
Interplay of Immunometabolism and Epithelial–Mesenchymal Transition in the Tumor Microenvironment

Abstract
:1. Introduction
2. Epithelial-Mesenchymal Transition and Functional Change of Immune Cells in Tumor Metastasis: The Mutually Regulatory Loop
2.1. EMT in Cancer
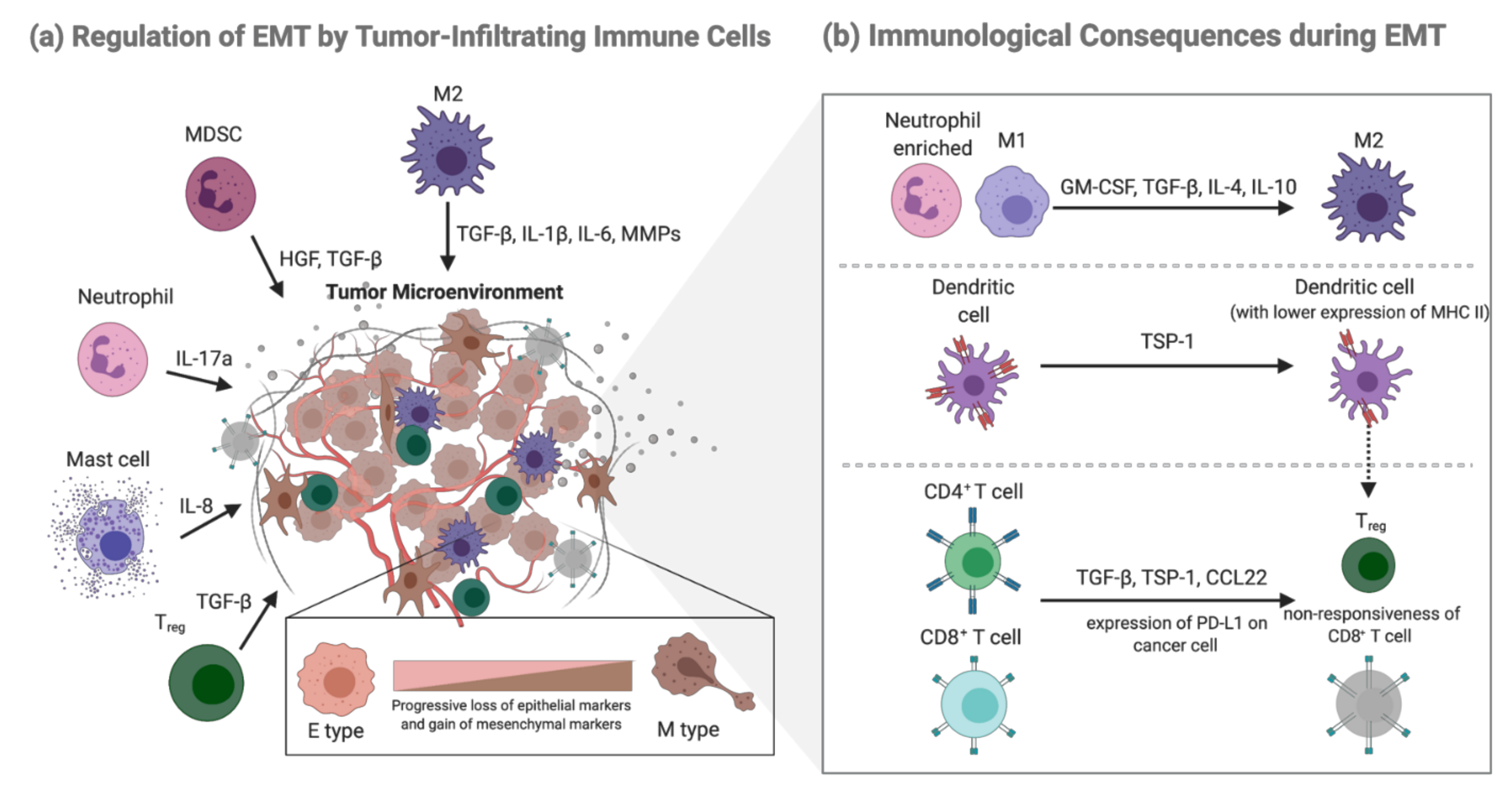
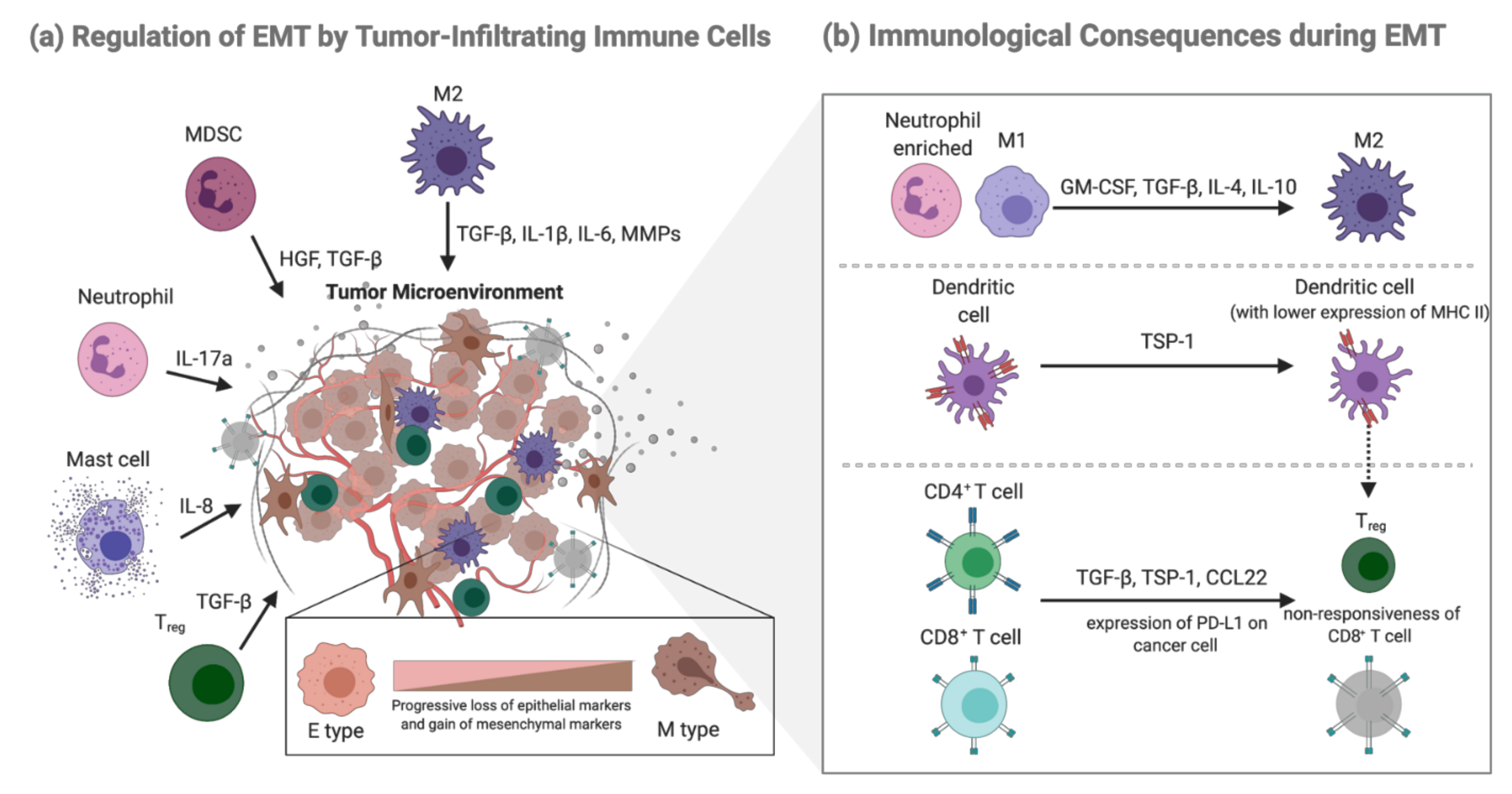
2.2. EMT-Mediated Immunological Consequences
2.3. Regulation of EMT by Tumor-Infiltrating Immune Cells
3. Interplay of Metabolic Reprogramming of Immune Cells and EMT
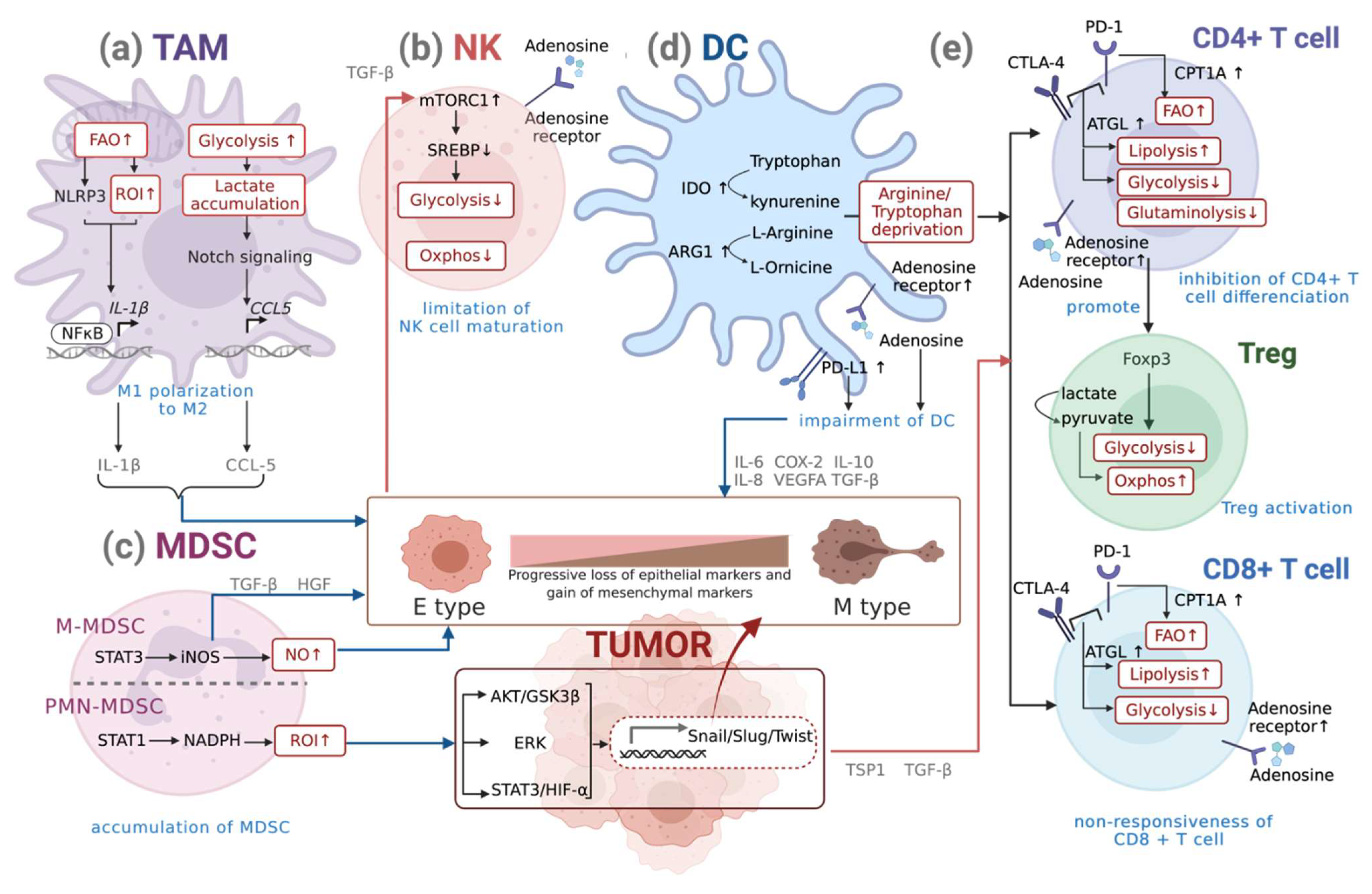
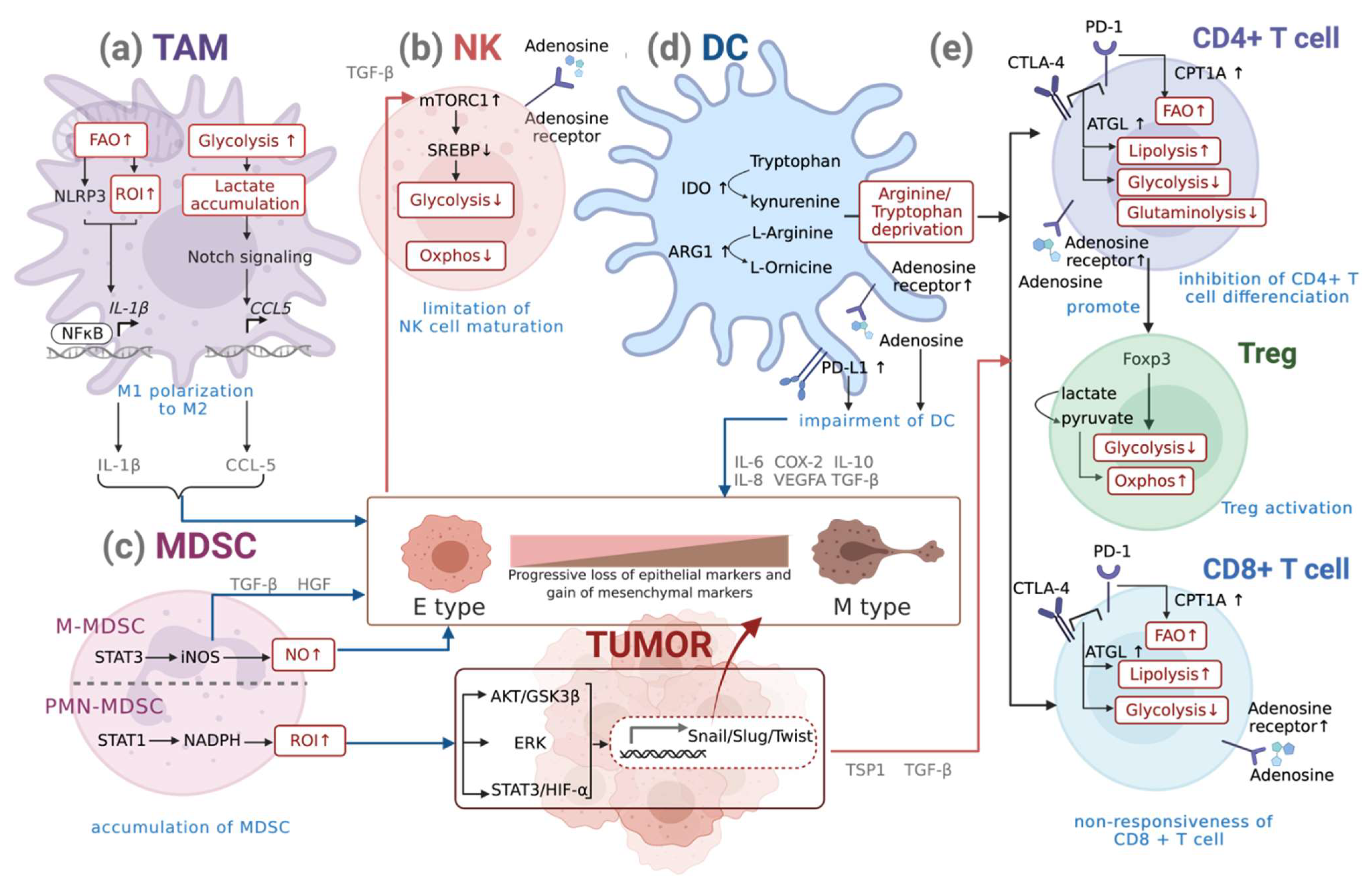
3.1. Metabolism of Macrophages
Metabolic Reprogramming of Macrophages during EMT
3.2. Metabolism of DCs
Metabolic Reprogramming of DCs during EMT
3.3. Metabolism of NK Cells
Metabolic Reprogramming of NKs during EMT
3.4. Metabolism of MDSCs
Metabolic Reprogramming of MDSCs during EMT
3.5. Metabolism of T Cells
Metabolic Reprogramming of T Cells during EMT
4. Targeting Immunometabolism: Challenges and Perspectives
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Bonnomet, A.; Brysse, A.; Tachsidis, A.; Waltham, M.; Thompson, E.W.; Polette, M.; Gilles, C. Epithelial-to-mesenchymal transitions and circulating tumor cells. J. Mammary Gland Biol. Neoplasia 2010, 15, 261–273. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Nieto, M.A.; Cano, A. The epithelial-mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Yang, W.H.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Tzeng, C.H.; Kao, S.Y.; Wu, K.J.; Hung, M.C.; Yang, M.H. RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef]
- Hwang, W.L.; Jiang, J.K.; Yang, S.H.; Huang, T.S.; Lan, H.Y.; Teng, H.W.; Yang, C.Y.; Tsai, Y.P.; Lin, C.H.; Wang, H.W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef]
- Hsu, D.S.; Hwang, W.L.; Yuh, C.H.; Chu, C.H.; Ho, Y.H.; Chen, P.B.; Lin, H.S.; Lin, H.K.; Wu, S.P.; Lin, C.Y.; et al. Lymphotoxin-beta Interacts with Methylated EGFR to Mediate Acquired Resistance to Cetuximab in Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 4388–4401. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Li, Y.; Wang, J.; Wen, Z.; Lai, M.; Zhang, H. Molecular mechanisms of microRNAs in regulating epithelial-mesenchymal transitions in human cancers. Cancer Lett. 2016, 371, 301–313. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Strutz, F.; Zeisberg, M.; Ziyadeh, F.N.; Yang, C.Q.; Kalluri, R.; Muller, G.A.; Neilson, E.G. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002, 61, 1714–1728. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef] [Green Version]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Tai, S.K.; Yang, M.H. Snail-overexpressing Cancer Cells Promote M2-Like Polarization of Tumor-Associated Macrophages by Delivering MiR-21-Abundant Exosomes. Neoplasia 2018, 20, 775–788. [Google Scholar] [CrossRef]
- Hsu, D.S.; Wang, H.J.; Tai, S.K.; Chou, C.H.; Hsieh, C.H.; Chiu, P.H.; Chen, N.J.; Yang, M.H. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell 2014, 26, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; Andre, P.; Dieu-Nosjean, M.C.; Alifano, M.; Regnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Lin, J.C.; Hwang, W.L.; Kuo, Y.J.; Chen, H.K.; Tai, S.K.; Lin, C.C.; Yang, M.H. Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 2018, 9, 3763. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Nordenfelt, E.; Hansson, B.G. Increasing heterogeneity of the ‘a’ determinant of HBsAg found in the presumed late phase of chronic hepatitis B virus infection. Scand. J. Infect. Dis. 1996, 28, 9–15. [Google Scholar] [CrossRef]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 6. [Google Scholar] [CrossRef] [Green Version]
- Martins-Cardoso, K.; Almeida, V.H.; Bagri, K.M.; Rossi, M.I.D.; Mermelstein, C.S.; Konig, S.; Monteiro, R.Q. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers 2020, 12, 1542. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Zou, X.; Yang, C.; Li, L.; Wang, B.; Li, R.; Li, H.; Xu, Z.; Huang, D.; Wu, Q. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelialmesenchymal transition. Int. J. Mol. Med. 2021, 48. [Google Scholar] [CrossRef]
- Visciano, C.; Liotti, F.; Prevete, N.; Cali, G.; Franco, R.; Collina, F.; de Paulis, A.; Marone, G.; Santoro, M.; Melillo, R.M. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene 2015, 34, 5175–5186. [Google Scholar] [CrossRef]
- Shi, C.; Chen, Y.; Chen, Y.; Yang, Y.; Bing, W.; Qi, J. CD4(+) CD25(+) regulatory T cells promote hepatocellular carcinoma invasion via TGF-beta1-induced epithelial-mesenchymal transition. OncoTargets Ther. 2019, 12, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, J.; Rychahou, P.G.; Qiu, S.; Evers, B.M.; Zhou, B.P. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009, 15, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, Y.; Du, Q.; Lu, P.; Fan, H.; Lu, J.; Hu, R. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp. Cell Res. 2016, 342, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Posokhova, E.N.; Khoshchenko, O.M.; Chasovskikh, M.I.; Pivovarova, E.N.; Dushkin, M.I. Lipid synthesis in macrophages during inflammation in vivo: Effect of agonists of peroxisome proliferator activated receptors alpha and gamma and of retinoid X receptors. Biochemistry 2008, 73, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, C.; Keast, D. Glutamine and macrophage function. Metabolism 1992, 41, 1016–1020. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D.; Nathan, C.; Hom, G.; Chartrain, N.; Fletcher, D.S.; Trumbauer, M.; Stevens, K.; Xie, Q.W.; Sokol, K.; Hutchinson, N.; et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995, 81, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.J.; Garg, N.J. Metabolic programming of macrophage functions and pathogens control. Redox Biol. 2019, 24, 101198. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Gunaltay, S.; McEntee, C.P.; Shuttleworth, E.E.; Smedley, C.; Houston, S.A.; Fenton, T.M.; Levison, S.; Mann, E.R.; Travis, M.A. Human monocytes and macrophages regulate immune tolerance via integrin alphavbeta8-mediated TGFbeta activation. J. Exp. Med. 2018, 215, 2725–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- de-Brito, N.M.; Duncan-Moretti, J.; da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118604. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.Y.; Wang, X.; Jiang, C. HIF1alpha-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [Green Version]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Bocca, C.; Ievolella, M.; Autelli, R.; Motta, M.; Mosso, L.; Torchio, B.; Bozzo, F.; Cannito, S.; Paternostro, C.; Colombatto, S.; et al. Expression of Cox-2 in human breast cancer cells as a critical determinant of epithelial-to-mesenchymal transition and invasiveness. Expert Opin. Ther. Targets 2014, 18, 121–135. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [Green Version]
- Mosca, M.; Polentarutti, N.; Mangano, G.; Apicella, C.; Doni, A.; Mancini, F.; De Bortoli, M.; Coletta, I.; Polenzani, L.; Santoni, G.; et al. Regulation of the microsomal prostaglandin E synthase-1 in polarized mononuclear phagocytes and its constitutive expression in neutrophils. J. Leukoc. Biol. 2007, 82, 320–326. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.E.; Hur, J.; Hong, E.B.; Choi, J.I.; Yang, J.M.; Kim, J.Y.; Kim, Y.C.; Cho, H.J.; Peters, J.M.; et al. M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARbeta/delta Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep. 2015, 10, 1614–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, G.; Xu, F.; Qin, T.; Zheng, Q.; Shi, D.; Xia, W.; Tian, Y.; Tang, Y.; Wang, J.; Xiao, X.; et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget 2015, 6, 41794–41808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.P.; Wang, Q.Y.; Li, C.H.; Li, X.W. COX-2 inhibition by celecoxib in epithelial ovarian cancer attenuates E-cadherin suppression through reduced Snail nuclear translocation. Chem.-Biol. Interact. 2018, 292, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Jessen, C.; Kress, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. (Camb.) 2019, 11, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin beta3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265–278. [Google Scholar] [CrossRef]
- de Araujo Junior, R.F.; Eich, C.; Jorquera, C.; Schomann, T.; Baldazzi, F.; Chan, A.B.; Cruz, L.J. Ceramide and palmitic acid inhibit macrophage-mediated epithelial-mesenchymal transition in colorectal cancer. Mol. Cell. Biochem. 2020, 468, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabon, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1beta secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Corinti, S.; Medaglini, D.; Cavani, A.; Rescigno, M.; Pozzi, G.; Ricciardi-Castagnoli, P.; Girolomoni, G. Human dendritic cells very efficiently present a heterologous antigen expressed on the surface of recombinant gram-positive bacteria to CD4+ T lymphocytes. J. Immunol. 1999, 163, 3029–3036. [Google Scholar] [PubMed]
- van Rijt, L.S.; Vos, N.; Willart, M.; Kleinjan, A.; Coyle, A.J.; Hoogsteden, H.C.; Lambrecht, B.N. Essential role of dendritic cell CD80/CD86 costimulation in the induction, but not reactivation, of TH2 effector responses in a mouse model of asthma. J. Allergy Clin. Immunol. 2004, 114, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Glasner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Pantel, A.; Teixeira, A.; Haddad, E.; Wood, E.G.; Steinman, R.M.; Longhi, M.P. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014, 12, e1001759. [Google Scholar] [CrossRef]
- Spirig, R.; Djafarzadeh, S.; Regueira, T.; Shaw, S.G.; von Garnier, C.; Takala, J.; Jakob, S.M.; Rieben, R.; Lepper, P.M. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS ONE 2010, 5, e0010983. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; van der Windt, G.J.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef]
- Li, H.; Shi, B. Tolerogenic dendritic cells and their applications in transplantation. Cell Mol. Immunol. 2015, 12, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013, 94, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Gorgun, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [Green Version]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-infiltrating dendritic cells in cancer pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Yang, M.; Ma, C.; Liu, S.; Shao, Q.; Gao, W.; Song, B.; Sun, J.; Xie, Q.; Zhang, Y.; Feng, A.; et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol. Cell Biol. 2010, 88, 165–171. [Google Scholar] [CrossRef]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.M.; Darcy, P.K.; Markby, J.L.; Godfrey, D.I.; Takeda, K.; Yagita, H.; Smyth, M.J. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat. Immunol. 2002, 3, 83–90. [Google Scholar] [CrossRef]
- Smyth, M.J.; Swann, J.; Cretney, E.; Zerafa, N.; Yokoyama, W.M.; Hayakawa, Y. NKG2D function protects the host from tumor initiation. J. Exp. Med. 2005, 202, 583–588. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 2341. [Google Scholar] [CrossRef] [PubMed]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Morandi, B.; Costa, R.; Frumento, G.; Forte, G.; Altavilla, G.; Ratto, G.B.; Mingari, M.C.; Moretta, L.; Ferlazzo, G. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 2008, 112, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Moller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [Green Version]
- Halfteck, G.G.; Elboim, M.; Gur, C.; Achdout, H.; Ghadially, H.; Mandelboim, O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J. Immunol. 2009, 182, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-beta Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Soto, A.; Huergo-Zapico, L.; Galvan, J.A.; Rodrigo, L.; de Herreros, A.G.; Astudillo, A.; Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 2013, 190, 4408–4419. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Andresen, L.; Hansen, K.A.; Jensen, H.; Pedersen, S.F.; Stougaard, P.; Hansen, H.R.; Jurlander, J.; Skov, S. Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J. Immunol. 2009, 183, 897–906. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.T.; Moncayo, G.; Hiron, T.K.; Jakobsen, N.A.; Valli, A.; Soga, T.; Adam, J.; O’Callaghan, C.A. Purine nucleotide metabolism regulates expression of the human immune ligand MICA. J. Biol. Chem. 2018, 293, 3913–3924. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Geschwind, J.F.; Karthikeyan, S.; Miller, E.; Kunjithapatham, R.; Wang, Z.; Ganapathy-Kanniappan, S. Metabolic perturbation sensitizes human breast cancer to NK cell-mediated cytotoxicity by increasing the expression of MHC class I chain-related A/B. Oncoimmunology 2015, 4, e991228. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, U.; Bronte, V. Control of immune response by amino acid metabolism. Immunol. Rev. 2010, 236, 243–264. [Google Scholar] [CrossRef]
- Saxena, V.; Ondr, J.K.; Magnusen, A.F.; Munn, D.H.; Katz, J.D. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J. Immunol. 2007, 179, 5041–5053. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.L.; Lan, H.Y.; Cheng, W.C.; Huang, S.C.; Yang, M.H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J. Hematol. Oncol. 2019, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Hammami, I.; Chen, J.; Bronte, V.; DeCrescenzo, G.; Jolicoeur, M. L-glutamine is a key parameter in the immunosuppression phenomenon. BioChem. Biophys. Res. Commun. 2012, 425, 724–729. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.S.; Tsai, R.K.; Chang, C.H.; Wang, S.; Wu, J.R.; Chang, Y.X. Reactive oxygen species mediated sustained activation of protein kinase C alpha and extracellular signal-regulated kinase for migration of human hepatoma cell Hepg2. Mol. Cancer Res. 2006, 4, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, N.; Parbin, S.; Kar, S.; Das, L.; Kirtana, R.; Suma Seshadri, G.; Sengupta, D.; Deb, M.; Kausar, C.; Patra, S.K. Epigenetic silencing of genes enhanced by collective role of reactive oxygen species and MAPK signaling downstream ERK/Snail axis: Ectopic application of hydrogen peroxide repress CDH1 gene by enhanced DNA methyltransferase activity in human breast cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Swamy, M.; Pathak, S.; Grzes, K.M.; Damerow, S.; Sinclair, L.V.; van Aalten, D.M.; Cantrell, D.A. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 2016, 17, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef] [PubMed]
- Raud, B.; Roy, D.G.; Divakaruni, A.S.; Tarasenko, T.N.; Franke, R.; Ma, E.H.; Samborska, B.; Hsieh, W.Y.; Wong, A.H.; Stuve, P.; et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab. 2018, 28, 504–515.e507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [Green Version]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Fedele, V.; Melisi, D. Permissive State of EMT: The Role of Immune Cell Compartment. Front. Oncol. 2020, 10, 587. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [Green Version]
- Araujo, L.; Khim, P.; Mkhikian, H.; Mortales, C.L.; Demetriou, M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 2017, 6, e21330. [Google Scholar] [CrossRef]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [Green Version]
- Sun, I.H.; Oh, M.H.; Zhao, L.; Patel, C.H.; Arwood, M.L.; Xu, W.; Tam, A.J.; Blosser, R.L.; Wen, J.; Powell, J.D. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3(+) Regulatory T Cells. J. Immunol. 2018, 201, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Li, J.; Li, R.Y.; Lan, T.; Xiao, C.; Gong, P. PD-L1 Expression Is Regulated By NF-kappaB During EMT Signaling In Gastric Carcinoma. OncoTargets Ther. 2019, 12, 10099–10105. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, M.; Allard, D.; Mittal, D.; Bareche, Y.; Buisseret, L.; Jose, V.; Pommey, S.; Delisle, V.; Loi, S.; Joensuu, H.; et al. CD73 Promotes Resistance to HER2/ErbB2 Antibody Therapy. Cancer Res. 2017, 77, 5652–5663. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.A.; de Carne Trecesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Xing, M.; Han, L.; Gan, S.; Ma, J.; Wu, F.; Huang, Y.; Chen, Y.; Tian, W.; An, C.; et al. High PDL1 expression drives glycolysis via an Akt/mTOR/HIF1alpha axis in acute myeloid leukemia. Oncol. Rep. 2020, 43, 999–1009. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Previte, D.M.; Martins, C.P.; O’Connor, E.C.; Marre, M.L.; Coudriet, G.M.; Beck, N.W.; Menk, A.V.; Wright, R.H.; Tse, H.M.; Delgoffe, G.M.; et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4(+) T Cells. Cell Rep. 2019, 27, 129–141.e124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8(+) T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Hasko, G. Anti-CD73 in cancer immunotherapy: Awakening new opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yao, F.; Davis, P.F.; Tan, S.T.; Hall, S.R.R. CD73, Tumor Plasticity and Immune Evasion in Solid Cancers. Cancers 2021, 13, 177. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, I.A.; Braga, T.T.; Almeida, D.C.; Batista de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haikala, H.M.; Anttila, J.M.; Marques, E.; Raatikainen, T.; Ilander, M.; Hakanen, H.; Ala-Hongisto, H.; Savelius, M.; Balboa, D.; Von Eyss, B.; et al. Pharmacological reactivation of MYC-dependent apoptosis induces susceptibility to anti-PD-1 immunotherapy. Nat. Commun. 2019, 10, 620. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; Alvarez, I.; Martinez, S.; Perez-Garcia, J.M.; Batista-Lopez, N.; Rodriguez-Sanchez, C.A.; Amillano, K.; Dominguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: The METTEN study. Oncotarget 2018, 9, 35687–35704. [Google Scholar] [CrossRef] [Green Version]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Di Marino, P.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef]
- Muller, A.J.; Manfredi, M.G.; Zakharia, Y.; Prendergast, G.C. Inhibiting IDO pathways to treat cancer: Lessons from the ECHO-301 trial and beyond. Semin. Immunopathol. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Murray, I.A.; Lee, S.; Hazlett, H.F.; Perdew, G.H.; Tomlinson, C.R. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors activate the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2017, 323, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Pathway | Drug | Function | Combined ICI | Cancer Type | Status |
|---|---|---|---|---|---|
| Adenosine pathway | Sym024 | CD73 antibody | Sym021 | Solid tumors | Phase I (NCT03835949) |
| AK119 | AK104 | Solid tumors | Phase I (NCT04572152) | ||
| TJ004309 | Atezolizumab | Solid tumors | Phase I (NCT03835949) | ||
| NZV930 | PDR001 | NSCLC, TNBC, PDAC, MSS-CRC, RCC, mCRPC, Ovarian cancer | Phase I (NCT03549000) | ||
| CPI-006 | Pembrolizumab | Solid tumors, Non-Hodgkin lymphoma | Phase I (NCT0345445) | ||
| MED19447 | Duvalumab | Ovarian cancer | Phase I (NCT03267589) | ||
| BMS-986179 | Nivolumab | Solid tumors | Phase I/II (NCT02754141) | ||
| Ciforadenant | Adenosine Receptor (A2A) antibody | Atezolizumab | RCC, Mcrpc | Phase I (NCT02655822) | |
| NIR178 | PDR001 | Solid tumors, Non-Hodgkin lymphoma | Phase II (NCT03207847) | ||
| Arginine metabolism | INCB001158 | Arginase inhibitor | Pembrolizumab | NSCLC, BLCA, MSI/MSS-CRC, GC, HNSCC, Melanoma, Mesothelioma | Phase II (NCT02903914) |
| Folate pathway | Pemetrexed | Pyrimidine and purine synthesis inhibitor | Nivolumab | HNSCC | Phase II (NCT04107103) |
| 5-fluorouracil | Biliary Tract Cancer | Phase Ib/II (NCT03785873) | |||
| Glucose metabolism | Metformin | Gluconeogenesis inhibitor | Pembrolizumab | Melanoma | Phase I (NCT03311308) |
| NSCLC, BLCA, MSI/NSS-CRC, GC, HNSCC, RCC, HCC, ESCA, Melanoma | Phase II (NCT04414540) (NCT04114136) | ||||
| Nivolumab | NSCLC, BLCA, MSI/NSS-CRC, GC, HNSCC, RCC, HCC, ESCA, Melanoma | Phase II (NCT03048500) (NCT03800602) (NCT04114136) | |||
| Sintilimab | SCLC | Phase II (NCT03994744) | |||
| Durvalumab | HNSCC | Phase I (NCT03618654) | |||
| Glutamine metabolism | CB-839 | Glutaminase inhibitor | Nivolumab | NSCLC, ccRCC, Melanoma | Phase II (NCT02771626) |
| IDO pathway | PD-L1/IDO peptite vaccine | IDO inhibitor | Nivolumab | Melanoma | Phase II (NCT03047928) |
| BMS-986205 | HCC | Phase II (NCT03695250) | |||
| Indoximod | Ipilimumab/Pembrolizumab/Nivolumab | Melanoma | Phase I/II (NCT02073123) | ||
| KHK2455 | Avelumab | BLCA | Phase I (NCT03915405) | ||
| Epacadostat | Ipilimumab/Pembrolizumab/Nivolumab/Lirilumab | Solid tumors | Phase II (NCT03291054) (NCT03414229) (NCT03347123) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, M.-Y.; Yang, M.-H. Interplay of Immunometabolism and Epithelial–Mesenchymal Transition in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 9878. https://doi.org/10.3390/ijms22189878
Chou M-Y, Yang M-H. Interplay of Immunometabolism and Epithelial–Mesenchymal Transition in the Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(18):9878. https://doi.org/10.3390/ijms22189878
Chicago/Turabian StyleChou, Ming-Yu, and Muh-Hwa Yang. 2021. "Interplay of Immunometabolism and Epithelial–Mesenchymal Transition in the Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 18: 9878. https://doi.org/10.3390/ijms22189878
APA StyleChou, M.-Y., & Yang, M.-H. (2021). Interplay of Immunometabolism and Epithelial–Mesenchymal Transition in the Tumor Microenvironment. International Journal of Molecular Sciences, 22(18), 9878. https://doi.org/10.3390/ijms22189878