
Human FoxP Transcription Factors as Tractable Models of the Evolution and Functional Outcomes of Three-Dimensional Domain Swapping



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
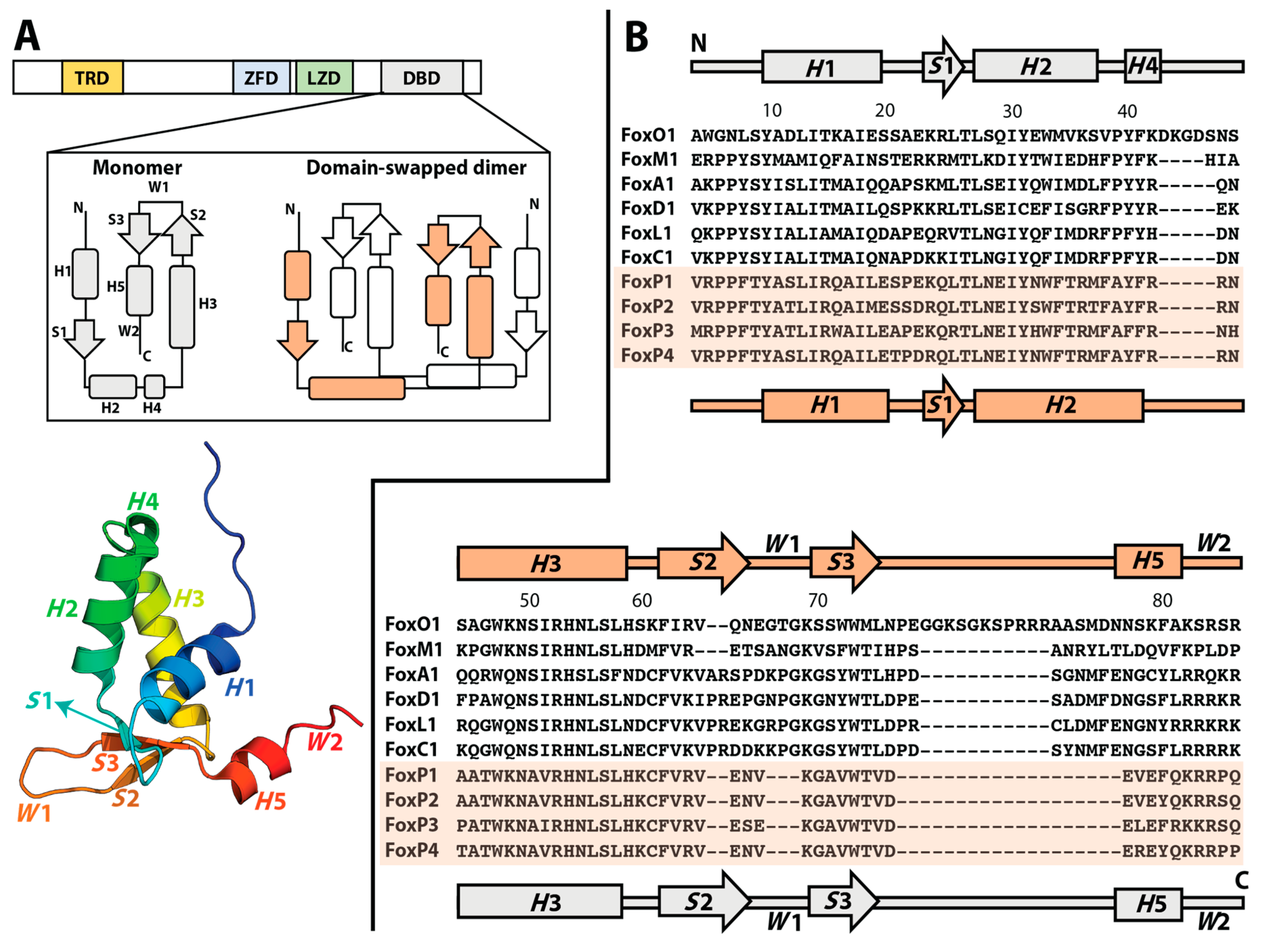
2. Molecular Evolution towards 3D-DS in Human FoxP Transcription Factors
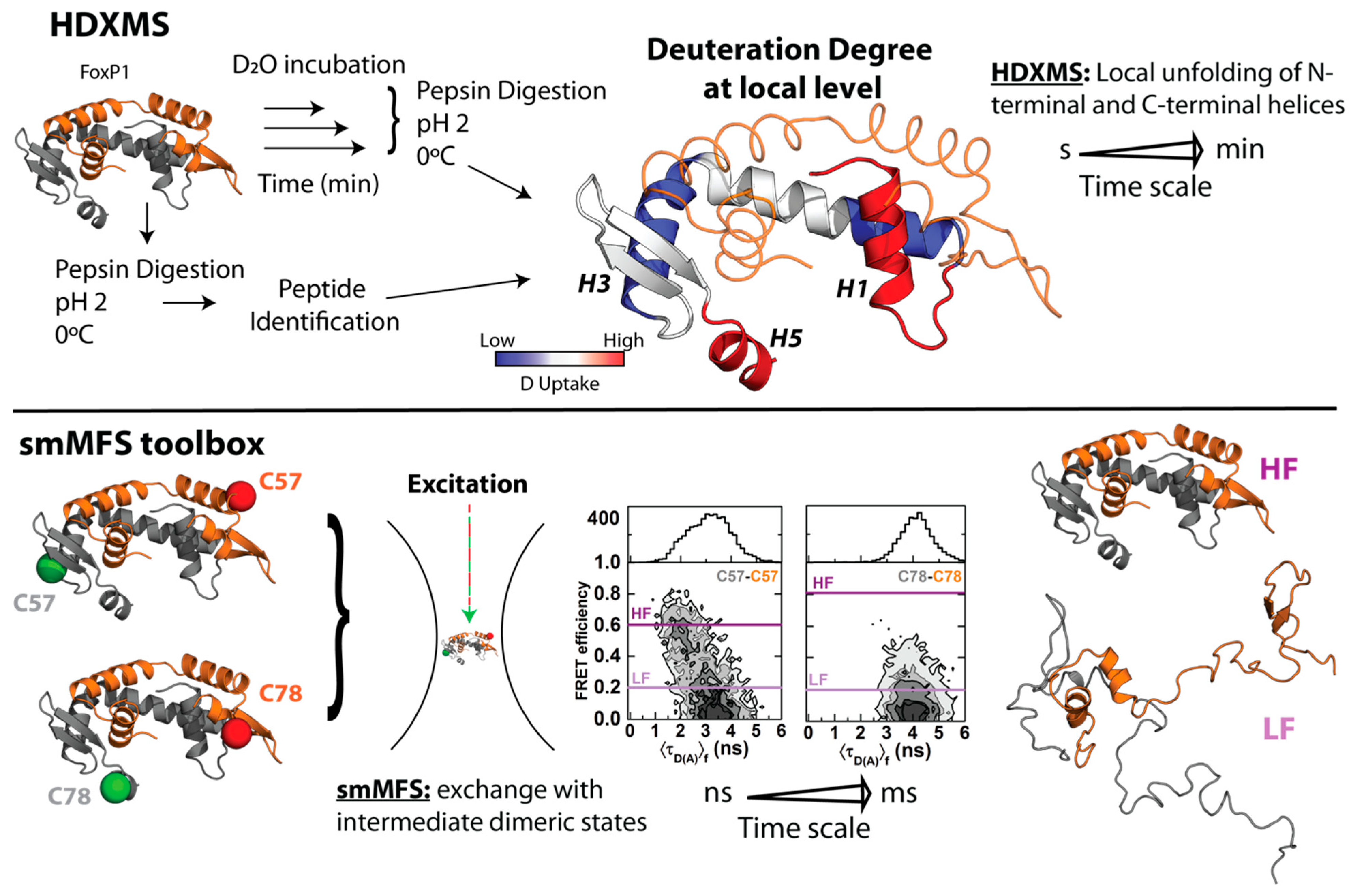
3. Biophysically Dissecting the Evolutionary Strategies of FoxP Proteins to Overcome the Thermodynamic Limitations of 3D-DS
4. The Molecular Mechanism of 3D-DS Explained at Near-Atomic Level
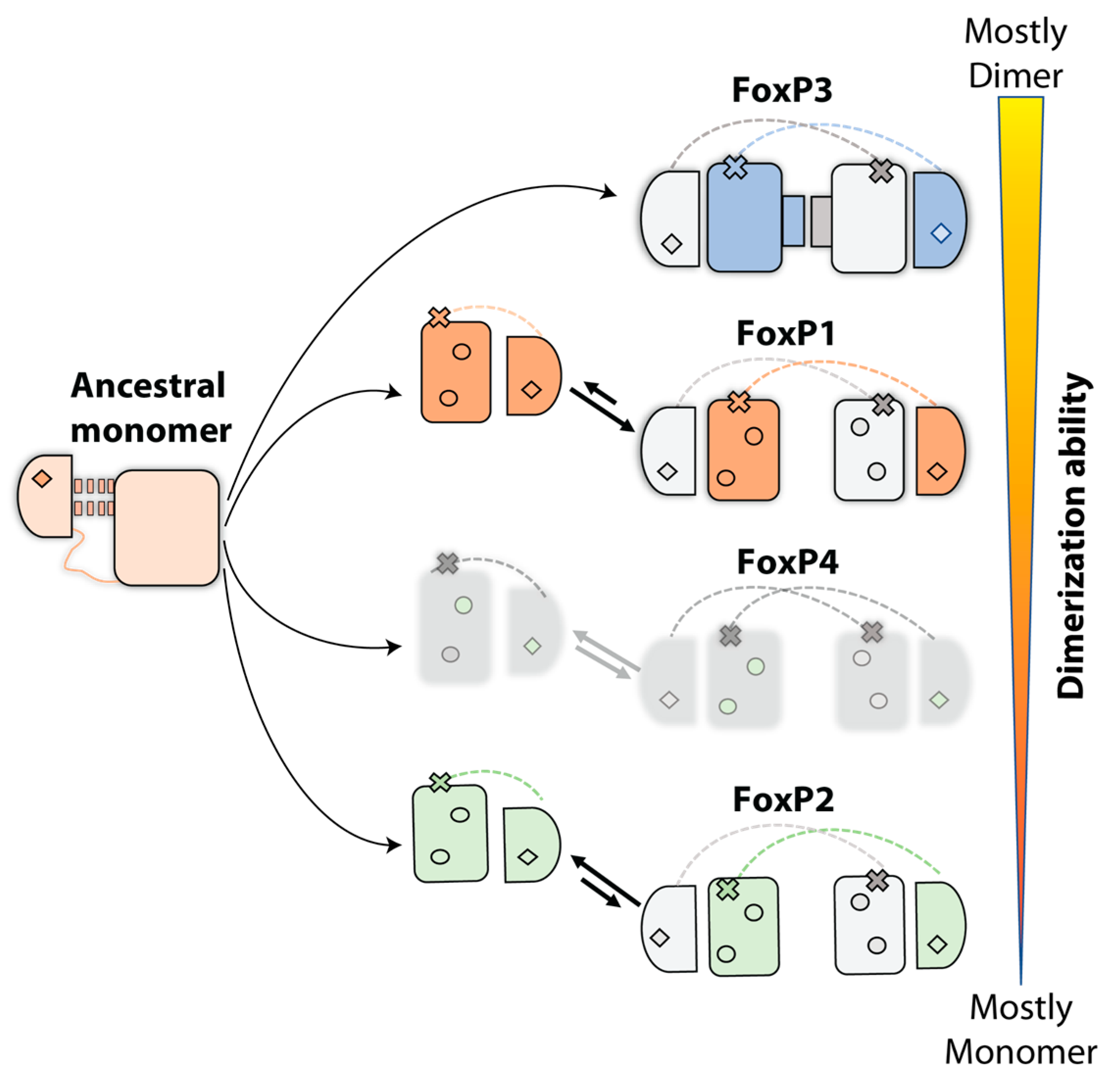
5. Evolution Pathway inside FoxP Subfamily and Their Impact in Functionality: From Homodimers to Heterodimers and Beyond
Funding
Acknowledgments
Conflicts of Interest
References
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The Power of Two: Protein Dimerization in Biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef]
- Qaiser Fatmi, M.; Chang, C.E.A. The Role of Oligomerization and Cooperative Regulation in Protein Function: The Case of Tryptophan Synthase. PLoS Comput. Biol. 2010, 6, e1000994. [Google Scholar] [CrossRef]
- Nooren, I.M.A.; Thornton, J.M. Structural Characterisation and Functional Significance of Transient Protein–Protein Interactions. J. Mol. Biol. 2003, 325, 991–1018. [Google Scholar] [CrossRef]
- Blundell, T.L.; Srinivasan, N. Symmetry, Stability, and Dynamics of Multidomain and Multicomponent Protein Systems. Proc. Natl. Acad. Sci. USA 1996, 93, 14243–14248. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Structural Symmetry and Protein Function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem. 2015, 84, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Protein-Protein Interactions: A Review of Protein Dimer Structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Gwyther, R.E.A.; Jones, D.D.; Worthy, H.L. Better Together: Building Protein Oligomers Naturally and by Design. Biochem. Soc. Trans. 2019, 47, 1773–1780. [Google Scholar] [CrossRef]
- Bennett, M.J.; Choe, S.; Eisenberg, D. Domain Swapping: Entangling Alliances between Proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef]
- Liu, Y.; Eisenberg, D. 3D Domain Swapping: As Domains Continue to Swap. Protein Sci. 2002, 11, 1285–1299. [Google Scholar] [CrossRef]
- Bennett, M.J.; Schlunegger, M.P.; Eisenberg, D. 3D Domain Swapping: A Mechanism for Oligomer Assembly. Protein Sci. 1995, 4, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Moudrianakis, E.N. The Histone Fold: A Ubiquitous Architectural Motif Utilized in DNA Compaction and Protein Dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Ammelburg, M.; Söding, J.; Lupas, A.N. On the Origin of the Histone Fold. BMC Struct. Biol. 2007, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Hadjithomas, M.; Moudrianakis, E.N. Experimental Evidence for the Role of Domain Swapping in the Evolution of the Histone Fold. Proc. Natl. Acad. Sci. USA 2011, 108, 13462–13467. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Schymkowitz, J.; Itzhaki, L.S. Implications of 3D Domain Swapping for Protein Folding, Misfolding and Function. Adv. Exp. Med. Biol. 2012, 747, 137–152. [Google Scholar]
- Liu, L.; Byeon, I.J.L.; Bahar, I.; Gronenborn, A.M. Domain Swapping Proceeds via Complete Unfolding: A 19F- and 1H-NMR Study of the Cyanovirin-N Protein. J. Am. Chem. Soc. 2012, 134, 4229–4235. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.W.; Wilkinson, H.R.; Itzhaki, L.S. Three-Dimensional Domain Swapping in p13suc1 Occurs in the Unfolded State and Is Controlled by Conserved Proline Residues. Proc. Natl. Acad. Sci. USA 2001, 98, 5596–5601. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.W.H.; Itzhaki, L.S. The Unfolding Story of Three-Dimensional Domain Swapping. Structure 2003, 11, 243–251. [Google Scholar] [CrossRef]
- Mazet, F.; Yu, J.K.; Liberles, D.A.; Holland, L.Z.; Shimeld, S.M. Phylogenetic Relationships of the Fox (Forkhead) Gene Family in the Bilateria. Gene 2003, 316, 79–89. [Google Scholar] [CrossRef]
- Pascual-Carreras, E.; Herrera-Úbeda, C.; Rosselló, M.; Garcia-Fernandez, J.; Saló, E.; Adell, T. Analysis of Fox Genes in Schmidtea Mediterranea Reveals New Families and a Conserved Role of Smed-foxO in Controlling Cell Death. Sci. Rep. 2021, 11, 2947. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, S.L. A Search for the Origins of Animals and Fungi: Comparing and Combining Molecular Data. Am. Nat. 1999, 154, S178–S188. [Google Scholar] [CrossRef]
- Mazet, F.; Amemiya, C.T.; Shimeld, S.M. An Ancient Fox Gene Cluster in Bilaterian Animals. Curr. Biol. 2006, 16, R314–R316. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead Transcription Factors: Key Players in Health and Disease. Trends Genet. 2011, 27, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Wotton, K.R.; Shimeld, S.M. Comparative Genomics of Vertebrate Fox Cluster Loci. BMC Genom. 2006, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-Crystal Structure of the HNF-3/fork Head DNA-Recognition Motif Resembles Histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Hergeth, S.P.; Schneider, R. The H1 Linker Histones: Multifunctional Proteins beyond the Nucleosomal Core Particle. EMBO Rep. 2015, 16, 1439–1453. [Google Scholar] [CrossRef]
- Kasinsky, H.E.; Lewis, J.D.; Dacks, J.B.; Ausió, J. Origin of H1 Linker Histones. FASEB J. 2001, 15, 34–42. [Google Scholar] [CrossRef]
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD-DNA complex at 1.9 Å resolution reveals new details of FOXO binding to the DNA. Acta. Crystallogr. D Biol. Crystallogr. 2010, 66, 1351–1357. [Google Scholar] [CrossRef]
- Li, J.; Dai, S.; Chen, X.; Liang, X.; Qu, L.; Jiang, L.; Guo, M.; Zhou, Z.; Wei, H.; Zhang, H.; et al. Mechanism of forkhead transcription factors binding to a novel palindromic DNA site. Nucleic Acids Res 2021, 49, 3573–3583. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pradhan, L.; Ashur, S.; Joshi, A.; Nam, H.-J. Crystal Structure of FOXC2 in Complex with DNA Target. ACS Omega 2019, 4, 10906–10914. [Google Scholar] [CrossRef]
- Chen, X.; Wei, H.; Li, J.; Liang, X.; Dai, S.; Jiang, L.; Guo, M.; Qu, L.; Chen, Z.; Chen, L.; et al. Structural Basis for DNA Recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764. [Google Scholar] [CrossRef]
- Nakagawa, S.; Gisselbrecht, S.S.; Rogers, J.M.; Hartl, D.L.; Bulyk, M.L. DNA-Binding Specificity Changes in the Evolution of Forkhead Transcription Factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12349–12354. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Brosens, J.J.; Gomes, A.R.; Koo, C.-Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Long, N.P.; Luu, T.-T.-T.; Anh, N.H.; Kwon, S.W.; Lee, S.K. The Dominant Role of Forkhead Box Proteins in Cancer. Int. J. Mol. Sci. 2018, 19, 3279. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Hwang, J.; Jung, J.H.; Lee, H.-J.; Lee, D.Y.; Kim, S.-H. Molecular Networks of FOXP Family: Dual Biologic Functions, Interplay with Other Molecules and Clinical Implications in Cancer Progression. Mol. Cancer 2019, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Huang, C.Y.; Chang, C.H.; Sun, Y.J.; Chuang, W.J.; Hsiao, C.D. Crystal Structure of the Human FOXK1a-DNA Complex and Its Implications on the Diverse Binding Specificity of Winged Helix/forkhead Proteins. J. Biol. Chem. 2006, 281, 17400–17409. [Google Scholar] [CrossRef]
- Littler, D.R.; Alvarez-Fernández, M.; Stein, A.; Hibbert, R.G.; Heidebrecht, T.; Aloy, P.; Medema, R.H.; Perrakis, A. Structure of the FoxM1 DNA-Recognition Domain Bound to a Promoter Sequence. Nucleic Acids Res. 2010, 38, 4527–4538. [Google Scholar] [CrossRef]
- Li, J.; Dantas Machado, A.C.; Guo, M.; Sagendorf, J.M.; Zhou, Z.; Jiang, L.; Chen, X.; Wu, D.; Qu, L.; Chen, Z.; et al. Structure of the Forkhead Domain of FOXA2 Bound to a Complete DNA Consensus Site. Biochemistry 2017, 56, 3745–3753. [Google Scholar] [CrossRef]
- Newman, J.A.; Aitkenhead, H.; Gavard, A.E.; Rota, I.A.; Handel, A.E.; Hollander, G.A.; Gileadi, O. The Crystal Structure of Human Forkhead Box N1 in Complex with DNA Reveals the Structural Basis for Forkhead Box Family Specificity. J. Biol. Chem. 2020, 295, 2948–2958. [Google Scholar] [CrossRef]
- Stroud, J.C.; Wu, Y.; Bates, D.L.; Han, A.; Nowick, K.; Paabo, S.; Tong, H.; Chen, L. Structure of the Forkhead Domain of FOXP2 Bound to DNA. Structure 2006, 14, 159–166. [Google Scholar] [CrossRef]
- Bandukwala, H.S.; Wu, Y.; Feuerer, M.; Chen, Y.; Barboza, B.; Ghosh, S.; Stroud, J.C.; Benoist, C.; Mathis, D.; Rao, A.; et al. Structure of a Domain-Swapped FOXP3 Dimer on DNA and Its Function in Regulatory T Cells. Immunity 2011, 34, 479–491. [Google Scholar] [CrossRef]
- Chu, Y.P.; Chang, C.H.; Shiu, J.H.; Chang, Y.T.; Chen, C.Y.; Chuang, W.J. Solution Structure and Backbone Dynamics of the DNA-Binding Domain of FOXP1: Insight into Its Domain Swapping and DNA Binding. Protein Sci. 2011, 20, 908–924. [Google Scholar] [CrossRef]
- Medina, E.; Córdova, C.; Villalobos, P.; Reyes, J.; Komives, E.A.; Ramírez-Sarmiento, C.A.; Babul, J. Three-Dimensional Domain Swapping Changes the Folding Mechanism of the Forkhead Domain of FoxP1. Biophys. J. 2016, 110, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Perumal, K.; Dirr, H.W.; Fanucchi, S. A Single Amino Acid in the Hinge Loop Region of the FOXP Forkhead Domain Is Significant for Dimerisation. Protein J. 2015, 34, 111–121. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Zhang, Z.; Liu, C.-C.; Johnson, M.E.; Espinoza, C.A.; Edsall, L.E.; Ren, B.; Zhou, X.J.; Grant, S.F.A.; et al. DNA Binding by FOXP3 Domain-Swapped Dimer Suggests Mechanisms of Long-Range Chromosomal Interactions. Nucleic Acids Res. 2015, 43, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Human FOX Gene Family (Review). Int. J. Oncol. 2004, 25, 1495–1500. [Google Scholar] [CrossRef]
- Medina, E.; Villalobos, P.; Coñuecar, R.; Ramírez-Sarmiento, C.A.; Babul, J. The Protonation State of an Evolutionarily Conserved Histidine Modulates Domain Swapping Stability of FoxP1. Sci. Rep. 2019, 9, 5441. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, Y. Evidences for the Unfolding Mechanism of Three-Dimensional Domain Swapping. Protein Sci. 2013, 22, 280–286. [Google Scholar] [CrossRef]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human Cystatin C, an Amyloidogenic Protein, Dimerizes through Three-Dimensional Domain Swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef][Green Version]
- Hafner-Bratkovic, I.; Bester, R.; Pristovsek, P.; Gaedtke, L.; Veranic, P.; Gaspersic, J.; Mancek-Keber, M.; Avbelj, M.; Polymenidou, M.; Julius, C.; et al. Globular Domain of the Prion Protein Needs to Be Unlocked by Domain Swapping to Support Prion Protein Conversion. J. Biol. Chem. 2011, 286, 12149–12156. [Google Scholar] [CrossRef]
- Ren, C.; Nagao, S.; Yamanaka, M.; Komori, H.; Shomura, Y.; Higuchi, Y.; Hirota, S. Oligomerization Enhancement and Two Domain Swapping Mode Detection for Thermostable Cytochrome c 552 via the Elongation of the Major Hinge Loop. Mol. Biosyst. 2015, 11, 3218–3221. [Google Scholar] [CrossRef]
- Picone, D.; Di Fiore, A.; Ercole, C.; Franzese, M.; Sica, F.; Tomaselli, S.; Mazzarella, L. The Role of the Hinge Loop in Domain Swapping. The Special Case of Bovine Seminal Ribonuclease. J. Biol. Chem. 2005, 280, 13771–13778. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Prutzman, K.C.; Campbell, S.L.; Dokholyan, N.V. Topological Determinants of Protein Domain Swapping. Structure 2006, 14, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pino, A.; Martinez-Rodriguez, S.; Wahni, K.; Wyns, L.; Loris, R.; Messens, J. Coupling of Domain Swapping to Kinetic Stability in a Thioredoxin Mutant. J. Mol. Biol. 2009, 385, 1590–1599. [Google Scholar] [CrossRef]
- Yang, S.; Cho, S.S.; Levy, Y.; Cheung, M.S.; Levine, H.; Wolynes, P.G.; Onuchic, J.N. Domain Swapping Is a Consequence of Minimal Frustration. Proc. Natl. Acad. Sci. USA 2004, 101, 13786–13791. [Google Scholar] [CrossRef]
- Cho, S.S.; Levy, Y.; Onuchic, J.N.; Wolynes, P.G. Overcoming Residual Frustration in Domain-Swapping: The Roles of Disulfide Bonds in Dimerization and Aggregation. Phys. Biol. 2005, 2, S44–S55. [Google Scholar] [CrossRef]
- O’Neill, J.W.; Kim, D.E.; Johnsen, K.; Baker, D.; Zhang, K.Y.J. Single-Site Mutations Induce 3D Domain Swapping in the B1 Domain of Protein L from Peptostreptococcus Magnus. Structure 2001, 9, 1017–1027. [Google Scholar] [CrossRef]
- Bhatt, A.N.; Khan, M.Y.; Bhakuni, V. The C-Terminal Domain of Dimeric Serine Hydroxymethyltransferase Plays a Key Role in Stabilization of the Quaternary Structure and Cooperative Unfolding of Protein: Domain Swapping Studies with Enzymes Having High Sequence Identity. Protein Sci. 2004, 13, 2184–2195. [Google Scholar] [CrossRef]
- Yang, S.; Levine, H.; Onuchic, J.N.; Cox, D.L. Structure of Infectious Prions: Stabilization by Domain Swapping. FASEB J. 2005, 19, 1778–1782. [Google Scholar] [CrossRef][Green Version]
- Nagao, S.; Ueda, M.; Osuka, H.; Komori, H.; Kamikubo, H.; Kataoka, M.; Higuchi, Y.; Hirota, S. Domain-Swapped Dimer of Pseudomonas Aeruginosa Cytochrome c551: Structural Insights into Domain Swapping of Cytochrome c Family Proteins. PLoS ONE 2015, 10, e0123653. [Google Scholar]
- Balasubramaniam, D.; Komives, E.A. Hydrogen-Exchange Mass Spectrometry for the Study of Intrinsic Disorder in Proteins. Biochim. Biophys. Acta 2013, 1834, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Sarmiento, C.A.; Komives, E.A. Hydrogen-Deuterium Exchange Mass Spectrometry Reveals Folding and Allostery in Protein-Protein Interactions. Methods 2018, 144, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.; Latham, D.R.; Sanabria, H. Unraveling Protein’s Structural Dynamics: From Configurational Dynamics to Ensemble Switching Guides Functional Mesoscale Assemblies. Curr. Opin. Struct. Biol. 2021, 66, 129–138. [Google Scholar] [CrossRef]
- Medina, E.; Villalobos, P.; Hamilton, G.L.; Komives, E.A.; Sanabria, H.; Ramírez-Sarmiento, C.A.; Babul, J. Intrinsically Disordered Regions of the DNA-Binding Domain of Human FoxP1 Facilitate Domain Swapping. J. Mol. Biol. 2020, 432, 5411–5429. [Google Scholar] [CrossRef] [PubMed]
- Tsytlonok, M.; Sanabria, H.; Wang, Y.; Felekyan, S.; Hemmen, K.; Phillips, A.H.; Yun, M.K.; Waddell, M.B.; Park, C.G.; Vaithiyalingam, S.; et al. Dynamic Anticipation by Cdk2/Cyclin A-Bound p27 Mediates Signal Integration in Cell Cycle Regulation. Nat. Commun. 2019, 10, 1676. [Google Scholar] [CrossRef]
- Dimura, M.; Peulen, T.O.; Hanke, C.A.; Prakash, A.; Gohlke, H.; Seidel, C.A. Quantitative FRET Studies and Integrative Modeling Unravel the Structure and Dynamics of Biomolecular Systems. Curr. Opin. Struct. Biol. 2016, 40, 163–185. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Resing, K.A.; Ahn, N.G. Protein Analysis by Hydrogen Exchange Mass Spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 1–25. [Google Scholar] [CrossRef]
- Markwick, P.R.L.; Peacock, R.B.; Komives, E.A. Accurate Prediction of Amide Exchange in the Fast Limit Reveals Thrombin Allostery. Biophys. J. 2019, 116, 49–56. [Google Scholar] [CrossRef]
- Noel, J.K.; Onuchic, J.N. The Many Faces of Structure-Based Potentials: From Protein Folding Landscapes to Structural Characterization of Complex Biomolecules. In Computational Modeling of Biological Systems; Springer: Boston, MA, USA, 2012; pp. 31–54. [Google Scholar]
- Sin, C.; Li, H.; Crawford, D.A. Transcriptional Regulation by FOXP1, FOXP2, and FOXP4 Dimerization. J. Mol. Neurosci. 2014, 55, 437–448. [Google Scholar] [CrossRef]
- Li, S.; Weidenfeld, J.; Morrisey, E.E. Transcriptional and DNA Binding Activity of the Foxp1/2/4 Family Is Modulated by Heterotypic and Homotypic Protein Interactions. Mol. Cell. Biol. 2004, 24, 809–822. [Google Scholar] [CrossRef]
- Mendoza, E.; Scharff, C.; Rácz, I.; Jarvis, E.D.; Wu, Y. Protein-Protein Interaction Among the FoxP Family Members and Their Regulation of Two Target Genes, VLDLR and CNTNAP2 in the Zebra Finch Song System. Front. Mol. Neurosci. 2017, 10, 112. [Google Scholar] [CrossRef]
- Mascarenhas, N.M.; Gosavi, S. Protein Domain-Swapping Can Be a Consequence of Functional Residues. J. Phys. Chem. B 2016, 120, 6929–6938. [Google Scholar] [CrossRef]
- Shingate, P.; Sowdhamini, R. Analysis of Domain-Swapped Oligomers Reveals Local Sequence Preferences and Structural Imprints at the Linker Regions and Swapped Interfaces. PLoS ONE 2012, 7, e39305. [Google Scholar] [CrossRef]
- Wright, C.F.; Teichmann, S.A.; Clarke, J.; Dobson, C.M. The Importance of Sequence Diversity in the Aggregation and Evolution of Proteins. Nature 2005, 438, 878–881. [Google Scholar] [CrossRef]
- Borgia, M.B.; Borgia, A.; Best, R.B.; Steward, A.; Nettels, D.; Wunderlich, B.; Schuler, B.; Clarke, J. Single-Molecule Fluorescence Reveals Sequence-Specific Misfolding in Multidomain Proteins. Nature 2011, 474, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Schafer, N.P.; Wolynes, P.G. Frustration in the Energy Landscapes of Multidomain Protein Misfolding. Proc. Natl. Acad. Sci. USA 2013, 110, 1680–1685. [Google Scholar] [CrossRef]
- Wang, B.; Lin, D.; Li, C.; Tucker, P. Multiple Domains Define the Expression and Regulatory Properties of Foxp1 Forkhead Transcriptional Repressors. J. Biol. Chem. 2003, 278, 24259–24268. [Google Scholar] [CrossRef]
- Struhl, K. Helix-Turn-Helix, Zinc-Finger, and Leucine-Zipper Motifs for Eukaryotic Transcriptional Regulatory Proteins. Trends Biochem. Sci. 1989, 14, 137–140. [Google Scholar] [CrossRef]
- Gamsjaeger, R.; Liew, C.K.; Loughlin, F.E.; Crossley, M.; Mackay, J.P. Sticky Fingers: Zinc-Fingers as Protein-Recognition Motifs. Trends Biochem. Sci. 2007, 32, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Cranz, S.; Berger, C.; Baici, A.; Jelesarov, I.; Bosshard, H.R. Monomeric and Dimeric bZIP Transcription Factor GCN4 Bind at the Same Rate to Their Target DNA Site. Biochemistry 2004, 43, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Onate-Sanchez, L.; Weltmeier, F.; Ehlert, A.; Diaz, I.; Dietrich, K.; Vicente-Carbajosa, J.; Droge-Laser, W. A Pivotal Role of the Basic Leucine Zipper Transcription Factor bZIP53 in the Regulation of Arabidopsis Seed Maturation Gene Expression Based on Heterodimerization and Protein Complex Formation. Plant Cell Online 2009, 21, 1747–1761. [Google Scholar] [CrossRef] [PubMed]
- Thulo, M.; Rabie, M.A.; Pahad, N.; Donald, H.L.; Blane, A.A.; Perumal, C.M.; Penedo, J.C.; Fanucchi, S. The Influence of Various Regions of the FOXP2 Sequence on Its Structure and DNA-Binding Function. Biosci. Rep. 2021, 41, BSR20202128. [Google Scholar] [CrossRef] [PubMed]
- Häußermann, K.; Young, G.; Kukura, P.; Dietz, H. Dissecting FOXP2 Oligomerization and DNA Binding. Angew. Chem. Int. Ed Engl. 2019, 58, 7662–7667. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalobos, P.; Ramírez-Sarmiento, C.A.; Babul, J.; Medina, E. Human FoxP Transcription Factors as Tractable Models of the Evolution and Functional Outcomes of Three-Dimensional Domain Swapping. Int. J. Mol. Sci. 2021, 22, 10296. https://doi.org/10.3390/ijms221910296
Villalobos P, Ramírez-Sarmiento CA, Babul J, Medina E. Human FoxP Transcription Factors as Tractable Models of the Evolution and Functional Outcomes of Three-Dimensional Domain Swapping. International Journal of Molecular Sciences. 2021; 22(19):10296. https://doi.org/10.3390/ijms221910296
Chicago/Turabian StyleVillalobos, Pablo, César A. Ramírez-Sarmiento, Jorge Babul, and Exequiel Medina. 2021. "Human FoxP Transcription Factors as Tractable Models of the Evolution and Functional Outcomes of Three-Dimensional Domain Swapping" International Journal of Molecular Sciences 22, no. 19: 10296. https://doi.org/10.3390/ijms221910296
APA StyleVillalobos, P., Ramírez-Sarmiento, C. A., Babul, J., & Medina, E. (2021). Human FoxP Transcription Factors as Tractable Models of the Evolution and Functional Outcomes of Three-Dimensional Domain Swapping. International Journal of Molecular Sciences, 22(19), 10296. https://doi.org/10.3390/ijms221910296