
The miR-133a, TPM4 and TAp63γ Role in Myocyte Differentiation Microfilament Remodelling and Colon Cancer Progression

 ,
, 
 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
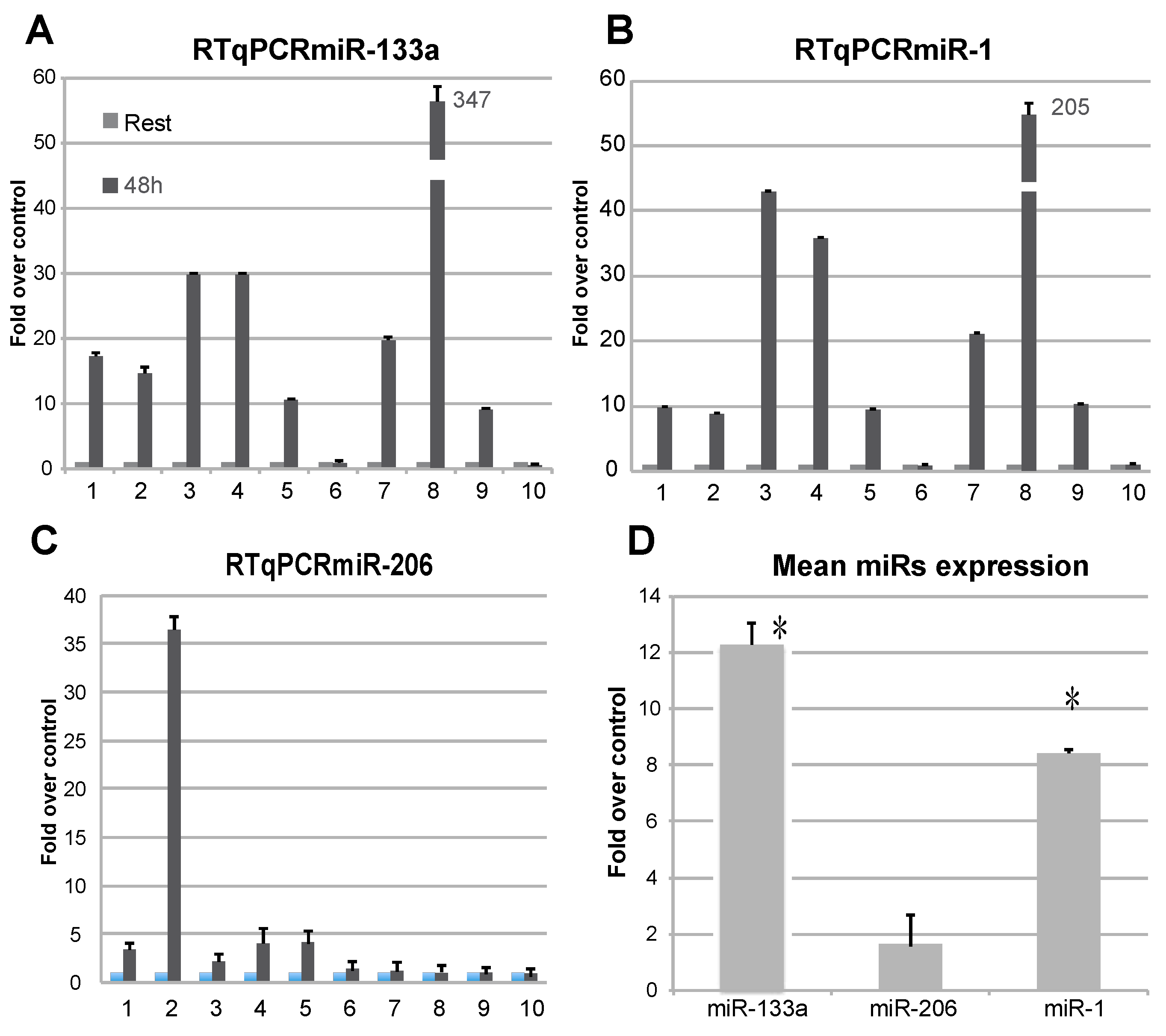
2.1. Blood Parameters Related to Physical Exercise and Analysis of C-MyomiRs in Plasma
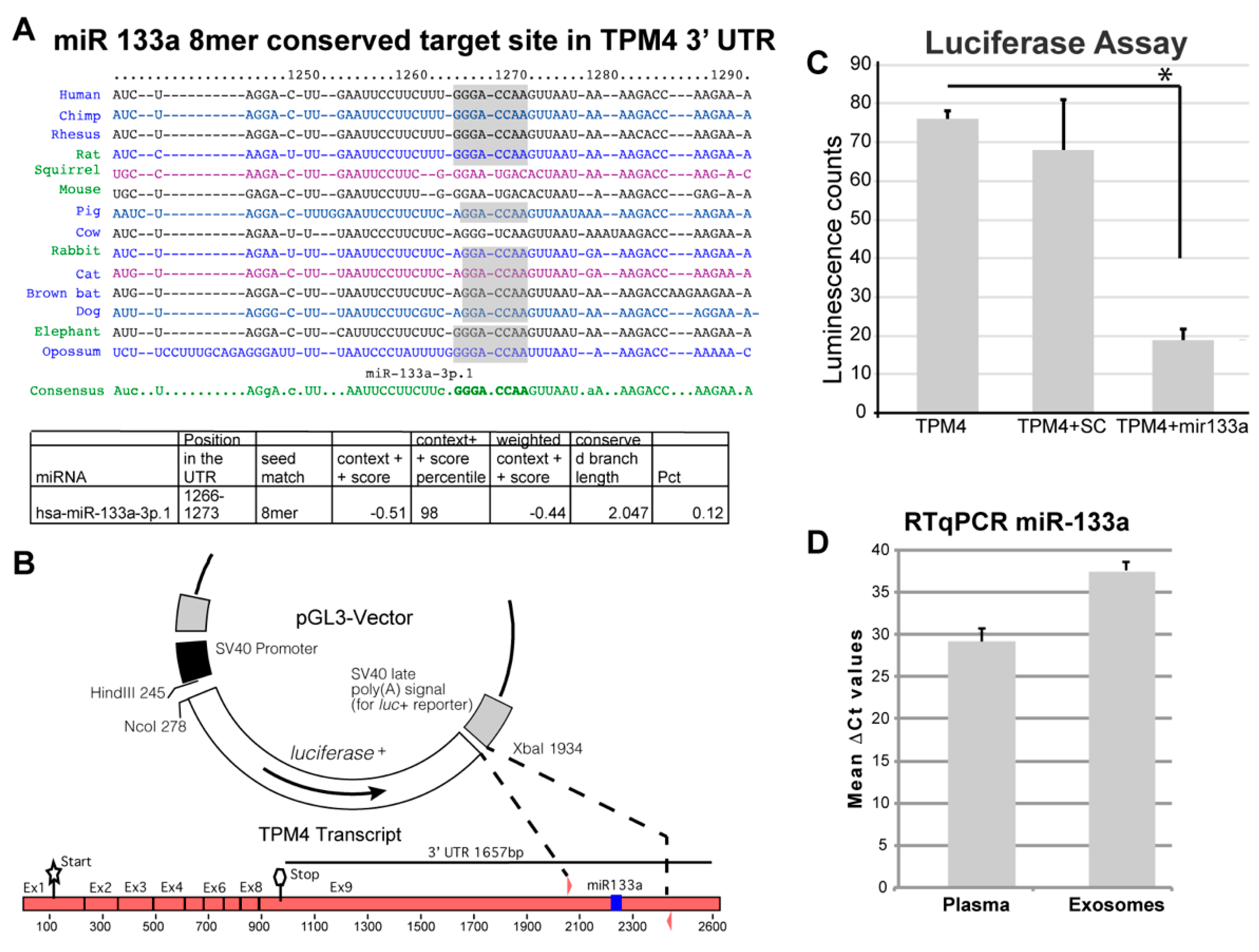
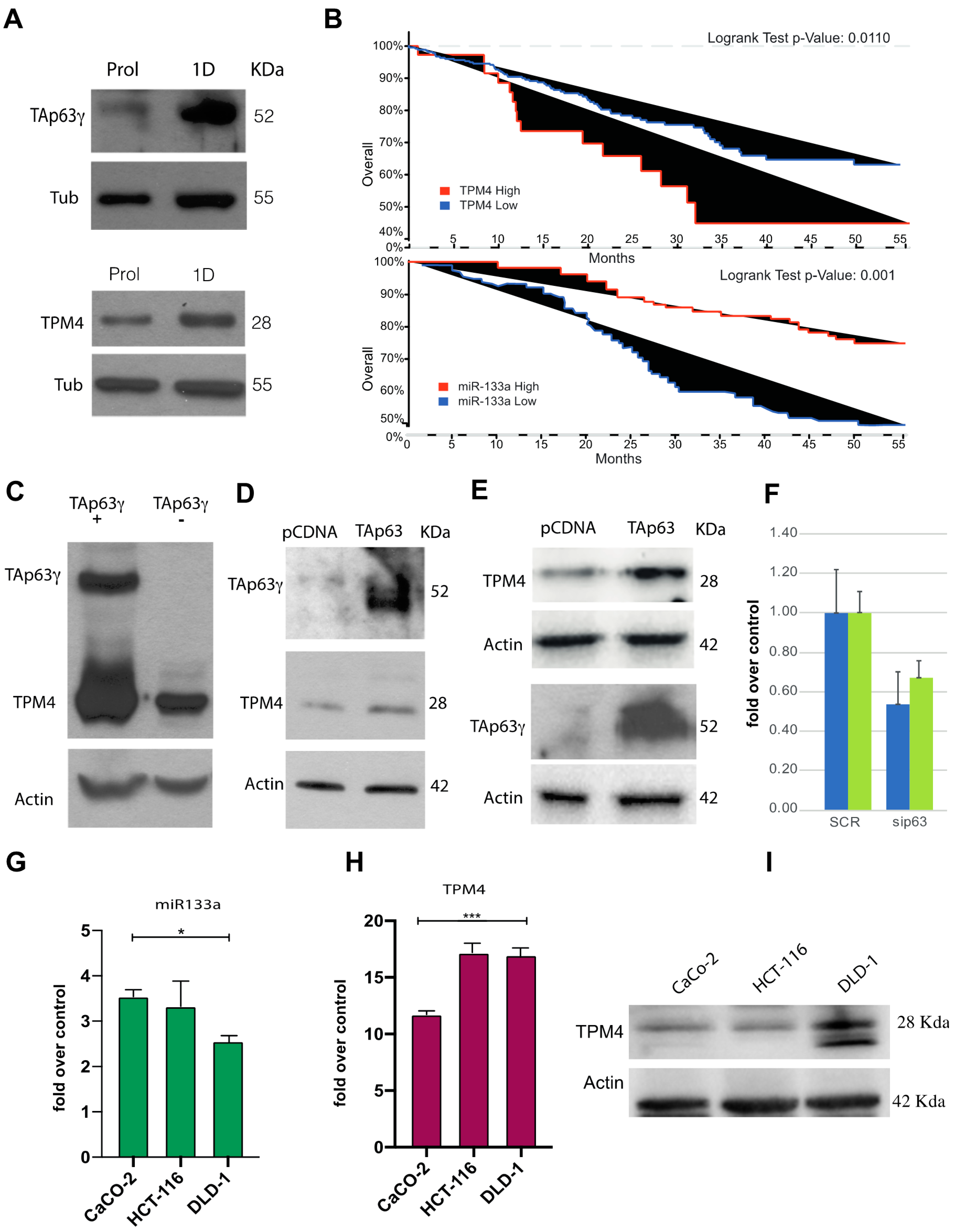
2.2. TPM4 as Possible miR-133a Target
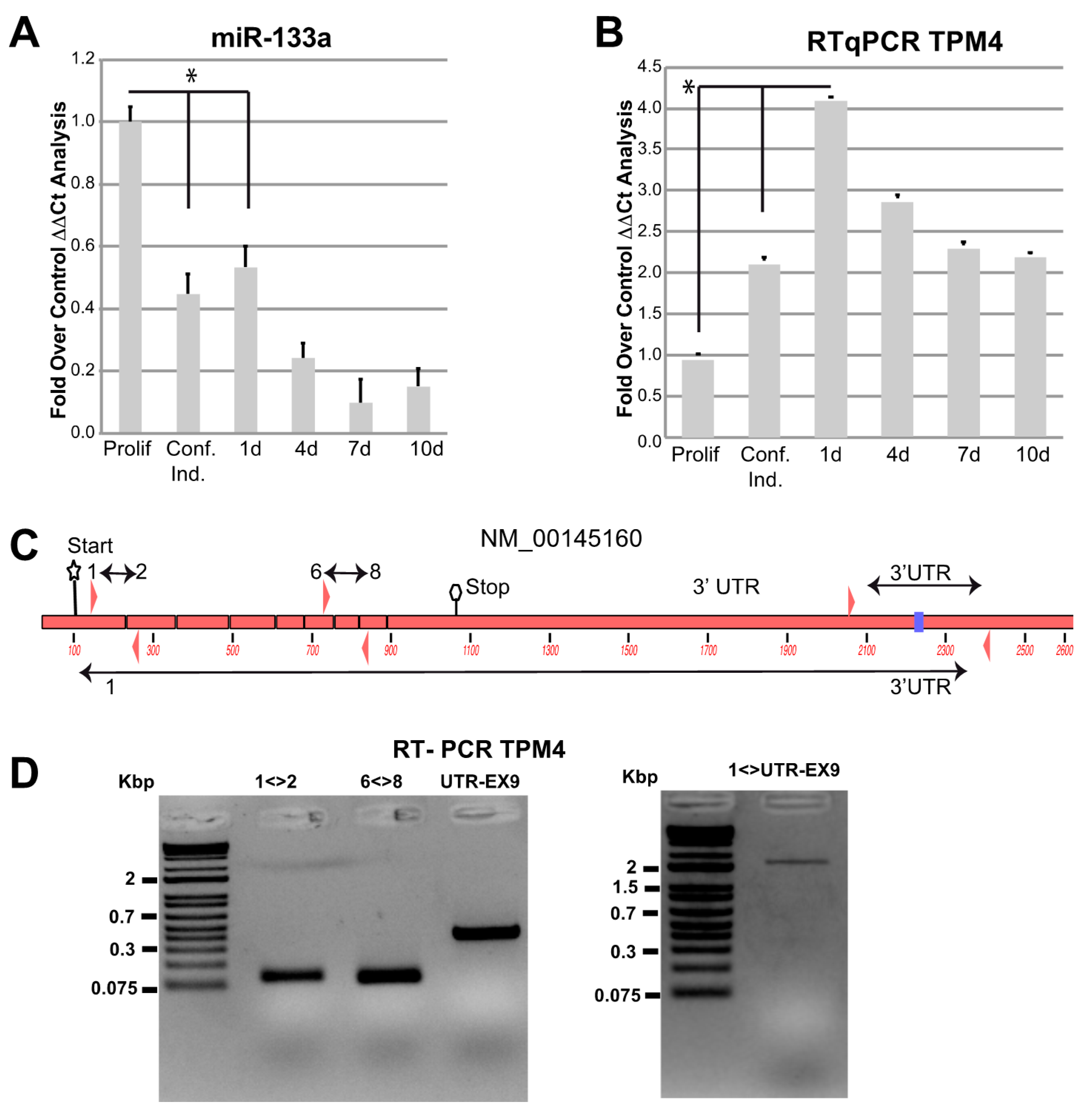
2.3. TPM4 and miR-133a in Myocyte Proliferation and Differentiation
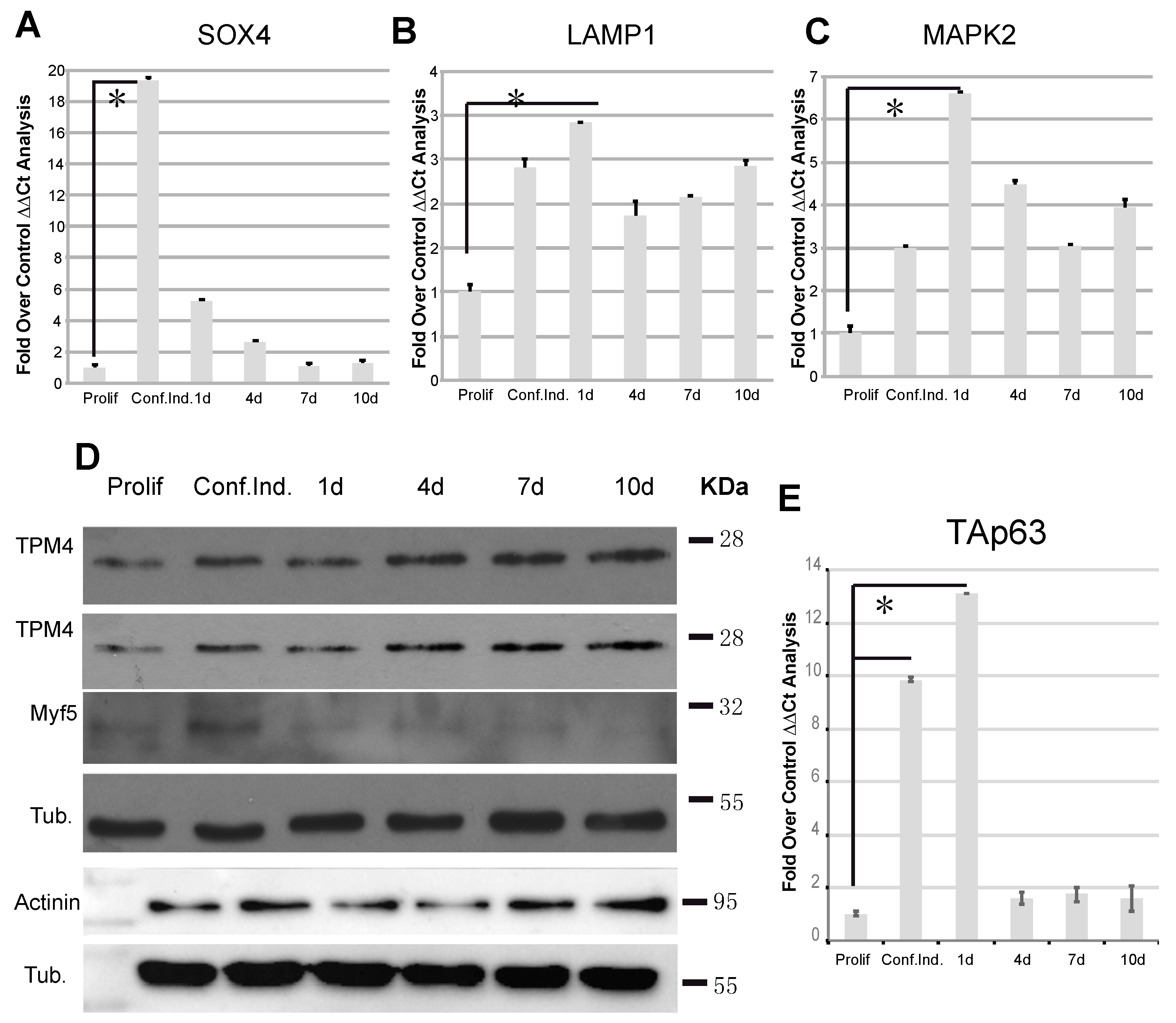
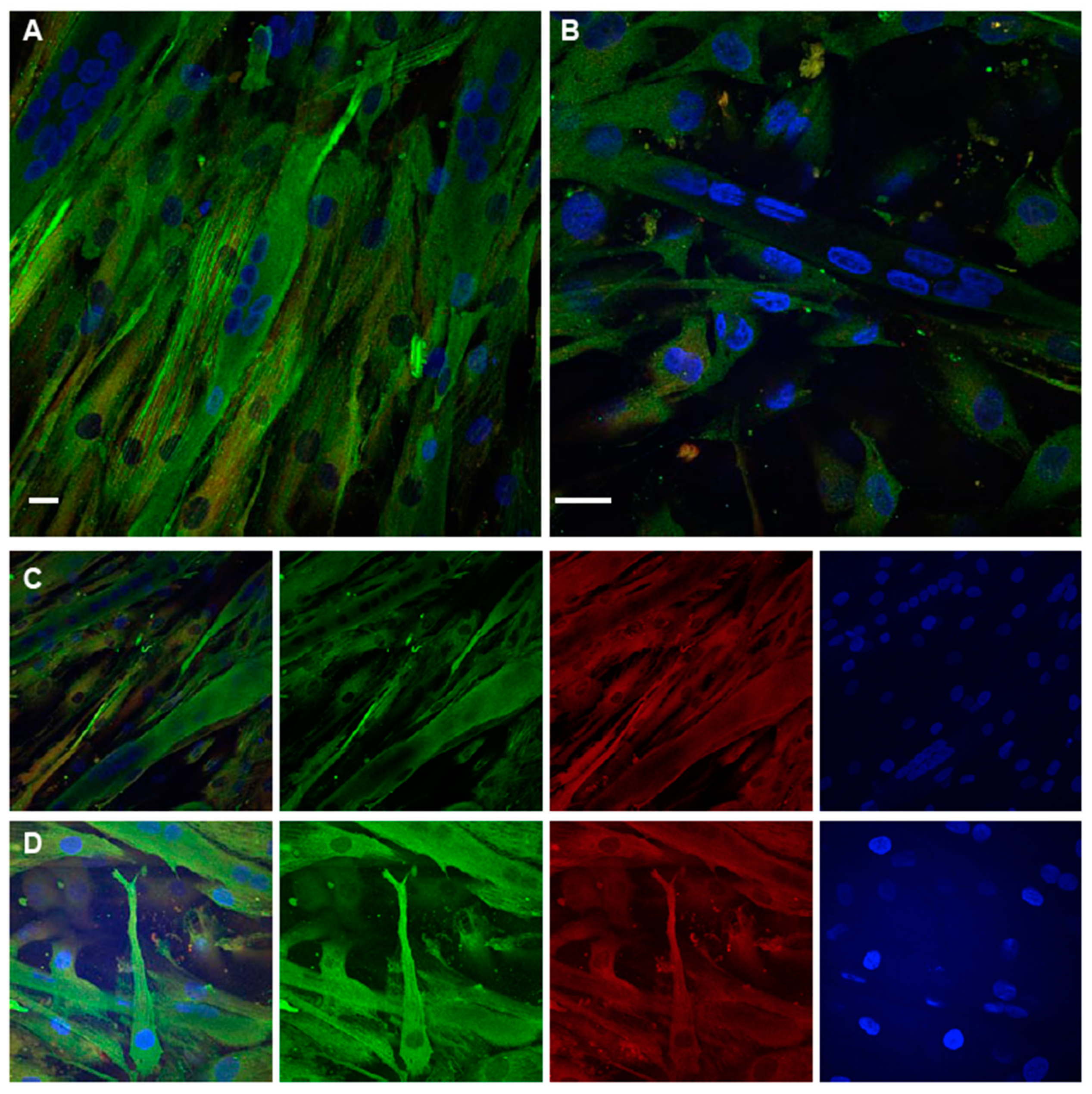
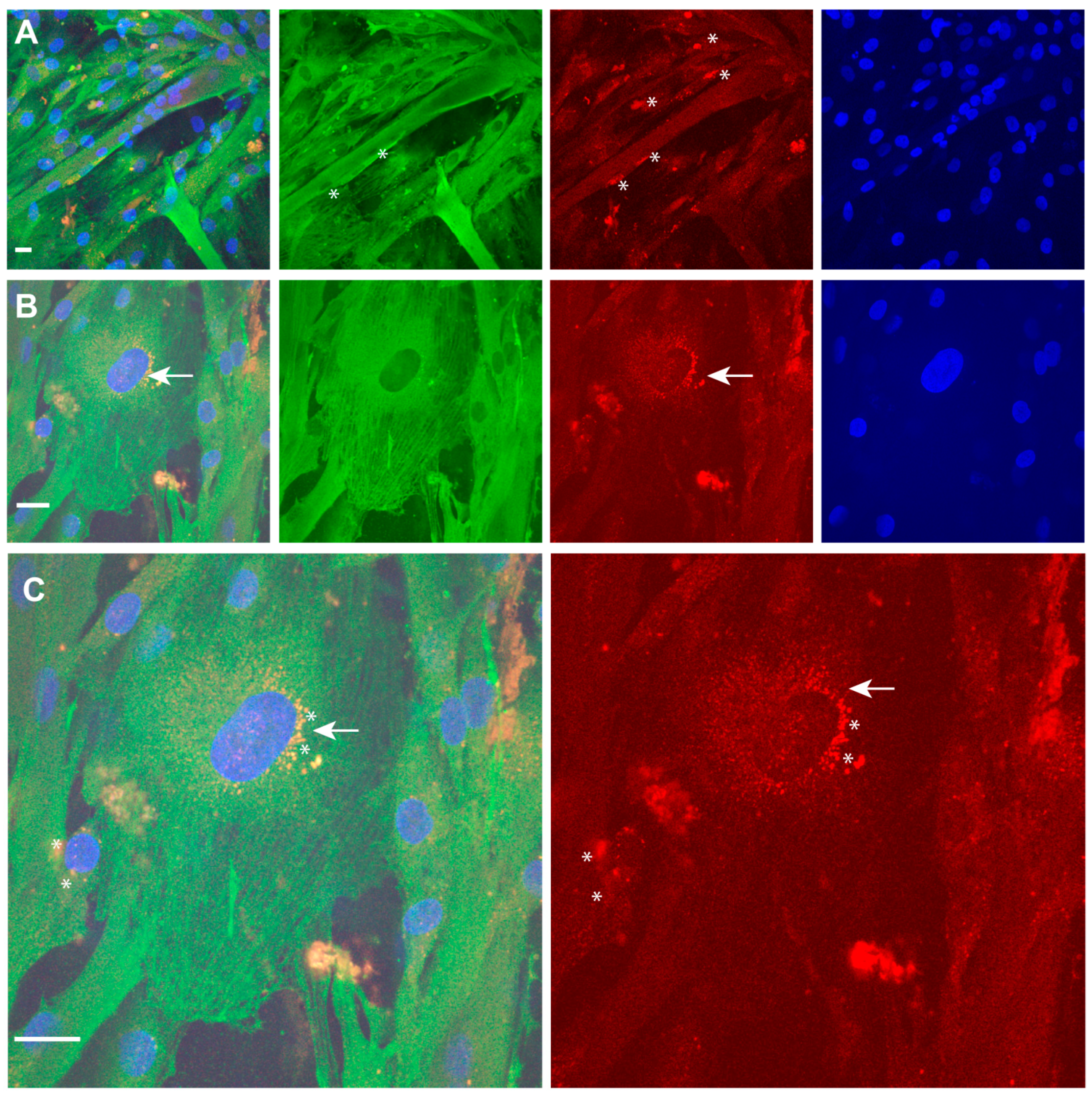
2.4. Immunofluorescence Analysis of TPM4 in Myocyte Differentiation
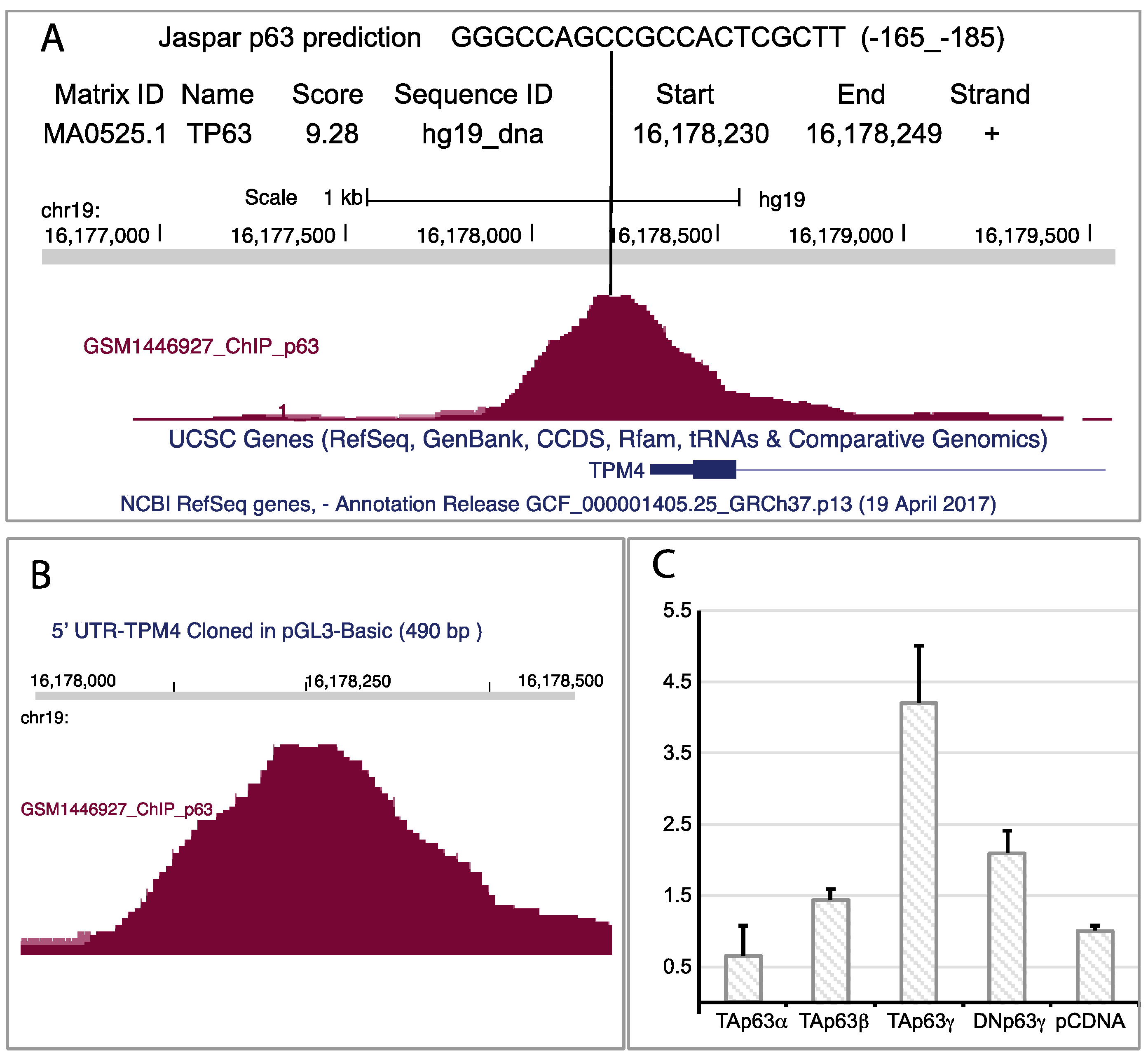
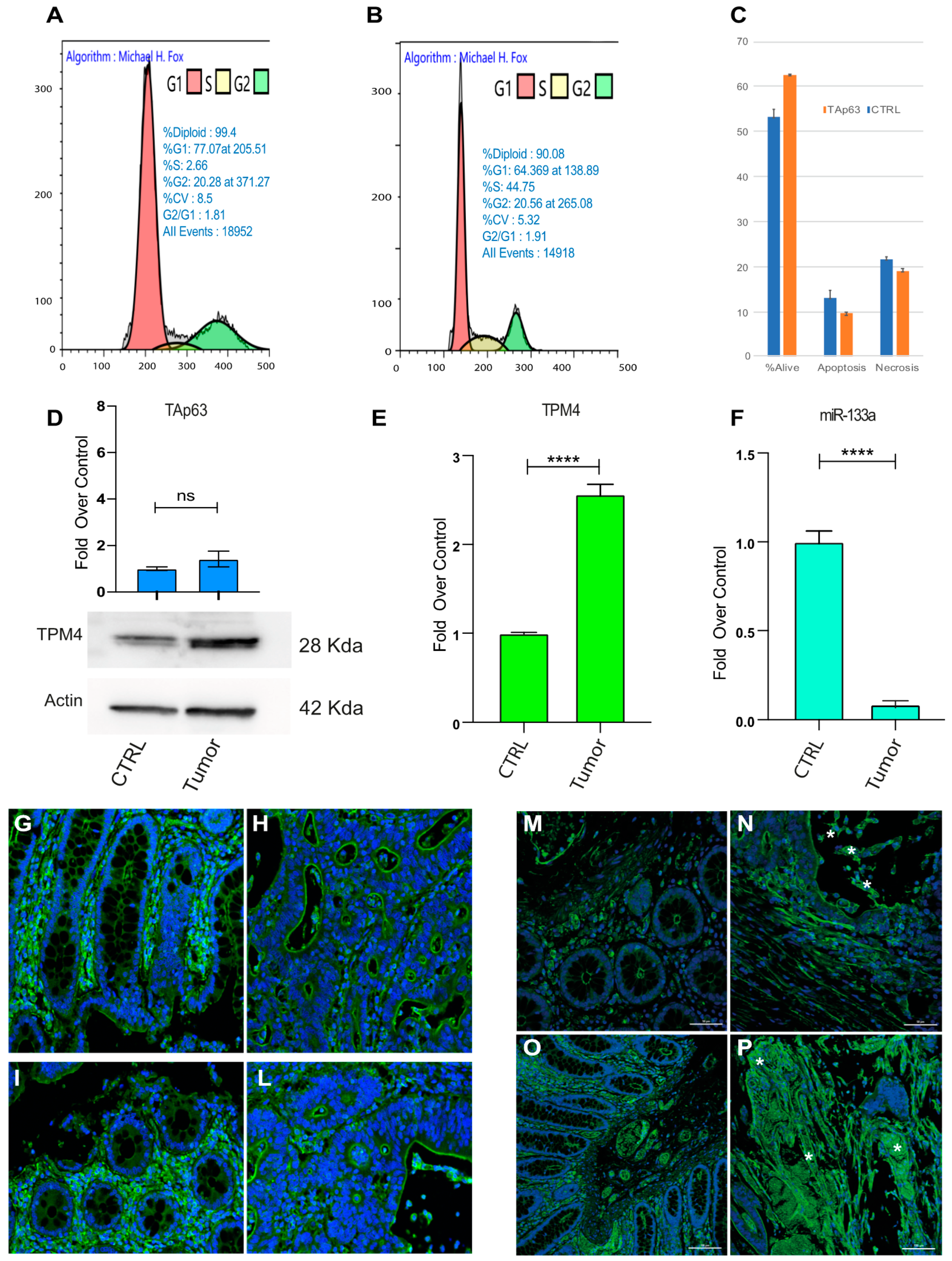
2.5. TAp63 Controls the Expression of TPM4 in Myocytes and in Colon Cancer Cells
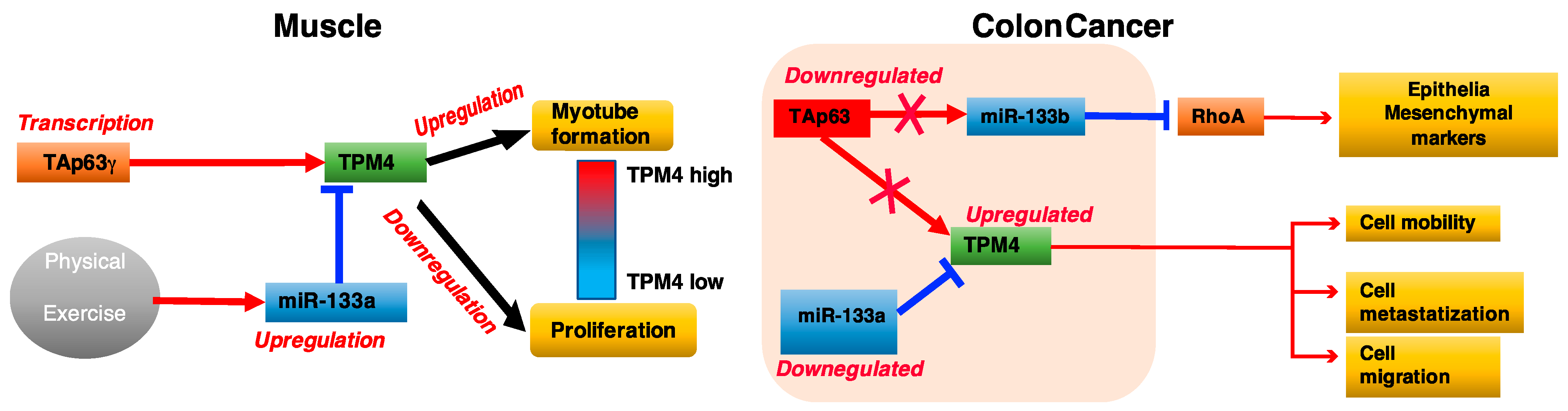
3. Discussion
4. Materials and Methods
4.1. Probands
4.2. Blood Sampling
4.3. MicroRNA Extraction from Plasma Samples
4.4. Exosome Isolation from Plasma
4.5. mRNA and miRNA Extraction from Cells
4.6. Protein Extraction
4.7. RT and Real Time PCR
4.8. Cloning and Luc-Assay
4.9. Isolation and Culture of Primary Human Muscle Cells
4.10. Culture Condition of Colon Cancer Cells
4.11. Cell Transfection
4.12. Cell Cycle Analysis
4.13. Immunofluorescence Analysis
4.14. Western Blotting
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Fife, C.M.; McCarroll, J.A.; Kavallaris, M. Movers and shakers: Cell cytoskeleton in cancer metastasis. Br. J. Pharm. 2014, 171, 5507–5523. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, G.; Helfman, D.M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 2001, 11, 41–47. [Google Scholar] [CrossRef]
- Rao, J.; Li, N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targets 2004, 4, 345–354. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers—Assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef]
- Zhao, X.; Jiang, M.; Wang, Z. TPM4 promotes cell migration by modulating F-actin formation in lung cancer. Onco. Targets 2019, 12, 4055–4063. [Google Scholar] [CrossRef]
- Sheng, Z.G.; Chen, M.H. TPM4 aggravates the malignant progression of hepatocellular carcinoma through negatively regulating SUSD2. Eur. Rev. Med. Pharm. Sci. 2020, 24, 4756–4765. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.Z.; Zhang, J.S.; Gong, J.X.; Wang, Y.H.; Zhang, C.L.; Chen, H.; Fang, X.T. Effects of microRNAs on skeletal muscle development. Gene 2018, 668, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. Causes and consequences of microRNA dysregulation. Cancer J. 2012, 18, 215–222. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.S.; El-Abd, Y.; Bui, K.; Hyun, Y.E.; Hughes, R.H.; Kraus, W.E.; Truskey, G.A. Conditions that promote primary human skeletal myoblast culture and muscle differentiation in vitro. Am. J. Physiol. Cell Physiol. 2014, 306, C385–C395. [Google Scholar] [CrossRef]
- Townley-Tilson, W.H.; Callis, T.E.; Wang, D. MicroRNAs 1, 133, and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int. J. Biochem. Cell Biol. 2010, 42, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Shelton, J.M.; Frisard, M.I.; Hulver, M.W.; McMillan, R.P.; Wu, Y.; Voelker, K.A.; Grange, R.W.; Richardson, J.A.; et al. Mice lacking microRNA 133a develop dynamin 2-dependent centronuclear myopathy. J. Clin. Investig. 2011, 121, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef]
- Cui, M.; Wang, H.; Yao, X.; Zhang, D.; Xie, Y.; Cui, R.; Zhang, X. Circulating MicroRNAs in Cancer: Potential and Challenge. Front. Genet. 2019, 10, 626. [Google Scholar] [CrossRef]
- Siracusa, J.; Koulmann, N.; Banzet, S. Circulating myomiRs: A new class of biomarkers to monitor skeletal muscle in physiology and medicine. J. Cachexia Sarcopenia Muscle 2018, 9, 20–27. [Google Scholar] [CrossRef]
- Hesari, A.; Azizian, M.; Darabi, H.; Nesaei, A.; Hosseini, S.A.; Salarinia, R.; Motaghi, A.A.; Ghasemi, F. Expression of circulating miR-17, miR-25, and miR-133 in breast cancer patients. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Zhang, C.; Yao, C.; Li, H.; Wang, G.; He, X. Serum levels of microRNA-133b and microRNA-206 expression predict prognosis in patients with osteosarcoma. Int. J. Clin. Exp. Pathol. 2014, 7, 4194–4203. [Google Scholar]
- Nohata, N.; Hanazawa, T.; Enokida, H.; Seki, N. microRNA-1/133a and microRNA-206/133b clusters: Dysregulation and functional roles in human cancers. Oncotarget 2012, 3, 9–21. [Google Scholar] [CrossRef]
- Deng, M.; Qin, Y.; Chen, X.; Wang, Q.; Wang, J. MiR-206 inhibits proliferation, migration, and invasion of gastric cancer cells by targeting the MUC1 gene. Onco. Targets 2019, 12, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yokobori, T.; Tanaka, N.; Sakai, M.; Sano, A.; Inose, T.; Sohda, M.; Nakajima, M.; Miyazaki, T.; Kato, H.; et al. CD47 expression regulated by the miR-133a tumor suppressor is a novel prognostic marker in esophageal squamous cell carcinoma. Oncol. Rep. 2012, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Chang, T.H.; Liu, Y.N.; Shih, J.Y. MicroRNA in Lung Cancer Metastasis. Cancers 2019, 11, 265. [Google Scholar] [CrossRef]
- Lan, D.; Zhang, X.; He, R.; Tang, R.; Li, P.; He, Q.; Chen, G. MiR-133a is downregulated in non-small cell lung cancer: A study of clinical significance. Eur. J. Med. Res. 2015, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Guo, Z.; Sun, J.; Li, J.; Dong, Z.; Zhang, Y.; Chen, J.; Kan, Q.; Yu, Z. MiR-133a acts as an anti-oncogene in Hepatocellular carcinoma by inhibiting FOSL2 through TGF-beta/Smad3 signaling pathway. Biomed. Pharm. 2018, 107, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Pan, J.; Huang, S.; Peng, X.; Zou, X.; Luo, Y.; Ren, D.; Zhang, X.; Li, R.; He, P.; et al. Downregulation of miR-133a-3p promotes prostate cancer bone metastasis via activating PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2018, 37, 160. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Dang, X.; Li, W.; Ma, Q. miR-133a functions as a tumor suppressor and directly targets FSCN1 in pancreatic cancer. Oncol. Res. 2013, 21, 353–363. [Google Scholar] [CrossRef]
- Navon, R.; Wang, H.; Steinfeld, I.; Tsalenko, A.; Ben-Dor, A.; Yakhini, Z. Novel rank-based statistical methods reveal microRNAs with differential expression in multiple cancer types. PLoS ONE 2009, 4, e8003. [Google Scholar] [CrossRef]
- Akcakaya, P.; Ekelund, S.; Kolosenko, I.; Caramuta, S.; Ozata, D.M.; Xie, H.; Lindforss, U.; Olivecrona, H.; Lui, W.O. miR-185 and miR-133b deregulation is associated with overall survival and metastasis in colorectal cancer. Int. J. Oncol. 2011, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Li, X.R.; Zhang, Y.; Hu, G.; Guo, Y.H.; Zhou, J.Y.; Du, J.; Lv, L.; Gao, K.; Zhang, Y.; et al. TAp63 suppress metastasis via miR-133b in colon cancer cells. Br. J. Cancer 2014, 110, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Y.; He, J.; Fu, Y.; Lin, C.; Li, X. MicroRNA-133b is regulated by TAp63 while no gene mutation is present in colorectal cancer. Oncol. Rep. 2017, 37, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Baggish, A.L.; Hale, A.; Weiner, R.B.; Lewis, G.D.; Systrom, D.; Wang, F.; Wang, T.J.; Chan, S.Y. Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J. Physiol. 2011, 589, 3983–3994. [Google Scholar] [CrossRef] [PubMed]
- Flowers, E.; Won, G.Y.; Fukuoka, Y. MicroRNAs associated with exercise and diet: A systematic review. Physiol. Genom. 2015, 47, 1–11. [Google Scholar] [CrossRef]
- Nielsen, S.; Scheele, C.; Yfanti, C.; Akerstrom, T.; Nielsen, A.R.; Pedersen, B.K.; Laye, M.J. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J. Physiol. 2010, 588, 4029–4037. [Google Scholar] [CrossRef] [PubMed]
- Polakovicova, M.; Musil, P.; Laczo, E.; Hamar, D.; Kyselovic, J. Circulating MicroRNAs as Potential Biomarkers of Exercise Response. Int. J. Mol. Sci. 2016, 17, 1553. [Google Scholar] [CrossRef]
- Cheng, G. Circulating miRNAs: Roles in cancer diagnosis, prognosis and therapy. Adv. Drug Deliv. Rev. 2015, 81, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Calabrese, C.; Basso, D.; Aita, A.; Caporali, S.; Plebani, M.; Bernardini, S. The circulating miRNAs as diagnostic and prognostic markers. Clin. Chem. Lab. Med. 2018. [Google Scholar] [CrossRef]
- Quadrilatero, J.; Hoffman-Goetz, L. Physical activity and colon cancer. A systematic review of potential mechanisms. J. Sports Med. Phys. Fit. 2003, 43, 121–138. [Google Scholar]
- Devin, J.L.; Hill, M.M.; Mourtzakis, M.; Quadrilatero, J.; Jenkins, D.G.; Skinner, T.L. Acute high intensity interval exercise reduces colon cancer cell growth. J. Physiol. 2019, 597, 2177–2184. [Google Scholar] [CrossRef]
- D’Ottavio, S.; Briotti, G.; Rosazza, C.; Partipilo, F.; Silvestri, A.; Calabrese, C.; Bernardini, A.; Gabrielli, P.R.; Ruscello, B. Effects of Two Modalities of Whole-Body Electrostimulation Programs and Resistance Circuit Training on Strength and Power. Int. J. Sports Med. 2019, 40, 831–841. [Google Scholar] [CrossRef]
- Basso, D.; Padoan, A.; Laufer, T.; Aneloni, V.; Moz, S.; Schroers, H.; Pelloso, M.; Saiz, A.; Krapp, M.; Fogar, P.; et al. Relevance of pre-analytical blood management on the emerging cardiovascular protein biomarkers TWEAK and HMGB1 and on miRNA serum and plasma profiling. Clin. Biochem. 2017, 50, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Tanihata, J.; Komaki, H.; Ishiyama, A.; Oya, Y.; Ruegg, U.; Takeda, S.I.; Hashido, K. Characterization and Functional Analysis of Extracellular Vesicles and Muscle-Abundant miRNAs (miR-1, miR-133a, and miR-206) in C2C12 Myocytes and mdx Mice. PLoS ONE 2016, 11, e0167811. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Lim, S.; Schevzov, G.; Gunning, P.W.; Helfman, D.M. Loss of Tpm4.1 leads to disruption of cell-cell adhesions and invasive behavior in breast epithelial cells via increased Rac1 signaling. Oncotarget 2017, 8, 33544–33559. [Google Scholar] [CrossRef]
- Geeves, M.A.; Hitchcock-DeGregori, S.E.; Gunning, P.W. A systematic nomenclature for mammalian tropomyosin isoforms. J. Muscle Res. Cell Motil. 2015, 36, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.K.; Dube, S.; Abbott, L.; Alshiekh-Nasany, R.; Mitschow, C.; Poiesz, B.J. Cloning, Sequencing, and the Expression of the Elusive Sarcomeric TPM4alpha Isoform in Humans. Mol. Biol. Int. 2016, 2016, 3105478. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.M.; Kim, J.W.; Kim, C.H.; An, J.H.; Johnson, A.; Song, P.I.; Rhee, S.; Choi, K.H. KAT5-mediated SOX4 acetylation orchestrates chromatin remodeling during myoblast differentiation. Cell Death Dis. 2015, 6, e1857. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qin, X.; Li, Y.; Zhang, X.; Niu, R.; Zhang, H.; Cui, A.; An, W.; Wang, X. MiR-133a suppresses the migration and invasion of esophageal cancer cells by targeting the EMT regulator SOX4. Am. J. Transl. Res. 2015, 7, 1390–1403. [Google Scholar]
- Jang, S.M.; Kim, C.H.; Kim, J.W.; Choi, K.H. Transcriptional regulatory network of SOX4 during myoblast differentiation. Biochem. Biophys. Res. Commun. 2015, 462, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef] [PubMed]
- Palombo, R.; Porta, G.; Bruno, E.; Provero, P.; Serra, V.; Neduri, K.; Viziano, A.; Alessandrini, M.; Micarelli, A.; Ottaviani, F.; et al. OTX2 regulates the expression of TAp63 leading to macular and cochlear neuroepithelium development. Aging 2015, 7, 928–936. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Terrinoni, A.; Serra, V.; Bruno, E.; Strasser, A.; Valente, E.; Flores, E.R.; van Bokhoven, H.; Lu, X.; Knight, R.A.; Melino, G. Role of p63 and the Notch pathway in cochlea development and sensorineural deafness. Proc. Natl. Acad. Sci. USA 2013, 110, 7300–7305. [Google Scholar] [CrossRef]
- Cefalu, S.; Lena, A.M.; Vojtesek, B.; Musaro, A.; Rossi, A.; Melino, G.; Candi, E. TAp63gamma is required for the late stages of myogenesis. Cell Cycle 2015, 14, 894–901. [Google Scholar] [CrossRef]
- Kouwenhoven, E.N.; Oti, M.; Niehues, H.; van Heeringen, S.J.; Schalkwijk, J.; Stunnenberg, H.G.; van Bokhoven, H.; Zhou, H. Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep. 2015, 16, 863–878. [Google Scholar] [CrossRef]
- Wang, L.L.; Du, L.T.; Li, J.; Liu, Y.M.; Qu, A.L.; Yang, Y.M.; Zhang, X.; Zheng, G.X.; Wang, C.X. Decreased expression of miR-133a correlates with poor prognosis in colorectal cancer patients. World J. Gastroenterol. 2014, 20, 11340–11346. [Google Scholar] [CrossRef]
- Sergeant, G.; van Eijsden, R.; Roskams, T.; Van Duppen, V.; Topal, B. Pancreatic cancer circulating tumour cells express a cell motility gene signature that predicts survival after surgery. BMC Cancer 2012, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, H.; Wang, B.; Liao, Q.; Li, W.; Jin, X.; Cui, S.; Zhang, Y.; Ding, Y.; Zhao, L. miR-133a represses tumour growth and metastasis in colorectal cancer by targeting LIM and SH3 protein 1 and inhibiting the MAPK pathway. Eur. J. Cancer 2013, 49, 3924–3935. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Xuan, Z.; Xu, P.; Wang, W.; Chen, Z.; Wang, S.; Sun, G.; Xu, J.; Xu, Z. Novel role of miR-133a-3p in repressing gastric cancer growth and metastasis via blocking autophagy-mediated glutaminolysis. J. Exp. Clin. Cancer Res. 2018, 37, 320. [Google Scholar] [CrossRef]
- Sui, Y.; Zhang, X.; Yang, H.; Wei, W.; Wang, M. MicroRNA-133a acts as a tumour suppressor in breast cancer through targeting LASP1. Oncol. Rep. 2018, 39, 473–482. [Google Scholar] [CrossRef]
- Hanson, J.; Lowy, J. The Structure of Actin Filaments and the Origin of the Axial Periodicity in the I-Substance of Vertebrate Striated Muscle. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1964, 160, 449–460. [Google Scholar] [CrossRef]
- Gateva, G.; Kremneva, E.; Reindl, T.; Kotila, T.; Kogan, K.; Gressin, L.; Gunning, P.W.; Manstein, D.J.; Michelot, A.; Lappalainen, P. Tropomyosin Isoforms Specify Functionally Distinct Actin Filament Populations In Vitro. Curr. Biol. 2017, 27, 705–713. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Schevzov, G.; Hotulainen, P.; Naumanen, P.; Martin, C.; Gunning, P.W.; Lappalainen, P. A molecular pathway for myosin II recruitment to stress fibers. Curr. Biol. 2011, 21, 539–550. [Google Scholar] [CrossRef]
- Hendricks, M.; Weintraub, H. Tropomyosin is decreased in transformed cells. Proc. Natl. Acad. Sci. USA 1981, 78, 5633–5637. [Google Scholar] [CrossRef]
- Mun, H.; Jeon, T.J. Regulation of actin cytoskeleton by Rap1 binding to RacGEF1. Mol. Cells 2012, 34, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.O.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.; Perez-Atayde, A.; Hibbard, M.K.; Rubin, B.P.; Dal Cin, P.; Pinkus, J.L.; Pinkus, G.S.; Xiao, S.; Yi, E.S.; Fletcher, C.D.; et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am. J. Pathol. 2000, 157, 377–384. [Google Scholar] [CrossRef]
- Pleines, I.; Woods, J.; Chappaz, S.; Kew, V.; Foad, N.; Ballester-Beltran, J.; Aurbach, K.; Lincetto, C.; Lane, R.M.; Schevzov, G.; et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J. Clin. Investig. 2017, 127, 814–829. [Google Scholar] [CrossRef]
- Dong, Y.; Zhao, J.; Wu, C.W.; Zhang, L.; Liu, X.; Kang, W.; Leung, W.W.; Zhang, N.; Chan, F.K.; Sung, J.J.; et al. Tumor suppressor functions of miR-133a in colorectal cancer. Mol. Cancer Res. 2013, 11, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Melino, G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011, 18, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, A.; Xiong, L.; Chen, T.; Tao, F.; Lu, Y.; He, Q.; Zhao, L.; Ou, R.; Xu, Y. miR-133a acts as a tumor suppressor in colorectal cancer by targeting eIF4A1. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, G. The role of miR-133a-3p/SP1/IGF1R axis in the progression of colorectal cancer. Ann. Oncol. 2020, 31, s1277. [Google Scholar] [CrossRef]
- Currier, M.A.; Stehn, J.R.; Swain, A.; Chen, D.; Hook, J.; Eiffe, E.; Heaton, A.; Brown, D.; Nartker, B.A.; Eaves, D.W.; et al. Identification of Cancer-Targeted Tropomyosin Inhibitors and Their Synergy with Microtubule Drugs. Mol. Cancer Ther. 2017, 16, 1555–1565. [Google Scholar] [CrossRef]
- Serra, V.; Castori, M.; Paradisi, M.; Bui, L.; Melino, G.; Terrinoni, A. Functional characterization of a novel TP63 mutation in a family with overlapping features of Rapp-Hodgkin/AEC/ADULT syndromes. Am. J. Med. Genet. A 2011, 155, 3104–3109. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K. A new program for investigating adult human skeletal muscle grown aneurally in tissue culture. Neurology 1975, 25, 58–67. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caporali, S.; Calabrese, C.; Minieri, M.; Pieri, M.; Tarantino, U.; Marini, M.; D’Ottavio, S.; Angeletti, S.; Mauriello, A.; Cortese, C.; et al. The miR-133a, TPM4 and TAp63γ Role in Myocyte Differentiation Microfilament Remodelling and Colon Cancer Progression. Int. J. Mol. Sci. 2021, 22, 9818. https://doi.org/10.3390/ijms22189818
Caporali S, Calabrese C, Minieri M, Pieri M, Tarantino U, Marini M, D’Ottavio S, Angeletti S, Mauriello A, Cortese C, et al. The miR-133a, TPM4 and TAp63γ Role in Myocyte Differentiation Microfilament Remodelling and Colon Cancer Progression. International Journal of Molecular Sciences. 2021; 22(18):9818. https://doi.org/10.3390/ijms22189818
Chicago/Turabian StyleCaporali, Sabrina, Cosimo Calabrese, Marilena Minieri, Massimo Pieri, Umberto Tarantino, Mario Marini, Stefano D’Ottavio, Silvia Angeletti, Alessandro Mauriello, Claudio Cortese, and et al. 2021. "The miR-133a, TPM4 and TAp63γ Role in Myocyte Differentiation Microfilament Remodelling and Colon Cancer Progression" International Journal of Molecular Sciences 22, no. 18: 9818. https://doi.org/10.3390/ijms22189818
APA StyleCaporali, S., Calabrese, C., Minieri, M., Pieri, M., Tarantino, U., Marini, M., D’Ottavio, S., Angeletti, S., Mauriello, A., Cortese, C., Bernardini, S., & Terrinoni, A. (2021). The miR-133a, TPM4 and TAp63γ Role in Myocyte Differentiation Microfilament Remodelling and Colon Cancer Progression. International Journal of Molecular Sciences, 22(18), 9818. https://doi.org/10.3390/ijms22189818