miRNA-29b-3p, miRNA-4454 | [273] |
| Plasma (EV) | sALS | miRNA-8089, miRNA-196a-5p, miRNA-3152-3p, miRNA-607, miRNA-3607-3p, miRNA-6825-3p, miRNA-7106-5p, miRNA-3976, miRNA-4492, miRNA-200a-3p, miRNA-205-5p, miRNA-6858-3p, miRNA-1273c, miRNA-6888.3p, miRNA-4302, miRNA-4634, miRNA-182-3p, miRNA-3160-3p, miRNA-1-3p, miRNA-200a-5p, miRNA-7704, miRNA-210-3p, miRNA-31-5p, miRNA-133a-3p, miRNA-34c-5p, miRNA-455-5p, miRNA-6842-5p, miRNA-3619-3p, miRNA-4279, miRNA-4508, miRNA-1469, miRNA-141-3p, miRNA-542-3p, miRNA-615-3p, miRNA-200c-3p, miRNA-4451, miRNA-18a-5p, miRNA-200b-3p, miRNA-184, miRNA-9-5p, miRNA-7c-5p, miRNA-6746-5p, miRNA-3195, miRNA-206, miRNA-6068 | miRNA-493-3p, 409-3p, miRNA-323b-3p, miRNA-6073, miRNA-432-5p, miRNA-134-5p, miRNA-330-3p, miRNA-625-3p, miRNA-4446-3p, miRNA-148b-3p, miRNA-370-3p, miRNA-584-5p, miRNA-224-5p, miRNA-381-3p, miRNA-199a-5p, miRNA-654-3p, miRNA-335-3p, miRNA-543, miRNA-4433b-5p, miRNA-130b-5p, miRNA-4286, miRNA-382-5p | [275] |
| Serum | sALS | let-7b-5p, miRNA-132-3p, miRNA-132-5p, miRNA-143-3p, miRNA-143-5p | [276] | |
| Serum | sALS | miR-338-3p | [277] | |
| Serum | fALS | miRNA-1825, miRNA-1915-3p, miRNA-3665, miRNA-4530, miRNA-4745-5p | [104] | |
| Serum | ALS | miRNA-106b, miRNA-206 | [165] | |
| Serum | sALS | miRNA-1234-3p, miRNA-1825 | [105] | |
| Serum | sALS | miRNA-143-3p, miRNA-206 | miRNA-374b-5p | [281] |
| Serum | sALS | miRNA-142-3p | miRNA-1249-3p | [283] |
| Serum | ALS | miRNA-1, miRNA-19a-3p, miRNA-133a-3p, miRNA-133b, miRNA-192-3p, miRNA-192-5p, miRNA-142-3p, miRNA-144-5p | let-7d-3p, miRNA-139-5p, miRNA-320a, miRNA-320b, miRNA-320c, miRNA-425-5p | [258] |
| Serum | sALS, fALS | miRNA-133a, miRNA-206 | miRNA-151a-5p, miRNA-199a-5p, miRNA-423-3p | [284] |
| Serum (EXO) | ALS | miRNA-27a-3p | [270] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laneve, P.; Tollis, P.; Caffarelli, E. RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. Int. J. Mol. Sci. 2021, 22, 10285. https://doi.org/10.3390/ijms221910285
Laneve P, Tollis P, Caffarelli E. RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. International Journal of Molecular Sciences. 2021; 22(19):10285. https://doi.org/10.3390/ijms221910285
Chicago/Turabian StyleLaneve, Pietro, Paolo Tollis, and Elisa Caffarelli. 2021. "RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective" International Journal of Molecular Sciences 22, no. 19: 10285. https://doi.org/10.3390/ijms221910285
APA StyleLaneve, P., Tollis, P., & Caffarelli, E. (2021). RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. International Journal of Molecular Sciences, 22(19), 10285. https://doi.org/10.3390/ijms221910285