
Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling through Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1)-Dependent EGFR Degradation

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
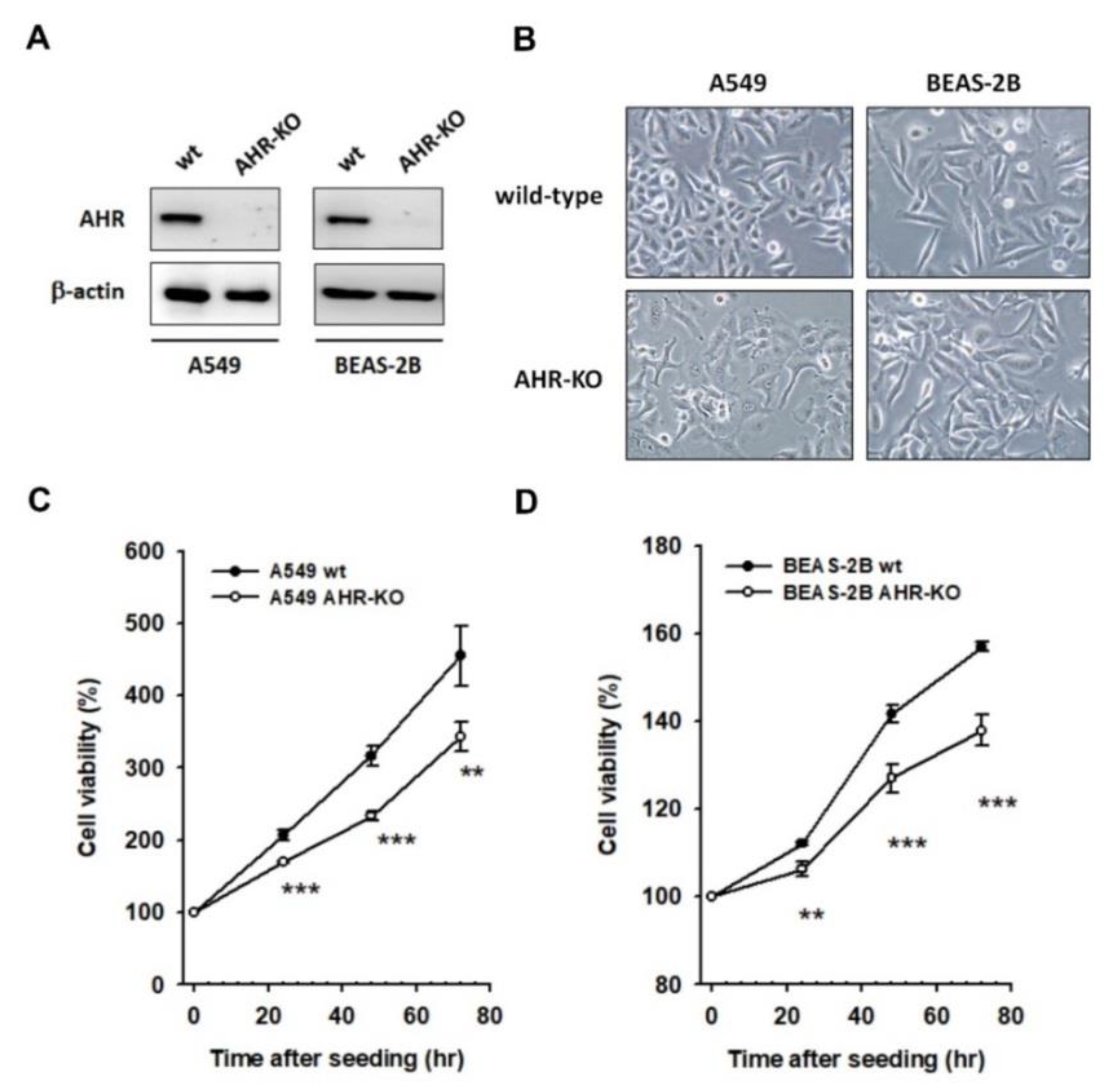
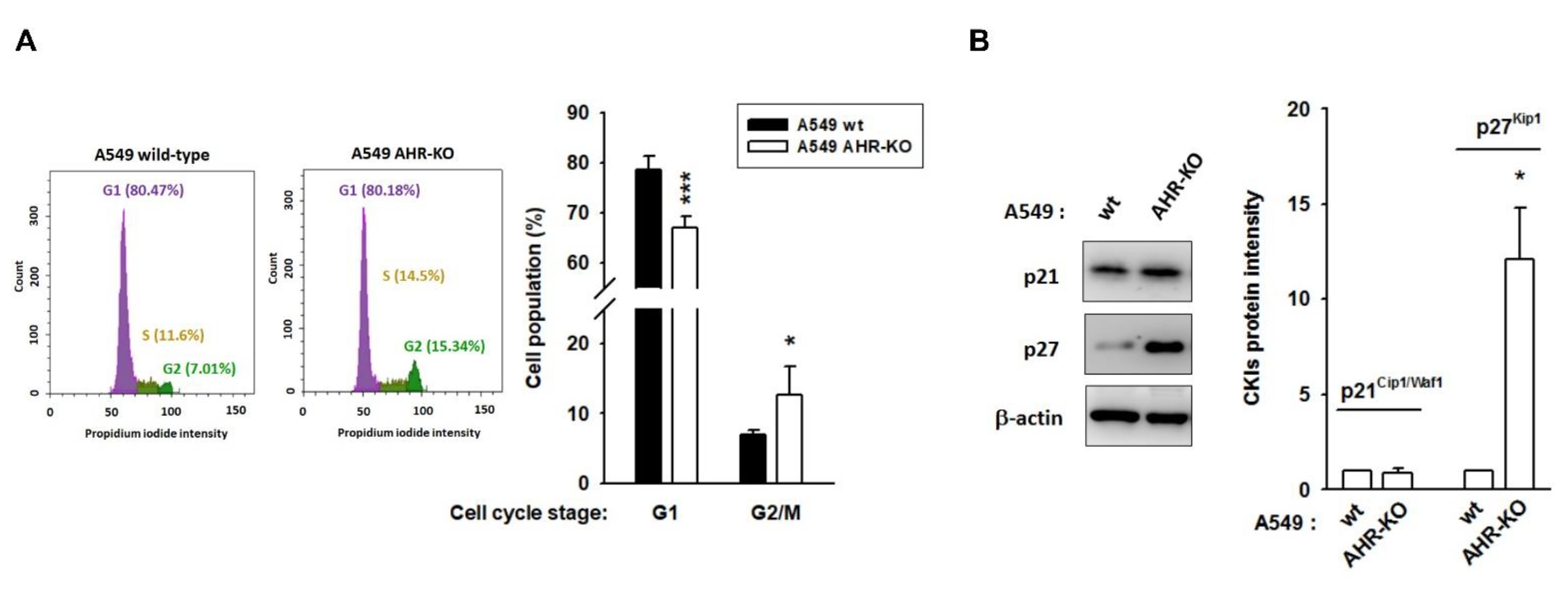
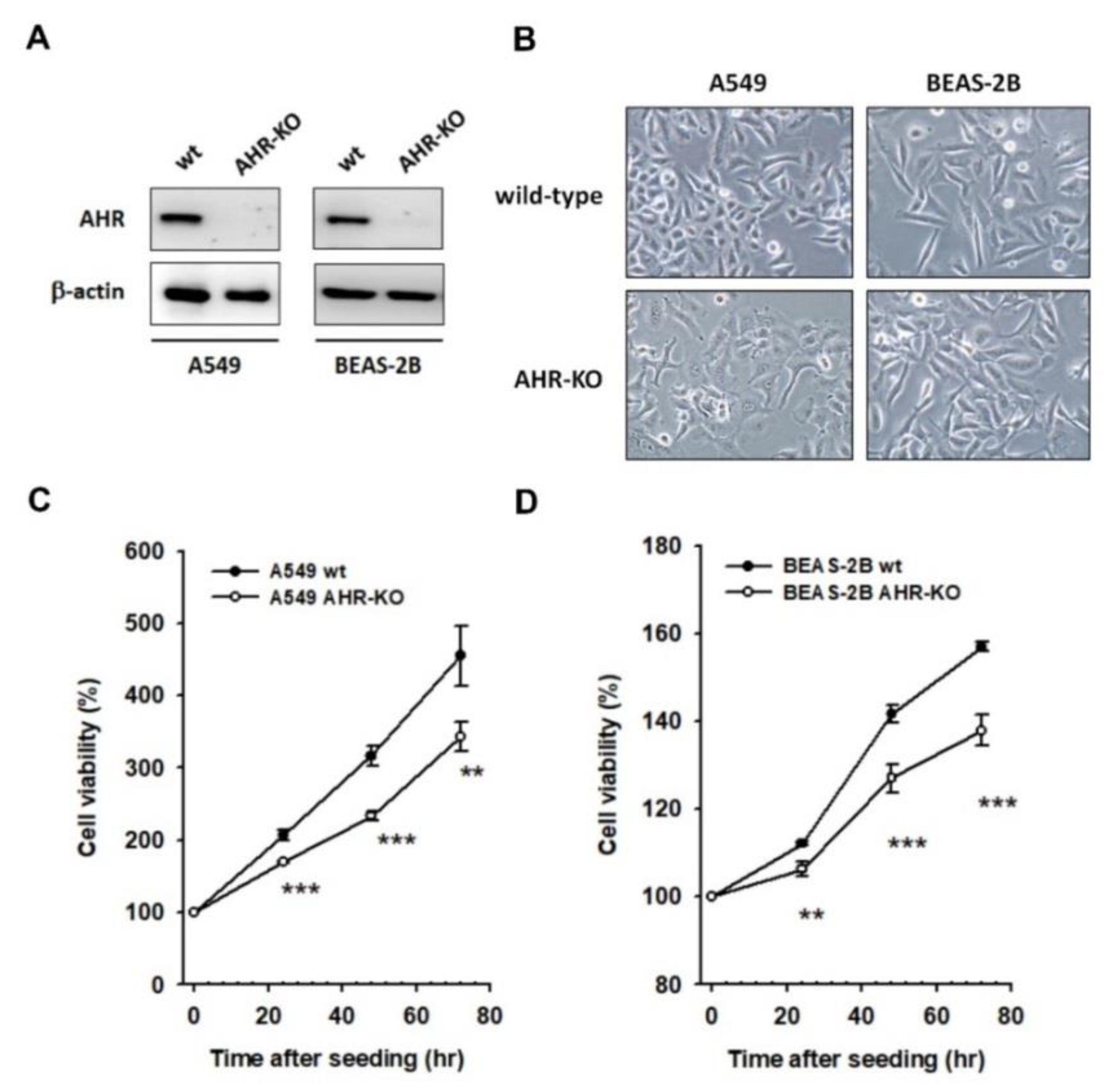
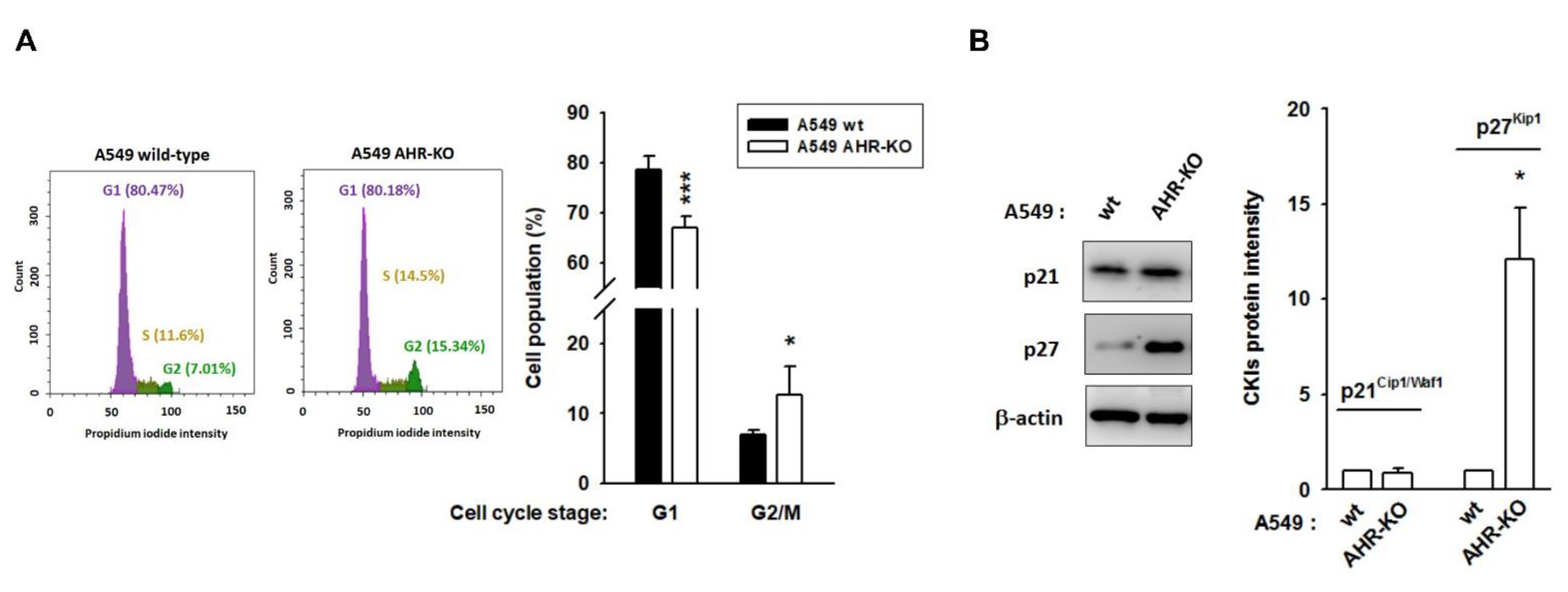
2.1. AHR-KO Clones Frequently Prolonged Cell Doubling-Time, Involving a Multitude of Changes of Cell Cycle Regulators
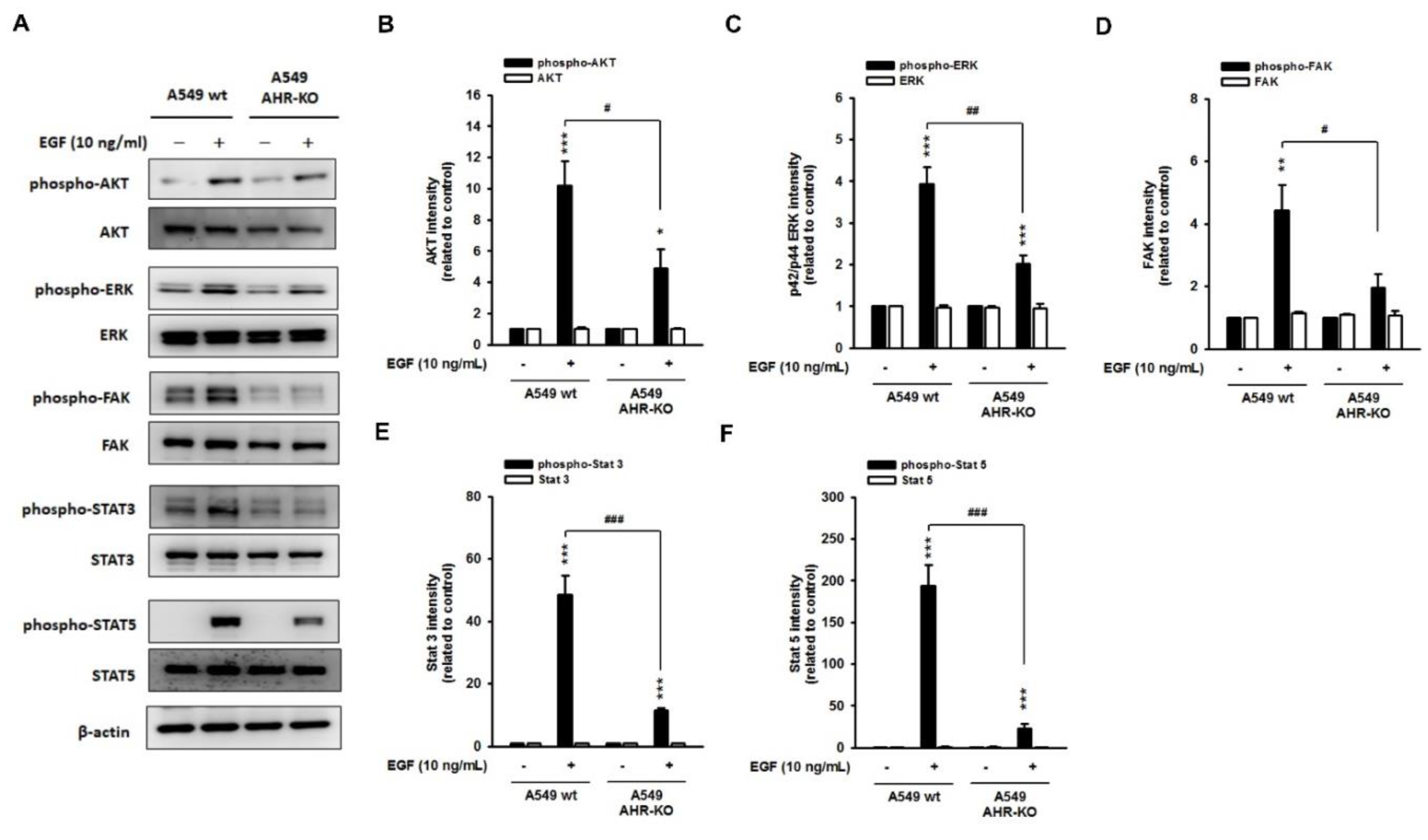
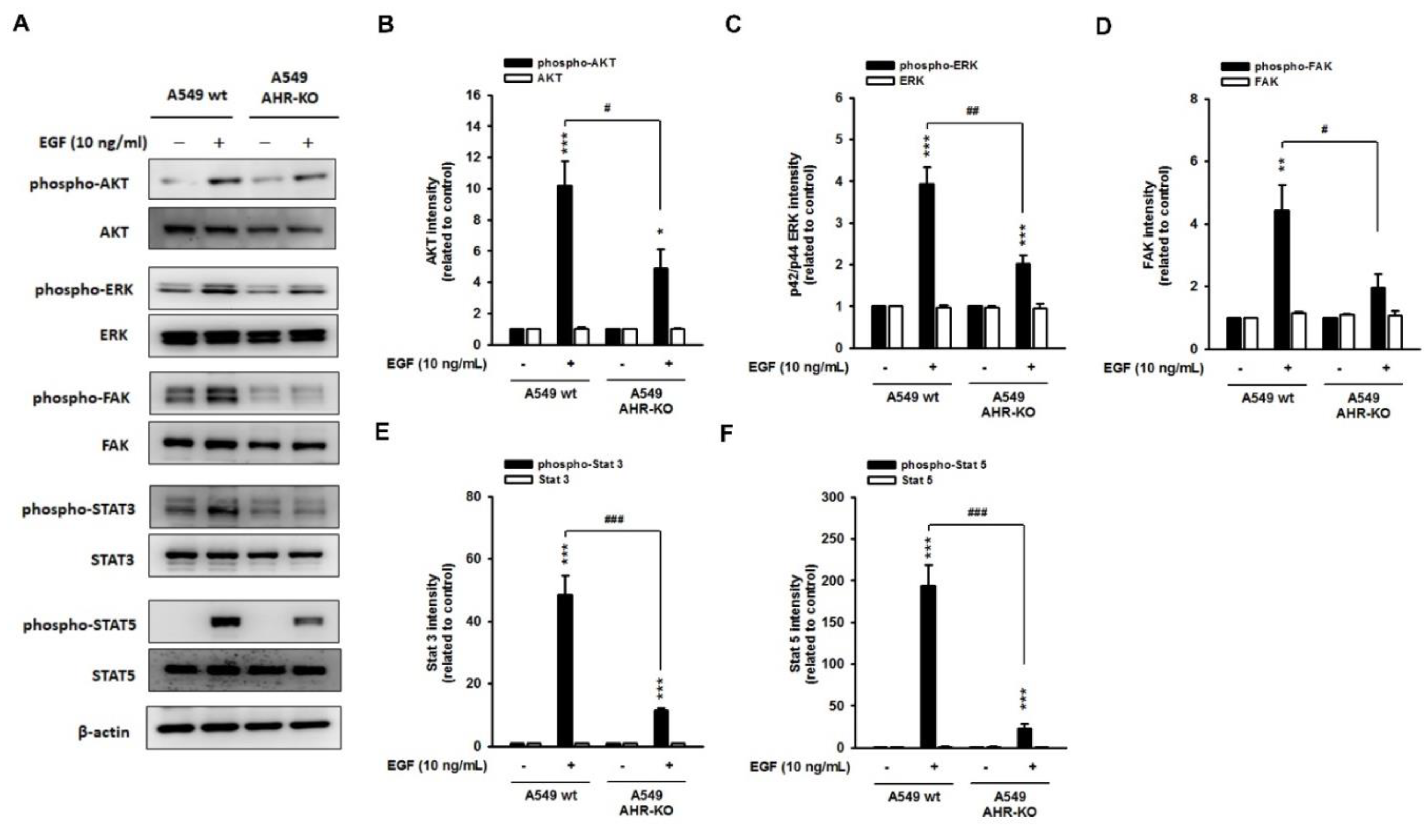
2.2. The Mitogen-Mediated Signaling Was Impaired in AHR-KO Clones
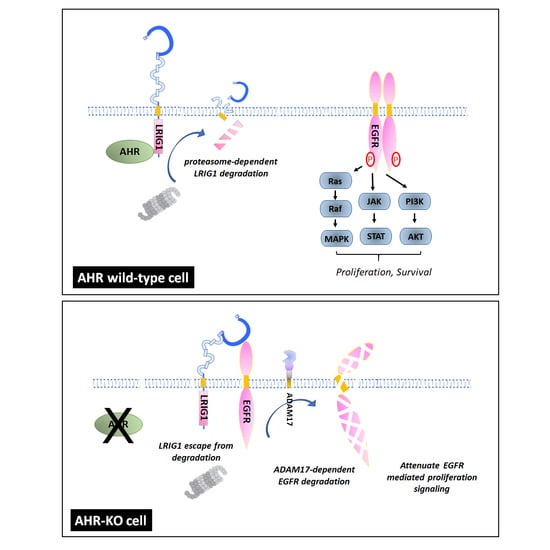
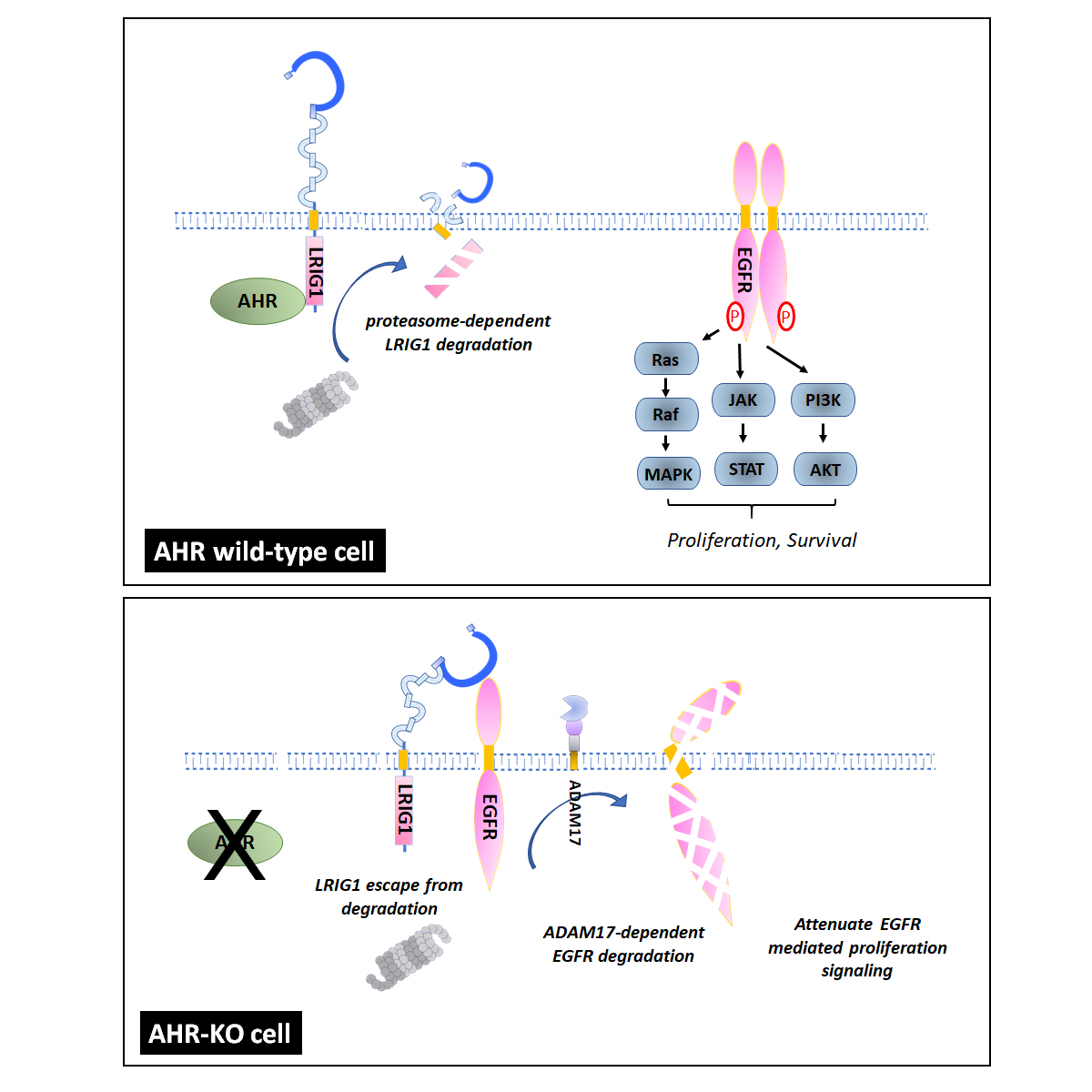
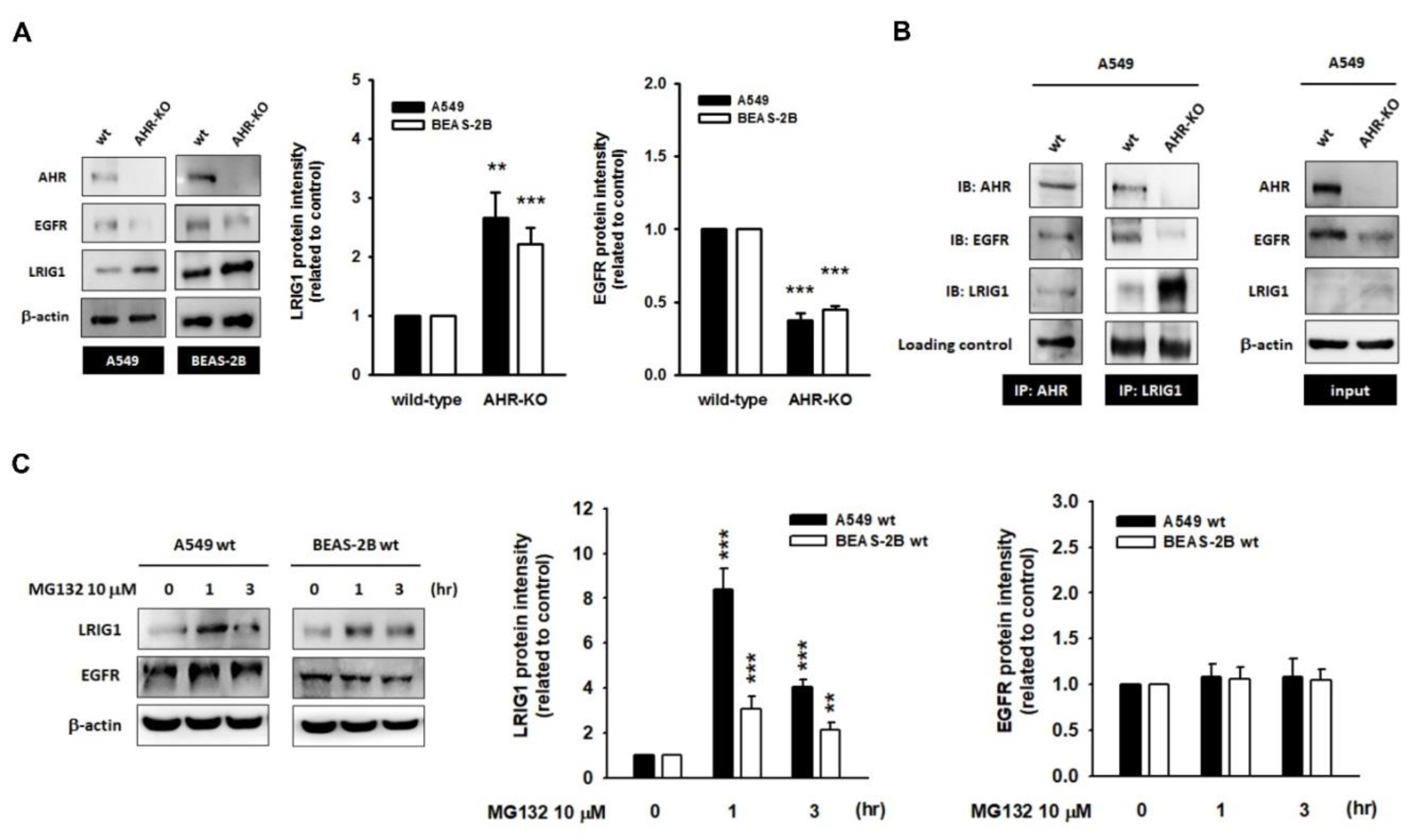
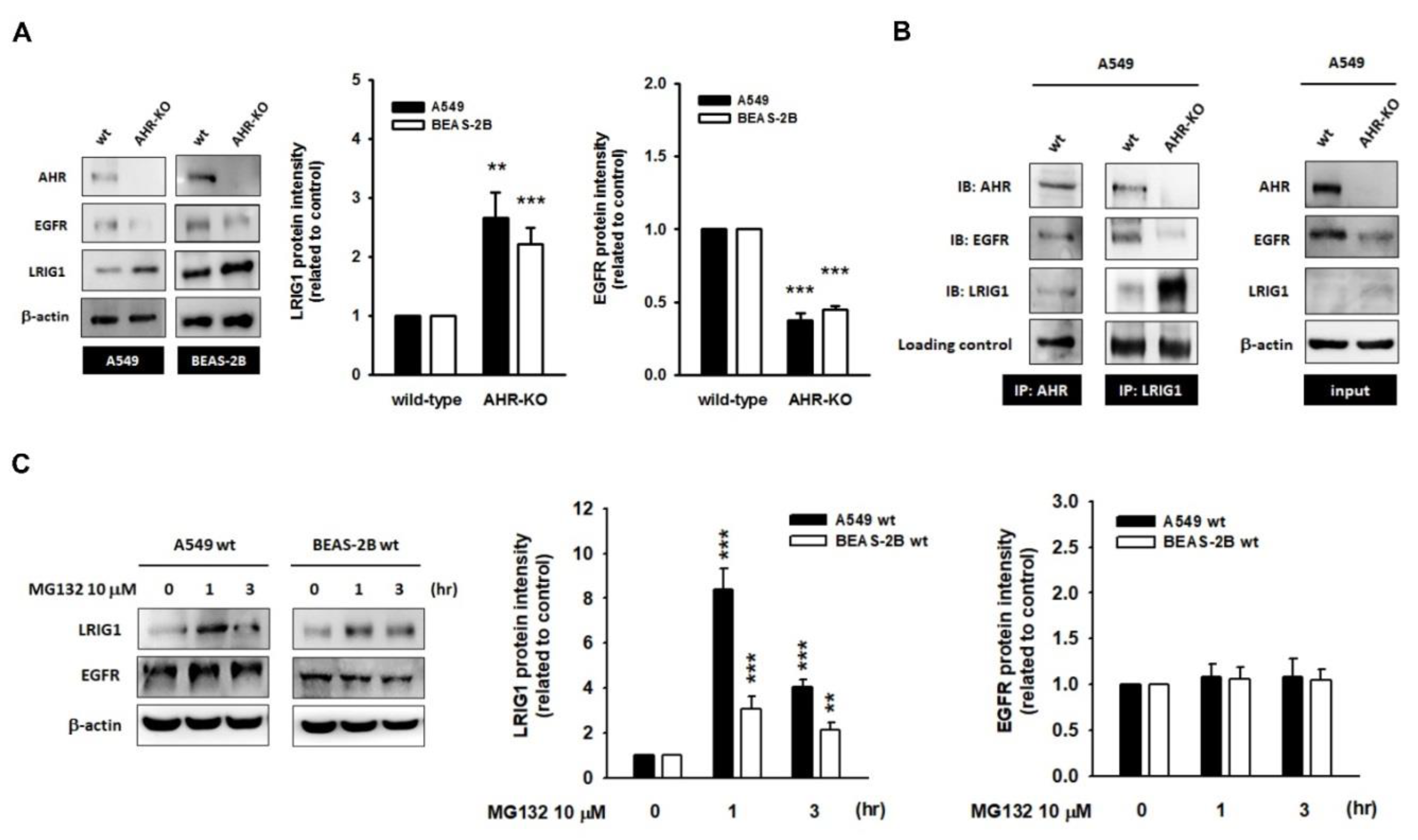
2.3. The Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1), an EGFR Repressor, Was Up-Regulated in AHR-KO Clones
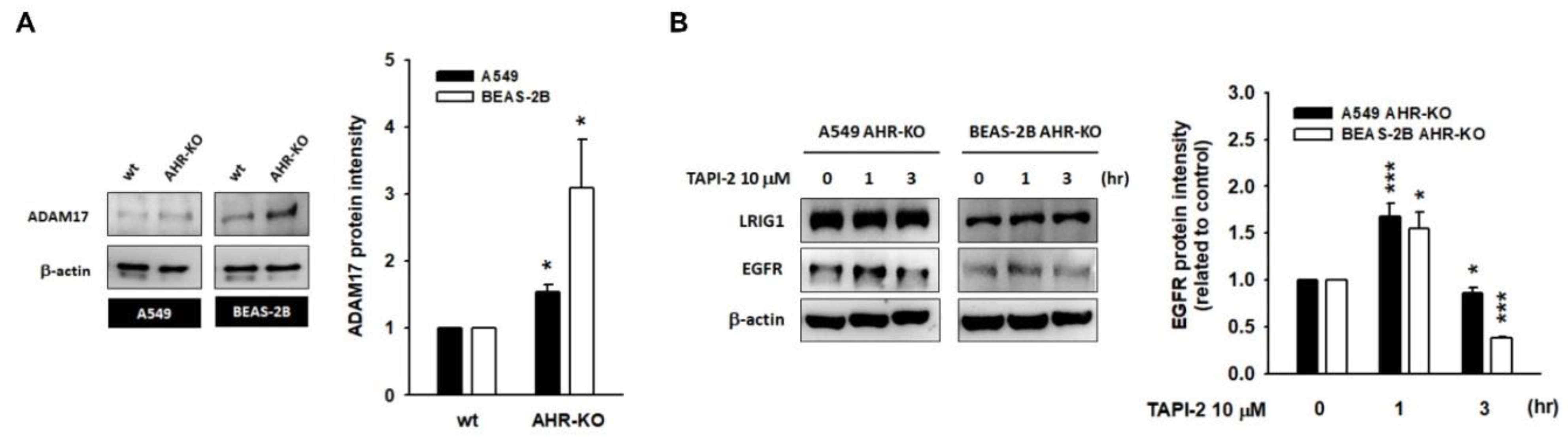
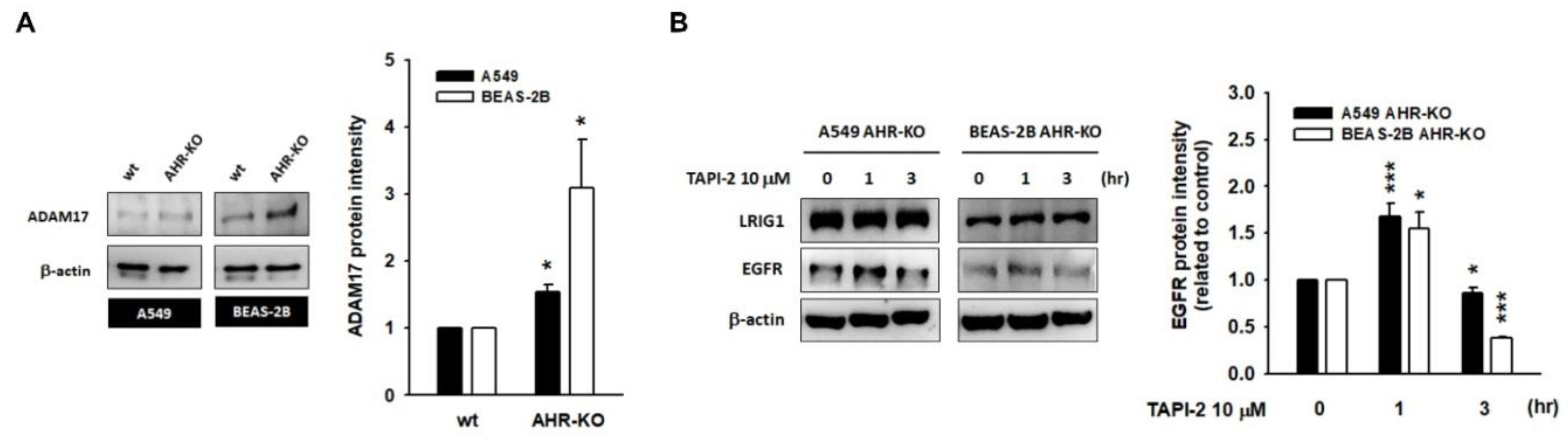
2.4. A Disintegrin and Metalloprotease 17 (ADAM17) Activity Is Required for LRIG1-Mediated EGFR Degradation
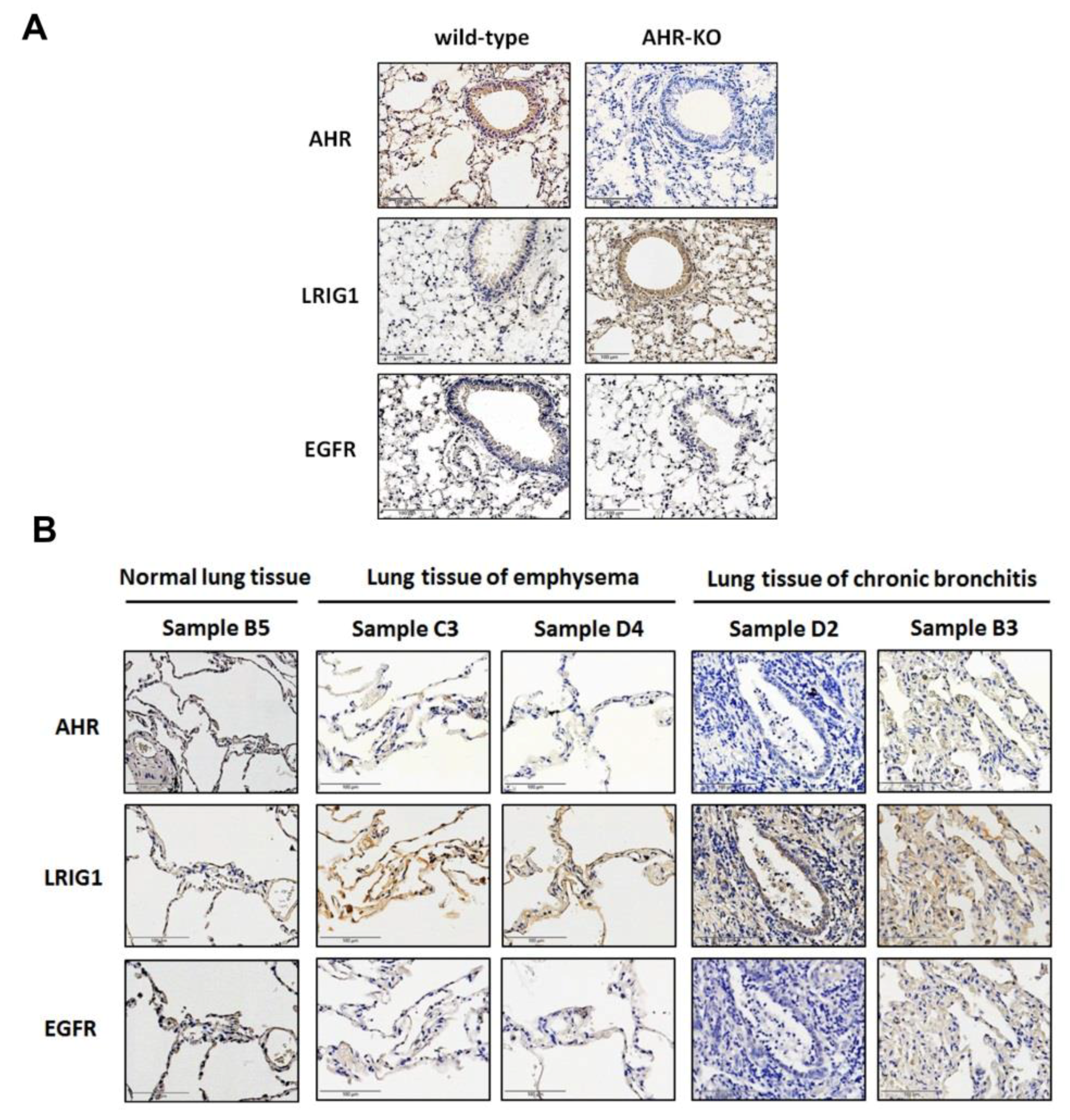
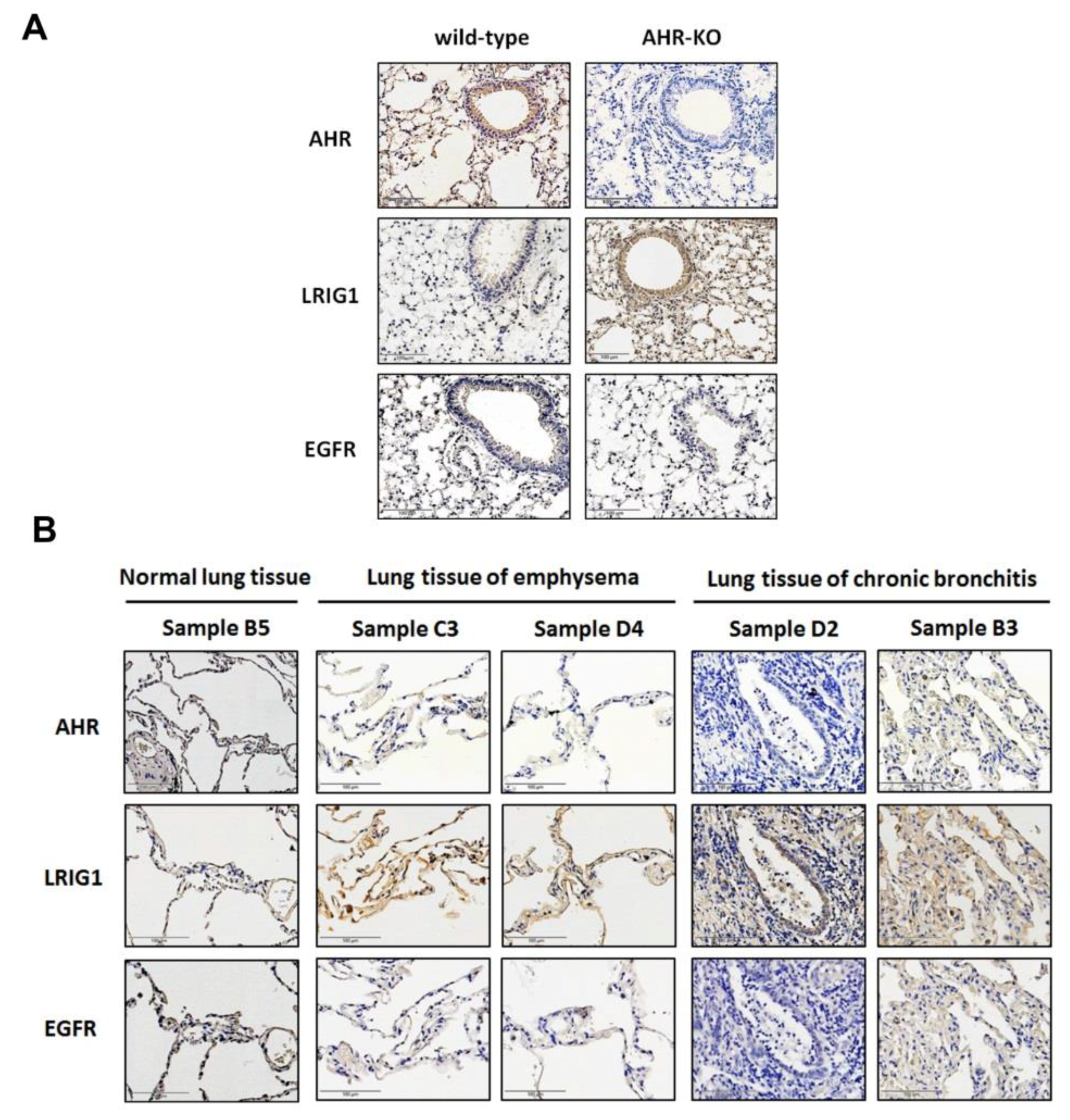
2.5. The Expression of AHR and LRIG1 Was Correlated Inversely In Vivo; Furthermore, an Enhanced LRIG1 Expression Was Found in Lung Tissues of COPD Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids, Transfection and Generating AHR Knockout Clones with CRISPR/Cas9 System
4.3. Cell Proliferation Curve Determination (MTT Assay)
4.4. Flow Cytometric Analysis of Cell Cycle
4.5. Protein Extraction and Western Blotting
4.6. Immunoprecipitation
4.7. Immunohistochemistry
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| MAPK | Mitogen-activated protein kinase |
| Akt | Protein kinase B |
| STAT | Signal transducer and activator of transcription |
| AHR | Aryl hydrocarbon receptor |
| LRIG1 | Leucine-rich repeats and immunoglobulin-like domains 1 |
| COPD | Chronic obstructive pulmonary disease |
| KO | Knock-out |
| ADAM17 | A disintegrin and metalloprotease 17 |
| CDK | Cyclin-dependent kinase |
References
- Lee, Y.; Ma, J.; Lyu, H.; Huang, J.; Kim, A.; Liu, B. Role of erbB3 receptors in cancer therapeutic resistance. Acta Biochim. Biophys. Sin. 2014, 46, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, S.; Langhans, S.A. Epidermal growth factor signaling in transformed cells. Int. Rev. Cell. Mol. Biol. 2015, 314, 1–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.L.; Gupta, R.D.; Goldstein, D.; Crowe, P.J. Significance of phosphorylated epidermal growth factor receptor and its signal transducers in human soft tissue sarcoma. Int. J. Mol. Sci. 2017, 18, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Poulain-Godefroy, O.; Bouté, M.; Carrard, J.; Alvarez-Simon, D.; Tsicopoulos, A.; de Nadai, P. The aryl hydrocarbon receptor in asthma: Friend or foe? Int. J. Mol. Sci. 2020, 21, 8797. [Google Scholar] [CrossRef] [PubMed]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The aryl hydrocarbon receptor and the maintenance of lung health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, J.; Liu, J.; Li, M.; Xie, J.; Lv, Q.; Deng, W.; Zhou, N.; Zhou, Y.; Song, J.; et al. Mucus production stimulated by IFN-AhR signaling triggers hypoxia of COVID-19. Cell. Res. 2020, 30, 1078–1087. [Google Scholar] [CrossRef]
- Weng, C.M.; Wang, C.H.; Lee, M.J.; He, J.R.; Huang, H.Y.; Chao, M.W.; Chung, K.F.; Kuo, H.P. Aryl hydrocarbon receptor activation by diesel exhaust particles mediates epithelium-derived cytokines expression in severe allergic asthma. Allergy 2018, 73, 2192–2204. [Google Scholar] [CrossRef]
- Tripathi, P.; Deng, F.; Scruggs, A.M.; Chen, Y.; Huang, S.K. Variation in doses and duration of particulate matter exposure in bronchial epithelial cells results in upregulation of different genes associated with airway disorders. Toxicol. Vitr. 2018, 51, 95–105. [Google Scholar] [CrossRef]
- Wong, P.S.; Vogel, C.F.; Kokosinski, K.; Matsumura, F. Arylhydrocarbon receptor activation in NCI-H441 cells and C57BL/6 mice: Possible mechanisms for lung dysfunction. Am. J. Respir. Cell Mol. Biol. 2010, 42, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.H.; Lin, H.T.; Suen, J.L.; Sheu, C.C.; Yokoyama, K.K.; Huang, S.K.; Cheng, C.M. Aryl hydrocarbon receptor-ligand axis mediates pulmonary fibroblast migration and differentiation through increased arachidonic acid metabolism. Toxicology 2016, 370, 116–126. [Google Scholar] [CrossRef]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-κB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Araújo, E.F.; Preite, N.W.; Veldhoen, M.; Loures, F.V.; Garcia Calich, V.L. Pulmonary paracoccidioidomycosis in AhR deficient hosts is severe and associated with defective Treg and Th22 responses. Sci. Rep. 2020, 10, 11312. [Google Scholar] [CrossRef]
- Michaudel, C.; Bataille, F.; Maillet, I.; Fauconnier, L.; Colas, C.; Sokol, H.; Straube, M.; Couturier-Maillard, A.; Dumoutier, L.; van Snick, J.; et al. Ozone-induced aryl hydrocarbon receptor activation controls lung inflammation via interleukin-22 modulation. Front Immunol. 2020, 11, 144. [Google Scholar] [CrossRef]
- Harb, H.; Alhamwe, B.A.; Garn, H.; Renz, H.; Potaczek, D.P. Recent developments in epigenetics of pediatric asthma. Curr. Opin. Pediatr. 2016, 28, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Harb, H.; Michel, S.; Alhamwe, B.A.; Renz, H.; Tost, J. Epigenetics and allergy: From basic mechanisms to clinical applications. Epigenomics 2017, 9, 539–571. [Google Scholar] [CrossRef]
- Burleson, J.D.; Siniard, D.; Yadagiri, V.K.; Chen, X.; Weirauch, M.T.; Ruff, B.P.; Brandt, E.B.; Hershey, G.K.K.; Ji, H. TET1 contributes to allergic airway inflammation and regulates interferon and aryl hydrocarbon receptor signaling pathways in bronchial epithelial cells. Sci. Rep. 2019, 9, 7361. [Google Scholar] [CrossRef] [PubMed]
- Pollenz, R.S.; Buggy, C. Ligand-dependent and -independent degradation of the human aryl hydrocarbon receptor (hAHR) in cell culture models. Chem. Biol. Interact. 2006, 164, 49–59. [Google Scholar] [CrossRef]
- Muku, G.E.; Lahoti, T.S.; Murray, I.A.; Podolsky, M.A.; Smith, K.J.; Hubbard, T.D.; Kuzu, G.; Gowda, K.; Amin, S.G.; Perdew, G.H. Ligand-mediated cytoplasmic retention of the Ah receptor inhibits macrophage-mediated acute inflammatory responses. Lab Investig. 2017, 97, 1471–1487. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kato, S. AHR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem. Pharmacol. 2009, 77, 474–484. [Google Scholar] [CrossRef]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A molecular mechanism to switch the aryl hydrocarbon receptor from a transcription factor to an E3 ubiquitin ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef] [Green Version]
- Tomkiewicz, C.; Herry, L.; Bui, L.C.; Métayer, C.; Bourdeloux, M.; Barouki, R.; Coumoul, X. The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway. Oncogene 2013, 32, 1811–1820. [Google Scholar] [CrossRef] [Green Version]
- Li, C.H.; Liu, C.W.; Tsai, C.H.; Peng, Y.J.; Yang, Y.H.; Liao, P.L.; Lee, C.C.; Cheng, Y.W.; Kang, J.J. Cytoplasmic AHR regulates glycogen synthase kinase 3 beta, accelerates vimentin degradation, and suppresses epithelial-mesenchymal transition in non-small cell lung cancer cells. Arch. Toxicol. 2017, 91, 2165–2178. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.H.; Li, C.H.; Cheng, Y.W.; Lee, C.C.; Liao, P.L.; Lin, C.H.; Huang, S.H.; Kang, J.J. The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 2017, 7, 41927. [Google Scholar] [CrossRef] [PubMed]
- Martey, C.A.; Baglole, C.J.; Gasiewicz, T.A.; Sime, P.J.; Phipps, R.P. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2005, 289, L391–L399. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Ferrer, I.; Cuartero, M.I.; Medina, V.; Ahedo-Quero, D.; Peña-Martínez, C.; Pérez-Ruíz, A.; Fernández-Valle, M.E.; Hernández-Sánchez, C.; Fernández-Salguero, P.M.; Lizasoain, I.; et al. Lack of the aryl hydrocarbon receptor accelerates aging in mice. FASEB J. 2019, 33, 12644–12654. [Google Scholar] [CrossRef] [Green Version]
- Zago, M.; Sheridan, J.A.; Traboulsi, H.; Hecht, E.; Zhang, Y.; Guerrina, N.; Matthews, J.; Nair, P.; Eidelman, D.H.; Hamid, Q.; et al. Low levels of the AhR in chronic obstructive pulmonary disease (COPD)-derived lung cells increases COX-2 protein by altering mRNA stability. PLoS ONE 2017, 12, e0180881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrina, N.; Traboulsi, H.; de Souza, A.R.; Bosse, Y.; Thatcher, T.H.; Robichaud, A.; Ding, J.; Li, P.Z.; Simon, L.; Pareek, S.; et al. Aryl hydrocarbon receptor deficiency causes the development of chronic obstructive pulmonary disease through the integration of multiple pathogenic mechanisms. FASEB J. 2021, 35, e21376. [Google Scholar] [CrossRef] [PubMed]
- Perotin, J.; Adam, D.; Vella-Boucaud, J.; Delepine, G.; Sandu, S.; Jonvel, A.; Prevost, A.; Berthiot, G.; Pison, C.; Lebargy, F.; et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir. Res. 2014, 15, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Hernández, A.; Nacarino-Palma, A.; Moreno-Marín, N.; Barrasa, E.; Paniagua-Quiñones, B.; Catalina-Fernández, I.; Alvarez-Barrientos, A.; Bustelo, X.R.; Merino, J.M.; Fernández-Salguero, P.M. Lung regeneration after toxic injury is improved in absence of dioxin receptor. Stem Cell Res. 2017, 25, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Fuentes, S.; Neganova, I.; Wadkin, L.E.; Baggaley, A.W.; Barrio, R.A.; Lako, M.; Shukurov, A.; Parker, N.G. Quantification of the morphological characteristics of hESC colonies. Sci. Rep. 2019, 9, 17569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliopanos, A.; Kleeff, J.; Xiao, Y.; Safe, S.; Zimmermann, A.; Büchler, M.W.; Friess, H. Increased aryl hydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene 2002, 21, 6059–6070. [Google Scholar] [CrossRef] [Green Version]
- Barhoover, M.A.; Hall, J.M.; Greenlee, W.F.; Thomas, R.S. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with CDK4. Mol. Pharmacol. 2010, 77, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahim, M.; Smith, R.; Safe, S. Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol. Pharmacol. 2003, 63, 1373–1381. [Google Scholar] [CrossRef]
- Kalmes, M.; Hennen, J.; Clemens, J.; Blömeke, B. Impact of AHR knockdown on cell cycle progression in human HaCaT keratinocytes. Biol. Chem. 2011, 392, 643–651. [Google Scholar] [CrossRef]
- Elizondo, G.; Fernandez-Salguero, P.; Sheikh, M.S.; Kim, G.Y.; Fornace, A.J.; Lee, K.S.; Gonzalez, F.J. Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 2000, 57, 1056–1063. [Google Scholar] [PubMed]
- Tohkin, M.; Fukuhara, M.; Elizondo, G.; Tomita, S.; Gonzalez, F.J. Aryl hydrocarbon receptor is required for p300-mediated induction of DNA synthesis by adenovirus E1A. Mol. Pharmacol. 2000, 58, 845–851. [Google Scholar] [CrossRef]
- Hecht, E.; Zago, M.; Sarill, M.; de Souza, A.R.; Gomez, A.; Matthews, J.; Hamid, O.; Eidelman, D.H.; Baglole, C.J. Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol. Appl. Pharmacol. 2014, 280, 511–525. [Google Scholar] [CrossRef]
- Yin, J.; Sheng, B.; Pu, A.; Han, B.; Yang, K.; Wang, Q.; Sun, L.; Yang, H. Keratinocyte growth factor regulation of aryl hydrocarbon receptor activation in colorectal cancer cells. Dig. Dis. Sci. 2016, 61, 444–452. [Google Scholar] [CrossRef]
- Tomblin, J.K.; Salisbury, T.B. Insulin like growth factor 2 regulation of aryl hydrocarbon receptor in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 443, 1092–1096. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, C.; Schneider, A.; Sherr, D.H.; Faller, D.V. Expression of the aryl hydrocarbon receptor is regulated by serum and mitogenic growth factors in murine 3T3 fibroblasts. J. Biol. Chem. 1996, 271, 25921–25927. [Google Scholar] [CrossRef] [Green Version]
- Stutz, M.A.; Shattuck, D.L.; Laederich, M.B.; Carraway, K.L., 3rd; Sweeney, C. LRIG1 negatively regulates the oncogenic EGF receptor mutant EGFRvIII. Oncogene 2008, 27, 5741–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledda, F.; Bieraugel, O.; Fard, S.S.; Vilar, M.; Paratcha, G. Lrig1 is an endogenous inhibitor of Ret receptor tyrosine kinase activation, downstream signaling, and biological responses to GDNF. J. Neurosci. 2008, 28, 39–49. [Google Scholar] [CrossRef]
- Lee, J.M.; Kim, B.; Lee, S.B.; Jeong, Y.; Oh, Y.M.; Song, Y.J.; Jung, S.; Choi, J.; Lee, S.; Cheong, K.H.; et al. Cbl-independent degradation of Met: Ways to avoid agonism of bivalent Met-targeting antibody. Oncogene 2014, 33, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Alsina, F.C.; Hita, F.J.; Fontanet, P.A.; Irala, D.; Hedman, H.; Ledda, F.; Paratcha, G. Lrig1 is a cell-intrinsic modulator of hippocampal dendrite complexity and BDNF signaling. EMBO Rep. 2016, 17, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Faraz, M.; Herdenberg, C.; Holmlund, C.; Henriksson, R.; Hedman, H. A protein interaction network centered on leucine-rich repeats and immunoglobulin-like domains 1 (LRIG1) regulates growth factor receptors. J. Biol. Chem. 2018, 293, 3421–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torigoe, H.; Yamamoto, H.; Sakaguchi, M.; Youyi, C.; Namba, K.; Sato, H.; Shien, K.; Soh, J.; Suzawa, K.; Tomida, S.; et al. Tumor-suppressive effect of LRIG1, a negative regulator of ErbB, in non-small cell lung cancer harboring mutant EGFR. Carcinogenesis 2018, 39, 719–727. [Google Scholar] [CrossRef]
- Kou, C.; Zhou, T.; Han, X.; Zhuang, H.; Qian, H. LRIG1, a 3p tumor suppressor, represses EGFR signaling and is a novel epigenetic silenced gene in colorectal cancer. Biochem. Biophys. Res. Commun. 2015, 464, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yu, Y.; Miao, L. Long non-coding RNA LINC00152 regulates cell proliferation and migration by epigenetically repressing LRIG1 expression in cholangiocarcinoma. Gastroenterology 2019, 156, S-770. [Google Scholar] [CrossRef]
- Rico de Souza, A.; Zago, M.; Pollock, S.J.; Sime, P.J.; Phipps, R.P.; Baglole, C.J. Genetic ablation of the aryl hydrocarbon receptor causes cigarette smoke-induced mitochondrial dysfunction and apoptosis. J. Biol. Chem. 2011, 286, 43214–43228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, J.; Xu, K.; Chen, Z.; Xu, X.; Xu, J.; Zheng, S.; Dai, M.; Yang, H. Helium protects against lipopolysaccharide-induced cardiac dysfunction in mice via suppressing Toll-like receptor 4-nuclear factor κB-tumor necrosis factor-alpha/interleukin-18 signaling. Chin. J. Physiol. 2020, 63, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.D.; Li, C.H.; Tsai, C.H.; Cheng, Y.W.; Kang, J.J.; Lee, C.C. Aryl hydrocarbon receptor deficiency enhanced airway inflammation and remodeling in a murine chronic asthma model. FASEB J. 2020, 34, 15300–15313. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-L.; Chen, H.-K.; Tsai, C.-H.; Liao, P.-L.; Chan, Y.-J.; Lee, Y.-C.; Lee, C.-C.; Li, C.-H. Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling through Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1)-Dependent EGFR Degradation. Int. J. Mol. Sci. 2021, 22, 9988. https://doi.org/10.3390/ijms22189988
Hsu H-L, Chen H-K, Tsai C-H, Liao P-L, Chan Y-J, Lee Y-C, Lee C-C, Li C-H. Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling through Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1)-Dependent EGFR Degradation. International Journal of Molecular Sciences. 2021; 22(18):9988. https://doi.org/10.3390/ijms22189988
Chicago/Turabian StyleHsu, Han-Lin, Hong-Kai Chen, Chi-Hao Tsai, Po-Lin Liao, Yen-Ju Chan, Yu-Cheng Lee, Chen-Chen Lee, and Ching-Hao Li. 2021. "Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling through Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1)-Dependent EGFR Degradation" International Journal of Molecular Sciences 22, no. 18: 9988. https://doi.org/10.3390/ijms22189988
APA StyleHsu, H.-L., Chen, H.-K., Tsai, C.-H., Liao, P.-L., Chan, Y.-J., Lee, Y.-C., Lee, C.-C., & Li, C.-H. (2021). Aryl Hydrocarbon Receptor Defect Attenuates Mitogen-Activated Signaling through Leucine-Rich Repeats and Immunoglobulin-like Domains 1 (LRIG1)-Dependent EGFR Degradation. International Journal of Molecular Sciences, 22(18), 9988. https://doi.org/10.3390/ijms22189988