
Acute Glucose Shift Induces the Activation of the NLRP3 Inflammasome in THP-1 Cells


Abstract
:1. Introduction
2. Results
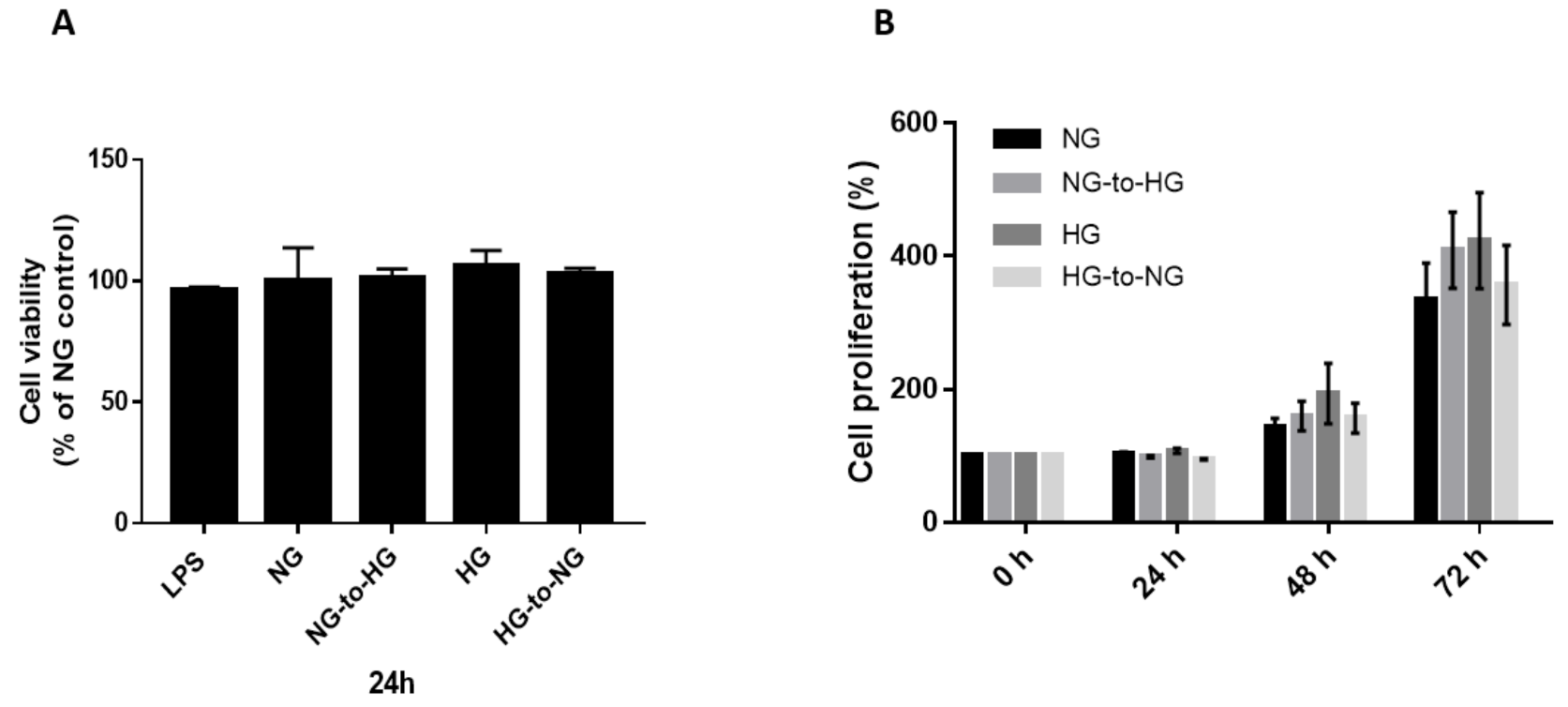
2.1. Acute Glucose Shift Did Not Alter the Viability and Proliferation of THP-1 Cells
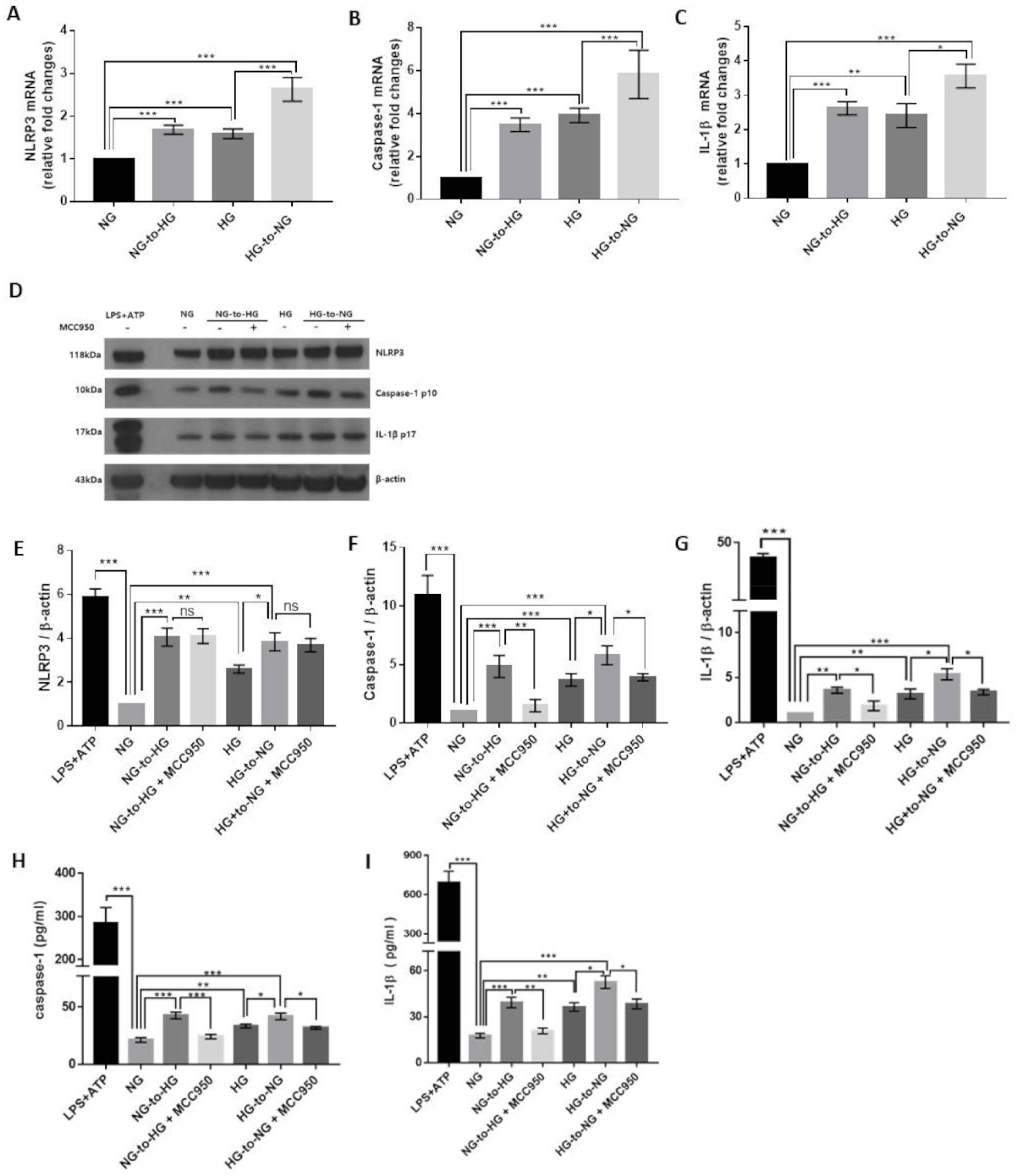
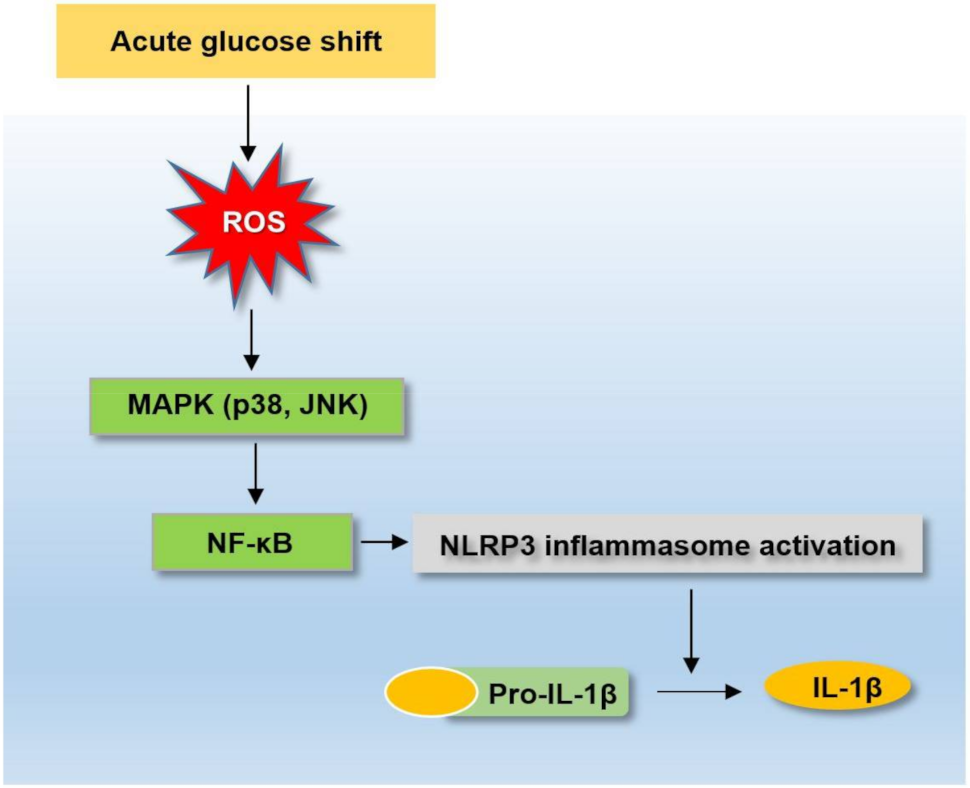
2.2. Acute Glucose Shift Induced the Activation of the NLRP3 Inflammasome in THP-1 Cells
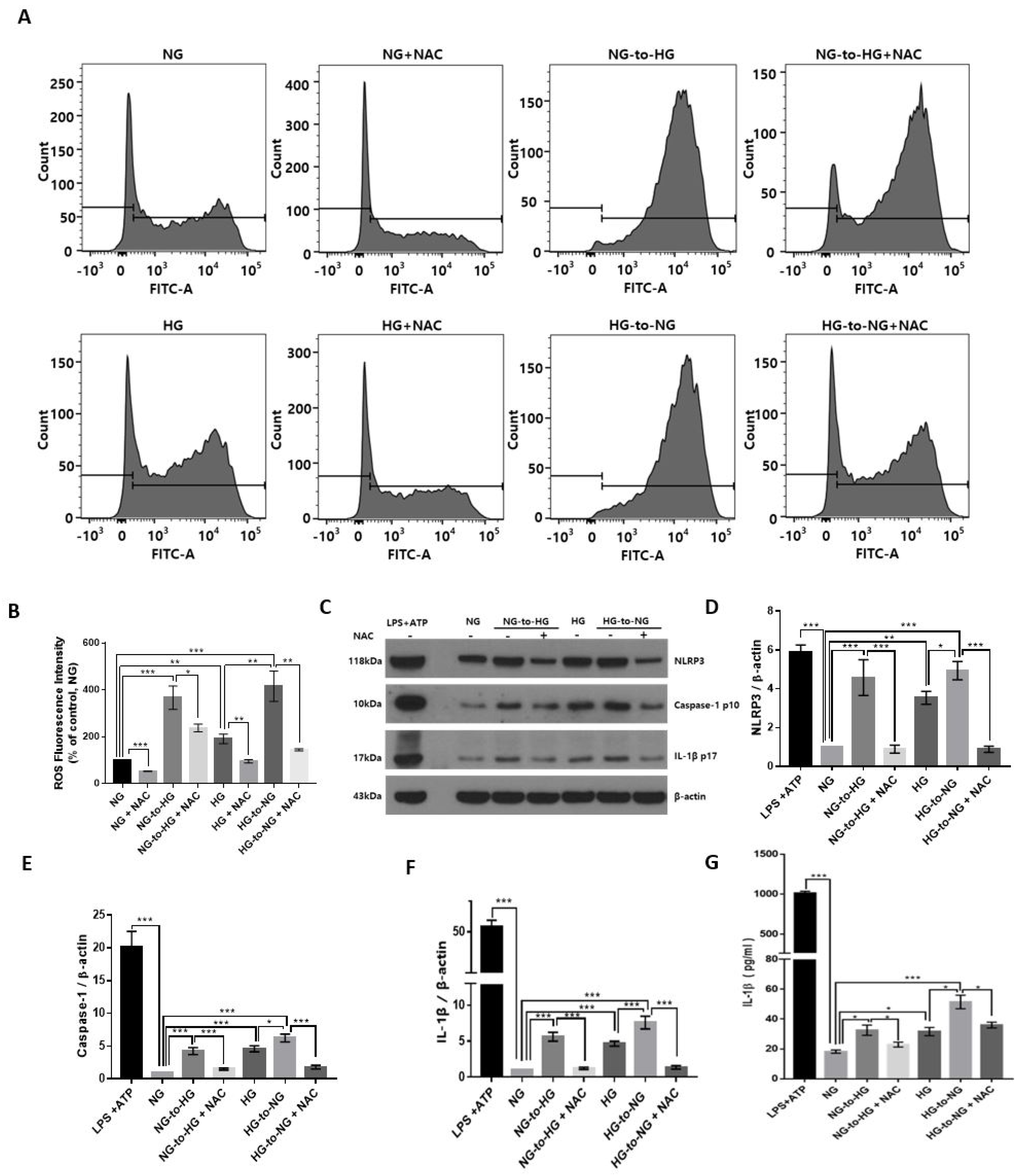
2.3. Acute Glucose Shift Activated the NLRP3 Inflammasome Mediated by ROS
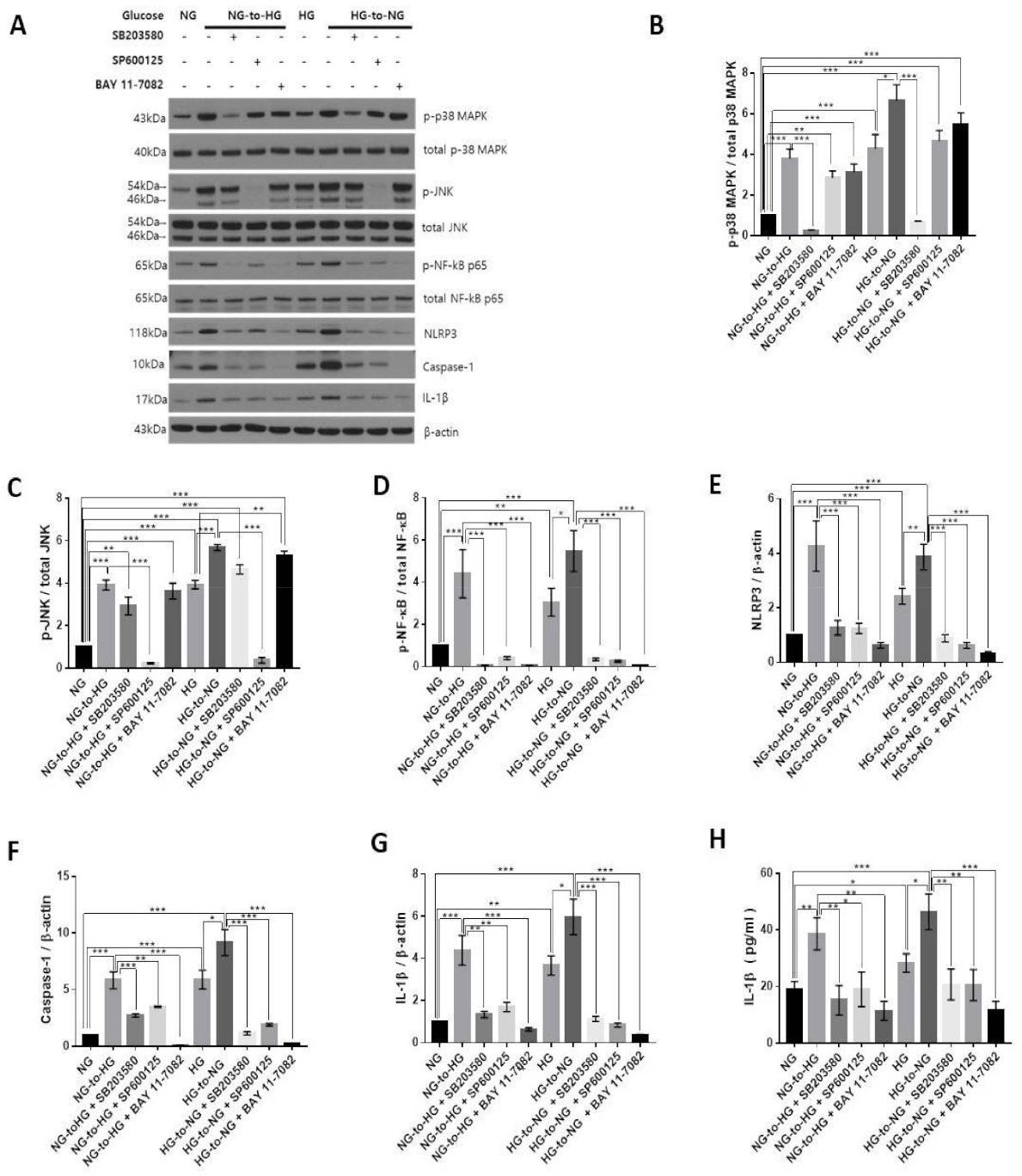
2.4. Acute Glucose Shift Activated the p38 MAPK, JNK and NF-κB Signaling Pathway Mediated by ROS
2.5. Acute Glucose Shift-Induced Activation of the NLRP3 Inflammasome Was p38 MAPK, JNK/NF-κB-Dependent
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cellular Treatments
4.2. Cell Viability Assay
4.3. ROS Measurement
4.4. Real-Time Quantitative PCR
4.5. Preparation of Protein Samples, Supernatants, and Western Blotting
4.6. ELISA
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cardoso, C.R.L.; Leite, N.C.; Moram, C.B.M.; Salles, G.F. Long-term visit-to-visit glycemic variability as predictor of micro- and macrovascular complications in patients with type 2 diabetes: The Rio de Janeiro Type 2 Diabetes Cohort Study. Cardiovasc. Diabetol. 2018, 17, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kim, M.K.; Rhee, E.J. Effects of Cardiovascular Risk Factor Variability on Health Outcomes. Endocrinol. Metab. 2020, 35, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Hirsch, I.B. Glycemic variability: A hemoglobin A1c-independent risk factor for diabetic complications. JAMA 2006, 295, 1707–1708. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Hibi, K.; Gohbara, M.; Kataoka, S.; Takano, K.; Akiyama, E.; Matsuzawa, Y.; Saka, K.; Maejima, N.; Endo, M.; et al. Association between blood glucose variability and coronary plaque instability in patients with acute coronary syndromes. Cardiovasc. Diabetol. 2015, 14, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, G.; Mi, S.; Tao, H.; Li, Z.; Yang, H.; Zheng, H.; Zhou, Y.; Ma, C. Association of glycemic variability and the presence and severity of coronary artery disease in patients with type 2 diabetes. Cardiovasc. Diabetol. 2011, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.H.; Han, K.; Park, S.; Lee, D.Y.; Nam, G.E.; Seo, J.A.; Kim, S.G.; Baik, S.H.; Park, Y.G.; Kim, S.M.; et al. Effects of long-term glycemic variability on incident cardiovascular disease and mortality in subjects without diabetes: A nationwide population-based study. Medicine 2019, 98, e16317. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.M.; Hsieh, C.J.; Huang, J.C.; Huang, I.C. Acute and chronic fluctuations in blood glucose levels can increase oxidative stress in type 2 diabetes mellitus. Acta Diabetol. 2012, 49 (Suppl. 1), S171–S177. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Shen, H.; Liu, H.; Wang, Y.; Bai, Y.; Han, P. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 109. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Kawamori, R.; Toyofuku, Y.; Kitahara, Y.; Sato, F.; Shimizu, T.; Miura, K.; Mine, T.; Tanaka, Y.; Mitsumata, M.; et al. Repetitive fluctuations in blood glucose enhance monocyte adhesion to the endothelium of rat thoracic aorta. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2275–2280. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1218. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef]
- Fusco, R.; Gugliandolo, E.; Biundo, F.; Campolo, M.; Di Paola, R.; Cuzzocrea, S. Inhibition of inflammasome activation improves lung acute injury induced by carrageenan in a mouse model of pleurisy. FASEB J. 2017, 31, 3497–3511. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.S.; Bruder-Nascimento, T.; Pereira, C.A.; Zanotto, C.Z.; Prado, D.S.; Silva, J.F.; Rassi, D.M.; Foss-Freitas, M.C.; Alves-Filho, J.C.; Carlos, D.; et al. NLRP3 Inflammasome and Mineralocorticoid Receptors Are Associated with Vascular Dysfunction in Type 2 Diabetes Mellitus. Cells 2019, 8, 1595. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, N.; Reddy, M.A.; Guha, M.; Natarajan, R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003, 52, 1256–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stienstra, R.; Tack, C.J.; Kanneganti, T.D.; Joosten, L.A.; Netea, M.G. The inflammasome puts obesity in the danger zone. Cell Metab. 2012, 15, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab. Syndr. Obes. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. NLRP3 Inflammasome Expression and Signaling in Human Diabetic Wounds and in High Glucose Induced Macrophages. J. Diabetes Res. 2017, 2017, 5281358. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.; Ergul, A. Relationship of endothelin-1 and NLRP3 inflammasome activation in HT22 hippocampal cells in diabetes. Life Sci 2016, 159, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, X.; Zhu, G.; Zhang, Y.; He, M.; Zhang, J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp. Cell Res. 2016, 341, 42–53. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zito, G.; Buscetta, M.; Cimino, M.; Dino, P.; Bucchieri, F.; Cipollina, C. Cellular Models and Assays to Study NLRP3 Inflammasome Biology. Int. J. Mol. Sci 2020, 21, 4294. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, Z.; Kiani, Z.; Afshari, M.; Kohan, F.; Dalvand, A.; Ghavami, S. Inflammasomes and type 2 diabetes: An updated systematic review. Immunol. Lett. 2017, 192, 97–103. [Google Scholar] [CrossRef]
- Schnackenberg, C.G. Oxygen radicals in cardiovascular-renal disease. Curr. Opin. Pharmacol. 2002, 2, 121–125. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- An, Y.; Zhang, H.; Wang, C.; Jiao, F.; Xu, H.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Tang, X.; et al. Activation of ROS/MAPKs/NF-kappaB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. 2019, 33, 12515–12527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Lee, H.J.; Choi, M.S.; Son, D.J.; Song, H.S.; Song, M.J.; Lee, J.M.; Han, S.B.; Kim, Y.; Hong, J.T. JNK pathway is involved in the inhibition of inflammatory target gene expression and NF-kappaB activation by melittin. J. Inflamm. (Lond.) 2008, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuyt, L.M.; Dokter, W.H.; Birkenkamp, K.; Koopmans, S.B.; Lummen, C.; Kruijer, W.; Vellenga, E. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J. Immunol. 1999, 162, 4893–4902. [Google Scholar] [PubMed]
- Wang, Y.; Cao, J.; Fan, Y.; Xie, Y.; Xu, Z.; Yin, Z.; Gao, L.; Wang, C. Artemisinin inhibits monocyte adhesion to HUVECs through the NF-kappaB and MAPK pathways in vitro. Int. J. Mol. Med. 2016, 37, 1567–1575. [Google Scholar] [CrossRef]
- Javelaud, D.; Besancon, F. NF-kappa B activation results in rapid inactivation of JNK in TNF alpha-treated Ewing sarcoma cells: A mechanism for the anti-apoptotic effect of NF-kappa B. Oncogene 2001, 20, 4365–4372. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.genome.jp/kegg-bin/show_pathway?ko04010+K04368 (accessed on 5 March 2021).
- Li, S.; Liu, R.; Xue, M.; Qiao, Y.; Chen, Y.; Long, G.; Tian, X.; Hu, Y.; Zhou, P.; Dong, X.; et al. Spleen tyrosine kinaseinduced JNKdependent NLRP3 activation is involved in diabetic cardiomyopathy. Int. J. Mol. Med. 2019, 43, 2481–2490. [Google Scholar] [CrossRef]
- Hsieh, C.F.; Liu, C.K.; Lee, C.T.; Yu, L.E.; Wang, J.Y. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci. Rep. 2019, 9, 840. [Google Scholar] [CrossRef] [Green Version]
- Quincozes-Santos, A.; Bobermin, L.D.; de Assis, A.M.; Goncalves, C.A.; Souza, D.O. Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1–14. [Google Scholar] [CrossRef]
- Piarulli, F.; Sartore, G.; Sechi, A.; Basso, D.; Fogar, P.; Greco, E.; Ragazzi, E.; Lapolla, A. Low Glucose Concentrations Induce a Similar Inflammatory Response in Monocytes from Type 2 Diabetic Patients and Healthy Subjects. Oxidative Med. Cell. Longev. 2017, 2017, 9185272. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GenBank Accession No. | Primer sequences (5′-3′) |
|---|---|---|
| NLRP3 | NM 001127462 | F: ACAGCCACCTCACTTCCAG |
| R: CCAACCACAATCTCCGAATG | ||
| Caspase-1 | NM 001257119 | F: GCACAAGACCTCTGACAGCA R: TTGGGCAGTTCTTGGTATTC |
| ASC | NM 145182 | F: TGGATGCTCTGTACGGGAAG R: TGGATGCTCTGTACGGGAAG |
| IL-1β | NM 000567 | F: GCCCTAAACAGATGAAGTGCTC R: GAACCAGCATCTTCC CAG |
| GAPDH | NM 002046 | F: CATGAGAAGTATGACAACAGC R: AGTCCTTCCACGATACCAAAGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.Y.; Kang, Y.; Kim, H.J.; Kim, D.J.; Lee, K.W.; Han, S.J. Acute Glucose Shift Induces the Activation of the NLRP3 Inflammasome in THP-1 Cells. Int. J. Mol. Sci. 2021, 22, 9952. https://doi.org/10.3390/ijms22189952
Lee JY, Kang Y, Kim HJ, Kim DJ, Lee KW, Han SJ. Acute Glucose Shift Induces the Activation of the NLRP3 Inflammasome in THP-1 Cells. International Journal of Molecular Sciences. 2021; 22(18):9952. https://doi.org/10.3390/ijms22189952
Chicago/Turabian StyleLee, Ji Yeon, Yup Kang, Hae Jin Kim, Dae Jung Kim, Kwan Woo Lee, and Seung Jin Han. 2021. "Acute Glucose Shift Induces the Activation of the NLRP3 Inflammasome in THP-1 Cells" International Journal of Molecular Sciences 22, no. 18: 9952. https://doi.org/10.3390/ijms22189952
APA StyleLee, J. Y., Kang, Y., Kim, H. J., Kim, D. J., Lee, K. W., & Han, S. J. (2021). Acute Glucose Shift Induces the Activation of the NLRP3 Inflammasome in THP-1 Cells. International Journal of Molecular Sciences, 22(18), 9952. https://doi.org/10.3390/ijms22189952